

Abstract
Alcohol abuse is a major cause of pancreatitis, a condition that can manifest as both acute necroinflammation and chronic damage (acinar atrophy and fibrosis). Pancreatic acinar cells can metabolize ethanol via the oxidative pathway, which generates acetaldehyde and involves the enzymes alcohol dehydrogenase and possibly cytochrome P4502E1. Additionally, ethanol can be metabolized via a nonoxidative pathway involving fatty acid ethyl ester synthases. Metabolism of ethanol by acinar and other pancreatic cells and the consequent generation of toxic metabolites, are postulated to play an important role in the development of alcohol-related acute and chronic pancreatic injury. This current work will review some recent advances in the knowledge about ethanol actions on the exocrine pancreas and its relationship to inflammatory disease and cancer.
Keywords: Pancreas, Calcium, Ethanol, Reactive oxygen species, Pancreatitis
INTRODUCTION
Pancreatitis is a potentially fatal disease in which the pancreas digests itself and its surroundings. An intracellular calcium (Ca2+) overload, as well as the generation of reactive oxygen species (ROS), have been indicated as the elements responsible for the initiation of the inflammatory process in the gland. One of the main causes of pancreatitis is alcohol abuse.
In the exocrine pancreas, the activation of phospholipase C (PLC)-linked receptors (R) by secretagogues produces an increase in the concentration of inositol 1,4,5-trisphosphate (IP3) in the cytosol. IP3 in turn releases Ca2+ from cytoplasmic stores, leading to an increase in cytosolic free calcium concentration ([Ca2+]i)[1]. Ca2+ signals are not only a result of its release from intracellular stores but also a co-ordinate influx from the extracellular space[2], Ca2+ extrusion across the plasma membrane[3] and Ca2+ uptake into intracellular organelles[4].
Ca2+ is a universal intracellular messenger that controls a wide range of cellular processes and does so by partitioning its actions in space and time. The coordination of the movement of Ca2+ is used to shape Ca2+ signals[5]. A rise in [Ca2+]i is an important early signal by which physiological secretagogues elicit the release of digestive enzymes from pancreatic acinar cells. An impairment of secretion would lead to intracellular activation of digestive enzymes which, in turn, could initiate auto-digestion of the gland and surrounding tissues and to the establishment of an inflammatory disease[6,7].
Mitogen-activated protein kinase (MAPK) family members mediate a wide variety of cellular processes in response to extracellular stimuli. Once MAPKs are activated, they phosphorylate target molecules in the cytoplasm and nucleus, resulting in the regulation of gene expression concerned with proliferation and differentiation[8]. On the other hand, Tumour Necrosis Factor (TNF) is a multifunctional proinflammatory cytokine with effects on lipid metabolism, coagulation, insulin resistance and endothelial function. Members of the TNF Receptor (TNFR) superfamily can send both survival and death signals to cells[9]. TNF family members play important roles in various physiological and pathological processes, including cell proliferation, differentiation, apoptosis and modulation of immune responses and induction of inflammation.
The exocrine pancreas can metabolize ethanol mainly via an oxidative pathway involving the enzymes alcohol dehydrogenase and cytochrome P4502E1, although a non-oxidative pathway involving fatty acid ethyl ester synthesis has been also proposed[10]. Therefore, metabolism of ethanol by pancreatic acinar cells and the consequent generation of toxic metabolites are postulated to play an important role in the development of alcohol-related pancreatic injury.
ETHANOL AND CALCIUM HOMEOSTASIS
The generation of repetitive local cytosolic Ca2+ signals in the apical pole of the pancreatic acinar cell is the starting point for the regulation of cellular function. Nevertheless, despite being one of the initial steps involved in cellular function, global and sustained changes in [Ca2+]i that are abnormal cytosolic Ca2+ signals, can result in necrosis. The release of Ca2+ through specific channels and the inhibition of Ca2+ pumps in intracellular stores, followed by entry of extracellular Ca2+, contribute to Ca2+ overload[11,12]. Additionally, it has been proposed that abnormally elevated [Ca2+]i is a shared phenomenon that could induce trypsin premature activation[13-15], which is a previous step that can trigger acute pancreatitis[12,16].
The actions of ethanol on Ca2+ homeostasis are currently under study. Its effects might be due to a direct action of ethanol on Ca2+ handling mechanisms or to an indirect effect, mediated by a production of ROS following ethanol metabolization or by non-oxidative metabolites of ethanol. We have shown that ethanol induces Ca2+ mobilization in mouse pancreatic acinar cells and that this mechanism is responsible for a ROS generation and a subsequent impairment of secretory function in this cell type[17].
A recent work shows that ethanol itself induces the release of Ca2+ from intracellular stores in the form of oscillations[18]. This effect was observed at doses ranging from 1 to 50 mmol/L. It has been shown that 50 mmol/L is a concentration within the range of blood alcohol levels in intoxicated humans[19]. Figure 1 shows an example of [Ca2+]i oscillations evoked by ethanol. In addition, it is possible that ethanol sensitizes the tissue to physiological agonists[20]. Therefore, a transformation of physiologically evoked oscillations in [Ca2+]i by agonists into a single transient signal[21] will be expected. As a consequence, ethanol leads to an increase in the total Ca2+ mobilization in response to the agonist[11].
Figure 1.
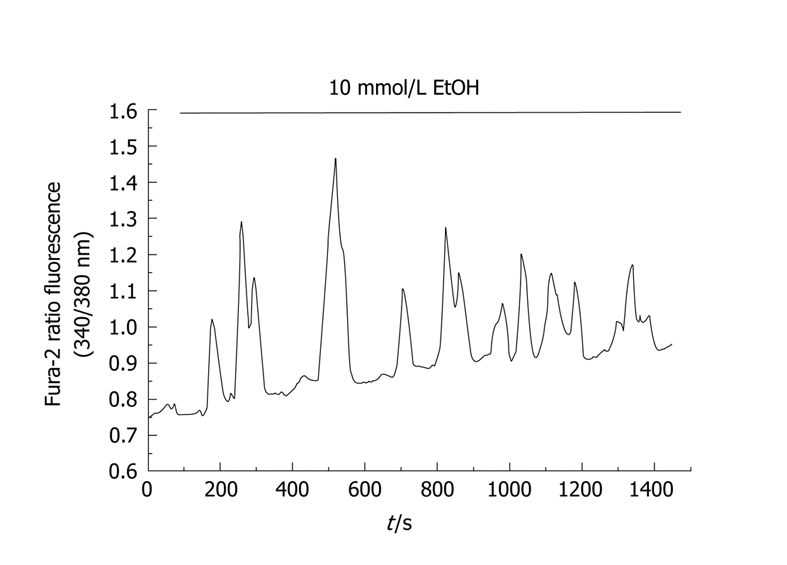
Time-course of changes in [Ca2+]i in response to ethanol. Cells were loaded with the fluorescent probe fura-2. Changes in fluorescence emitted by the fluorophore reflect changes in [Ca2+]i. In this setup, pancreatic acinar cells were stimulated with 10 mmol/L ethanol, which induced an oscillatory pattern in [Ca2+]i. The horizontal bar indicates the time during which ethanol was applied to the cells. (nm, nanometers).
In this sense, ethanol might present a direct action on the Ca2+ releasing mechanisms, whereas, on the other hand, it might be reducing the action of the pumps that extrude Ca2+ from the cytosol i.e. the plasma membrane Ca2+-ATPase (PMCA) and the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA). A slowing down of the activity of these pumps would lead to delayed Ca2+ extrusion out from the cytosol and, therefore, to an accumulation of the ion within the cytosol. This inhibition of Ca2+ pumping activity is probably due to the generation of ROS by ethanol metabolism[11,18], as will be discussed in the following section.
On the other hand, the actions of ethanol could be directed towards the mechanisms involved in Ca2+ influx. Further evidences for a cytosolic Ca2+ overload in the presence of ethanol come from the work by Del Castillo-Vaquero et al[21], who show that Ca2+ entry into the cells is increased in the presence of ethanol. This effect is also due to the generation of free radicals. This increased Ca2+ influx into the cells might be responsible for the potentiation of [Ca2+]i signals in response to physiological concentrations of cholecystokinin.
However, controversy exists and there are works that show little or no effect of ethanol on Ca2+ signalling. These studies propose an indirect action of ethanol on Ca2+ homeostasis. In this case, non-oxidative metabolites of ethanol (fatty acid ethyl esters and fatty acids) are those who evoke repetitive short-lasting [Ca2+]i spikes. In addition, fatty acids elicit a marked reduction in the cytosolic adenosine triphosphate (ATP) level, pointing towards the mitochondria as the putative point of action[12,22]. More recently, it has been proposed that these metabolites of ethanol release Ca2+ from the thapsigargin-sensitive ER as well as from a bafilomycin-sensitive acid compartment, which is localized exclusively in the apical granular pole. The emptying of this acidic compartment is linked to intracellular activation of digestive enzymes[14].
Putting all these observations together it can be seen that the actions of ethanol on pancreatic acinar cells create a situation potentially leading to a Ca2+ overload, a critical process in the initiation of alcohol-related acute pancreatitis.
ETHANOL AND REACTIVE OXYGEN SPECIES
ROS are a molecular group that can be produced in the course of different physiological processes and can react with a large variety of oxidizable cellular components. Thus, ROS have been considered as pathogenic factors in a variety of tissues and cells, including the pancreas[23,24]. Now there is growing evidence indicating that the exocrine pancreas is vulnerable to damage from ROS generated by ethanol metabolism[25].
It has been proposed that ethanol induces generation of oxygen radicals in pancreatic acinar cells[11,26,27]. Indeed, ethanol leads to an increase in fluorescence of CM-H2DCFDA, reflecting an increase in oxidation. A decrease in oxidation was observed in the absence of extracellular Ca2+ and in the presence of the Ca2+ chelator ethylene glycol-bis (2-aminoethylether)-N,N,N´,N´-tetraacetic acid (EGTA), indicating a Ca2+-mediated process for ethanol-evoked ROS generation. Similarly, when the cells were challenged in the presence of the intracellular Ca2+ chelator 1,2-Bis (2-amino-5-methylphenoxy) ethane-N,N,N´,N´-tetraacetic acid (BAPTA) and in the absence of extracellular Ca2+, the responses to ethanol were reduced. Thus, ethanol might exert its deleterious effects, at least in part, via generation of ROS[17]. Recently, it has been shown that oxidative metabolization of ethanol by alcohol-dehydrogenase leads to the generation of free radicals in pancreatic acinar cells[18,21]. Additionally, ethanol leads to a delayed extrusion of Ca2+ from the cytosol via generation of ROS[11,18]. However, the involvement of other enzymes and other metabolical routes in ethanol-derived free radicals generation cannot be excluded. Involvement of NADPH oxidase activity in ethanol-mediated ROS generation has been demonstrated. Evidence regarding this point derives from the work by Hu et al[28], who show that ethanol augments the activation of the cell’s NADPH oxidase system stimulated by platelet-derived growth factor PDGF and causes proliferation of stellate cells. Activated stellate cells are considered the principal mediators of chronic alcoholic pancreatitis/fibrosis.
Figure 2 shows the time course of changes in CM-H2DCFDA-derived fluorescence in response to ethanol. CM-H2DCFDA is a stable nonfluorescent molecule that passively diffuses into cells where the acetate can be cleaved by intracellular esterases to produce a polar diol that is well retained within the cells. The diol can then be oxidized by ROS to a fluorescent form. This dye has been proved to be an excellent probe for determination of ROS production[29,30].
Figure 2.
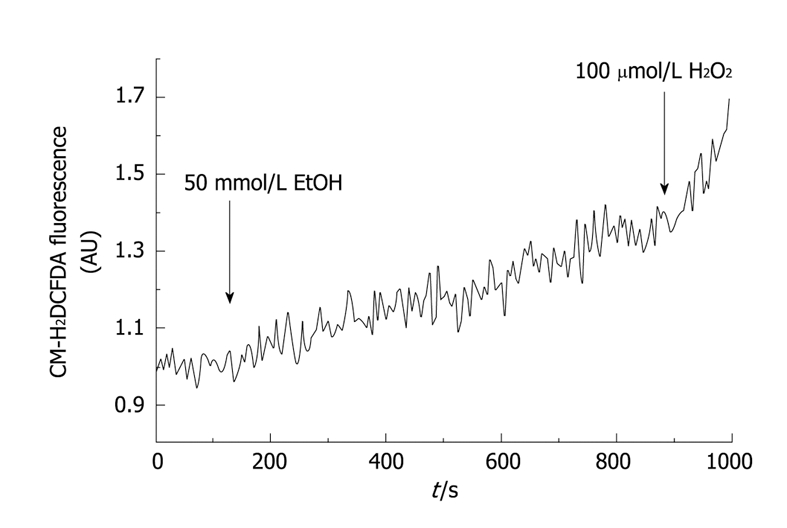
Time-course of ethanol-evoked reactive oxygen species (ROS) production in mouse pancreatic acinar cells. Pancreatic acinar cells were loaded with CM-H2DCFDA, a stable non-fluorescent molecule that yields a polar diol that is well retained within the cells. The diol can then be oxidized by ROS to a fluorescent form; therefore this dye has been proved to be an excellent probe for determination of ROS production. In this setup, stimulation of cells with 50 mmol/L ethanol (EtOH) led to a significant increase in ROS generation. At the end of the experiment H2O2 (100 μmol/L) was added, as a positive control for oxidation. a.u., absolute units of fluorescence.
ETHANOL AND CELLULAR PROLIFERATION
Moderate and high alcohol intake levels over a lifetime might increase cancer risk[31]. MAPK family members, including extracellular signal-regulated protein kinase (Erk), p38MAPK (p38) and c-jun NH2-terminal kinase (JNK), mediate a wide variety of cellular processes in response to extracellular stimuli. Once MAPKs are activated, they phosphorylate target molecules in the cytoplasm and nucleus, resulting in the regulation of gene expression concerned with proliferation, migration, extracellular matrix degradation and differentiation[8]. JNK and p38MAPK family members function in a cell context-specific and cell type-specific manner to integrate signals that affect these events. Consistent with the importance of these processes in tumorigenesis, JNK and p38MAPK signalling is associated with cancer[32].
To date there are several studies that implicate MAPK pathway as a critical regulator of the effects of ethanol and its metabolite, acetaldehyde, on acinar cells. Implication of the MAPK pathway as a critical regulator of the effects of ethanol and acetaldehyde on acinar cells has been proposed[33]. Ethanol and acetaldehyde increased the activation of all 3 subfamilies (ERK 1/2, JNK and p38 kinase) of the MAPK pathway. Treatment of cells with the inhibitor SB203 580 abolished the ethanol- and acetaldehyde-induced increase in p38 MAPK activity[34]. Furthermore, ethanol- and acetaldehyde-induced activation of MAPs was blocked by the antioxidant N-acetyl-cysteine, suggesting a role of oxidative stress in the signal transduction[35].
One study suggests a potential role for these pathways in contributing to the development of alcohol-related pancreatic carcinogenesis. In this study, ethanol stimulation of cell proliferation was inhibited by inhibition of mitogen-activated protein kinase (ERK1/2) and by blocking epidermal growth factor receptor-specific tyrosine kinase[36].
On the other hand, the intracellular signalling mechanisms regulating ethanol-induced cellular activation include the MAPK pathway and the factors responsible for mediating cell activation include ethanol itself, its metabolite acetaldehyde, oxidant stress and cytokines released during episodes of alcohol-induced pancreatic necroinflammation[37].
ETHANOL AND ENZYME SECRETION
The premature activation of digestive proenzymes, specifically proteases, within the pancreatic acinar cell is an early and critical event during acute pancreatitis. One of the early events leading to alcoholic pancreatitis seems to be the effect of ethanol on stimulus-secretion coupling mechanisms. In pancreatic acinar cells, a number of PLC-acting secretagogues, such as acetylcholine and cholecystokinin, regulate secretion via activation of a number of kinases concomitantly with the generation of repetitive local cytosolic Ca2+ signals in the apical pole. This leads to the fusion of the secretory vesicles with the apical membrane of the acinar cell and the exocytosis of the content into the extracellular space[38].
Classically, it is known that ethanol causes a dose-dependent inhibition of enzyme synthesis without affecting exocytosis of preformed or newly synthesized protein. This is a direct inhibitory effect of ethanol and is not mediated by its metabolic processing[39]. However, treatment of pancreatic acini with ethanol does not induce any significant effect on amylase release at a wide range of concentrations (1-50 mmol/L)[17,39]. These results indicate that ethanol likely lacks a direct role in secretion although it decreases the enzymatic synthesis.
In contrast, ethanol can modulate the secretagogue-induced secretion. Incubation of cells with 50 mmol/L ethanol clearly reduces amylase release stimulated by CCK-8. The inhibitory effect of ethanol on CCK-8-induced amylase secretion was abolished by dithiothreitol, a sulfhydryl reducing agent, suggesting a ROS-mediated acton on ethanol effects[17]. The effect of ethanol in modulating the secretory response to CCK-8 could be related to the ability of ethanol to modulate the inflammatory response of the pancreas to low concentrations of CCK-8 (the molecular mechanism involved will be discussed further in the next section).
Data on the effects of ethanol on pancreatitis induced by high (supramaximal) concentration of CCK-8 are contradictory, with it being reported that alcohol can worsen[40] or produce no effect on fully developed pancreatitis[41]. However, the effect of ethanol treatment on the ability of low doses of CCK-8 to produce pancreatic damage has been clearly demonstrated[42]. The idea that ethanol sensitizes the pancreas to the action of low doses of the hormone agrees with the results of in vitro experiments. Pancreatic acinar cells isolated from rats fed ethanol for 9-12 mo were found more susceptible to cerulein-induced activation of trypsinogen and chymotrypsinogen than pancreatic acini from pair-fed control rats[42,43]. In another study, ethanol with a low dose of CCK-8 but not ethanol alone was found to generate zymogen conversion that was 6-fold higher than that caused by CCK-8 alone[44]. In summary, all these studies show that ethanol diet sensitizes rats to the development of hormone-induced pancreatitis.
ETHANOL AND INFLAMMATION
Over the past several years, evidence has been accumulating on the involvement of inflammatory cytokines and chemokines in the development of pancreatitis[20,42,45,46]. It has been reported that the levels of IL-6 and TNF-α were up-regulated in pancreas from rats with experimental pancreatitis and that the blockade of these cytokines attenuates the disease[45-49]. Furthermore, a strong correlation was observed between the IL-6 level in serum and the severity of human pancreatitis[50,51].
It has been reported that ethanol acts to sensitize the pancreas to the deleterious effects of other stimuli such as the physiological agonist CCK-8, which then leads to an inflammatory response and pancreatitis. This effect is, in part, mediated by augmenting activation of proinflammatory factors[20,42]. It has been shown that rat cerulein pancreatitis is associated with rapid NF-κB activation and that NF-κB activation mediates intrapancreatic up-regulation of IL-6[49]. Interestingly, ethanol diet potentiates the ability of CCK-8 to activate NF-κB which in turns causes an increase in the cytokine expression, suggesting that activation of NF-κB can be one of the mechanisms for ethanol-induced cytokine up-regulation in the CCK-treated animals[20,42,45].
Another observed effect of ethanol consumption is that it alone attenuated pancreatic NF-κB and decreased the expression of IL-6, iNOS and MIP-2, all of which are regulated by NF-κB[42]. Furthermore, a decrease in the levels of prostaglandin E2 has been reported and could also be involved in alcohol-induced injury in the pancreas[52]. In summary, ethanol diet causes sensitization to CCK-8-induced activation of pancreatic NF-κB and cytokine/chemokine mRNA expression, and ethanol itself causes down-regulation of NF-κB activity and mRNA levels for certain cytokines and chemokines. Both mechanisms i.e. hormone sensitization and down-regulation of NF-κB, cytokines and chemokines, could be involved in the development of the pro-inflammatory effect of ethanol in the pancreas.
CONCLUSION
Pancreatic acinar cells and other pancreatic cells can metabolize ethanol and the consequent generation of toxic metabolites are postulated to play an important role in the development of alcohol-related acute and chronic pancreatic injury. Ethanol may itself, or through its oxidative or non-oxidative metabolites, lead to Ca2+ mobilization from intracellular stores, sensitization of the tissue to Ca2+-mobilizing agonists and/or decrease the activity of Ca2+ transport mechanisms. As a consequence, ethanol leads to cytosolic Ca2+ overload. Intracellular Ca2+ overload has been related to ROS over production which, in turn, can further increase cytosolic Ca2+ accumulation because oxidants impair Ca2+ handling by the cell. In addition, ethanol or its metabolites inhibit secretagogue-induced secretion of enzymes, that will then accumulate within the cell. Inhibition of zymogen secretion can lead to its intracellular activation, setting the starting point for autodigestion of the gland and a consequent inflammatory process. Moreover, ethanol causes an increase in the cytokine expression in response to agonists which represents a crosstalk with inflammation pathways. On the other hand, moderate and high alcohol intake levels over a lifetime might increase cancer risk through activation of a wide variety of cellular processes in response to extracellular stimuli that can led to tumorigenesis. The putative mechanisms of action of ethanol on pancreatic acinar cells physiology are summarised in Figure 3. In conclusion, ethanol impairs the exocrine pancreas function, creating a situation potentially leading to the development of pancreatic diseases.
Figure 3.
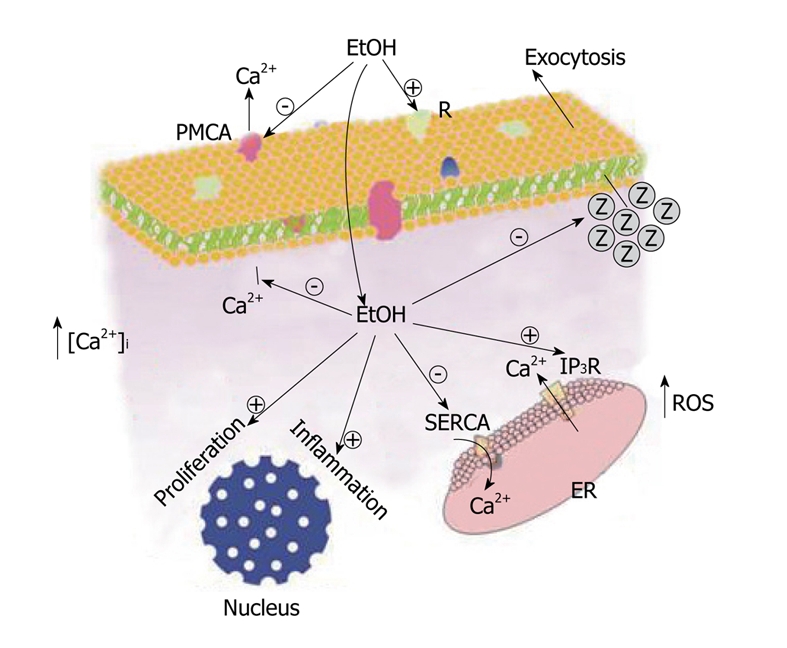
Putative mechanisms of action of ethanol on pancreatic acinar cells physiology. EtOH may, either itself or through its metabolites, sensitize the exocrine pancreas to physiological agonists. The point of action of ethanol may be at the R on the cell surface or intracellular. A stimulated Ca2+ release from the ER and a reduction in Ca2+ extrusion from the cytosol by the SERCA and/or the PMCA, will lead to accumulation of Ca2+ within the cytosol. This may lead to an overproduction of ROS which, in turn, will augment cytosolic Ca2+ accumulation, apart from other cellular effects. Cytosolic Ca2+ overload, together with ROS generation, may inhibit exocytosis of digestive enzymes (Z) that will accumulate inside the cell. Intracellular trapped digestive enzymes may be activated, and may initiate the autodigestion of the gland, establishing an inflammatory process. On the other hand, ethanol and/or its metabolites can activate intracellular routes for inflammation and/or cell proliferation, contributing to the impairment of cell function and to an uncontrolled cell growth. ER: Endoplasmic reticulum; SERCA: Sarco-endoplasmic reticulum Ca2+-ATPase; PMCA: Plasma membrane Ca2+-ATPase; IP3R: Inositol 1,4,5-trisphosphate.
Footnotes
Supported by Junta de Extremadura-FEDER, PRI08A018
Peer reviewer: Gianpiero Manes, MD, Department of Gastroenterology, University Hospital L. Sacco, Via G.B. Grassi 74, Milano I-20157, Italy
S- Editor Li LF L- Editor Roemmele A E- Editor Yang C
References
- 1.Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:67–69. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- 2.Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 3.Carafoli E. Calcium pump of the plasma membrane. Physiol Rev. 1991;71:129–153. doi: 10.1152/physrev.1991.71.1.129. [DOI] [PubMed] [Google Scholar]
- 4.González A, Camello PJ, Pariente JA, Salido GM. Free cytosolic calcium levels modify intracellular pH in rat pancreatic acini. Biochem Biophys Res Commun. 1997;230:652–656. doi: 10.1006/bbrc.1996.6026. [DOI] [PubMed] [Google Scholar]
- 5.Laude AJ, Simpson AW. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009;276:1800–1816. doi: 10.1111/j.1742-4658.2009.06927.x. [DOI] [PubMed] [Google Scholar]
- 6.Krüger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol. 2000;157:43–50. doi: 10.1016/S0002-9440(10)64515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci USA. 2000;97:13126–13131. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, Iwashita S, Kako K, Kishi T, Kasuya Y, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Kawasaki H, Onuki R, Suyama E, Taira K. Identification of genes that function in the TNF-alpha-mediated apoptotic pathway using randomized hybrid ribozyme libraries. Nat Biotechnol. 2002;20:376–380. doi: 10.1038/nbt0402-376. [DOI] [PubMed] [Google Scholar]
- 10.Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci USA. 2004;101:10738–10743. doi: 10.1073/pnas.0403431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.González A, Pariente JA, Salido GM. Ethanol impairs calcium homeostasis following CCK-8 stimulation in mouse pancreatic acinar cells. Alcohol. 2008;42:565–573. doi: 10.1016/j.alcohol.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006;27:113–120. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Ding YX, Yang K, Chin WC. Ethanol augments elevated-[Ca2+]C induced trypsin activation in pancreatic acinar zymogen granules. Biochem Biophys Res Commun. 2006;350:593–597. doi: 10.1016/j.bbrc.2006.09.086. [DOI] [PubMed] [Google Scholar]
- 14.Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Gerasimenko OV, Petersen OH. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci USA. 2009;106:10758–10763. doi: 10.1073/pnas.0904818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah AU, Sarwar A, Orabi AI, Gautam S, Grant WM, Park AJ, Shah AU, Liu J, Mistry PK, Jain D, et al. Protease Activation during in vivo Pancreatitis is Dependent upon Calcineurin Activation. Am J Physiol Gastrointest Liver Physiol. 2009:Epub ahead of print. doi: 10.1152/ajpgi.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward JB, Petersen OH, Jenkins SA, Sutton R. Is an elevated concentration of acinar cytosolic free ionised calcium the trigger for acute pancreatitis? Lancet. 1995;346:1016–1019. doi: 10.1016/s0140-6736(95)91695-4. [DOI] [PubMed] [Google Scholar]
- 17.González A, Núñez AM, Granados MP, Pariente JA, Salido GM. Ethanol impairs CCK-8-evoked amylase secretion through Ca2+-mediated ROS generation in mouse pancreatic acinar cells. Alcohol. 2006;38:51–57. doi: 10.1016/j.alcohol.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Fernández-Sánchez M, del Castillo-Vaquero A, Salido GM, González A. Ethanol exerts dual effects on calcium homeostasis in CCK-8-stimulated mouse pancreatic acinar cells. BMC Cell Biol. 2009;10:77. doi: 10.1186/1471-2121-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamarche F, Gonthier B, Signorini N, Eysseric H, Barret L. Impact of ethanol and acetaldehyde on DNA and cell viability of cultured neurones. Cell Biol Toxicol. 2004;20:361–374. doi: 10.1007/s10565-004-0087-9. [DOI] [PubMed] [Google Scholar]
- 20.Pandol SJ, Gukovsky I, Satoh A, Lugea A, Gukovskaya AS. Emerging concepts for the mechanism of alcoholic pancreatitis from experimental models. J Gastroenterol. 2003;38:623–628. doi: 10.1007/s00535-003-1134-7. [DOI] [PubMed] [Google Scholar]
- 21.Del Castillo-Vaquero A, Salido GM, González A. Increased calcium influx in the presence of ethanol in mouse pancreatic acinar cells. Int J Exp Pathol. 2009:Epub ahead of print. doi: 10.1111/j.1365-2613.2009.00691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Criddle DN, Sutton R, Petersen OH. Role of Ca2+ in pancreatic cell death induced by alcohol metabolites. J Gastroenterol Hepatol. 2006;21 Suppl 3:S14–S17. doi: 10.1111/j.1440-1746.2006.04577.x. [DOI] [PubMed] [Google Scholar]
- 23.Schoenberg MH, Büchler M, Beger HG. Oxygen radicals in experimental acute pancreatitis. Hepatogastroenterology. 1994;41:313–319. [PubMed] [Google Scholar]
- 24.Weber H, Roesner JP, Nebe B, Rychly J, Werner A, Schröder H, Jonas L, Leitzmann P, Schneider KP, Dummler W. Increased cytosolic Ca2+ amplifies oxygen radical-induced alterations of the ultrastructure and the energy metabolism of isolated rat pancreatic acinar cells. Digestion. 1998;59:175–185. doi: 10.1159/000007486. [DOI] [PubMed] [Google Scholar]
- 25.Palmieri VO, Grattagliano I, Palasciano G. Ethanol induces secretion of oxidized proteins by pancreatic acinar cells. Cell Biol Toxicol. 2007;23:459–464. doi: 10.1007/s10565-007-9007-0. [DOI] [PubMed] [Google Scholar]
- 26.Wilson JS, Apte MV. Role of alcohol metabolism in alcoholic pancreatitis. Pancreas. 2003;27:311–315. doi: 10.1097/00006676-200311000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Wittel UA, Bachem M, Siech M. Oxygen radical production precedes alcohol-induced acute pancreatitis in rats. Pancreas. 2003;26:e74–e80. doi: 10.1097/00006676-200305000-00018. [DOI] [PubMed] [Google Scholar]
- 28.Hu R, Wang YL, Edderkaoui M, Lugea A, Apte MV, Pandol SJ. Ethanol augments PDGF-induced NADPH oxidase activity and proliferation in rat pancreatic stellate cells. Pancreatology. 2007;7:332–340. doi: 10.1159/000105499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hempel SL, Buettner GR, O'Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2',7'-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2',7'-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic Biol Med. 1999;27:146–159. doi: 10.1016/s0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 30.Possel H, Noack H, Augustin W, Keilhoff G, Wolf G. 2,7-Dihydrodichlorofluorescein diacetate as a fluorescent marker for peroxynitrite formation. FEBS Lett. 1997;416:175–178. doi: 10.1016/s0014-5793(97)01197-6. [DOI] [PubMed] [Google Scholar]
- 31.Benedetti A, Parent ME, Siemiatycki J. Lifetime consumption of alcoholic beverages and risk of 13 types of cancer in men: results from a case-control study in Montreal. Cancer Detect Prev. 2009;32:352–362. doi: 10.1016/j.canep.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 33.Apte M, McCarroll J, Pirola R, Wilson J. Pancreatic MAP kinase pathways and acetaldehyde. Novartis Found Symp. 2007;285:200–211; discussion 211-216. doi: 10.1002/9780470511848.ch15. [DOI] [PubMed] [Google Scholar]
- 34.McCarroll JA, Phillips PA, Park S, Doherty E, Pirola RC, Wilson JS, Apte MV. Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas. 2003;27:150–160. doi: 10.1097/00006676-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 35.Masamune A, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J Pharmacol Exp Ther. 2002;302:36–42. doi: 10.1124/jpet.302.1.36. [DOI] [PubMed] [Google Scholar]
- 36.Askari MD, Tsao MS, Cekanova M, Schuller HM. Ethanol and the tobacco-specific carcinogen, NNK, contribute to signaling in immortalized human pancreatic duct epithelial cells. Pancreas. 2006;33:53–62. doi: 10.1097/01.mpa.0000226883.55828.e9. [DOI] [PubMed] [Google Scholar]
- 37.Apte MV, Pirola RC, Wilson JS. Battle-scarred pancreas: role of alcohol and pancreatic stellate cells in pancreatic fibrosis. J Gastroenterol Hepatol. 2006;21 Suppl 3:S97–S101. doi: 10.1111/j.1440-1746.2006.04587.x. [DOI] [PubMed] [Google Scholar]
- 38.Gardner JD, Jensen RT. Receptors and cell activation associated with pancreatic enzyme secretion. Annu Rev Physiol. 1986;48:103–117. doi: 10.1146/annurev.ph.48.030186.000535. [DOI] [PubMed] [Google Scholar]
- 39.Chapman BA, Pattinson NR. The effect of ethanol on enzyme synthesis and secretion in isolated rat pancreatic lobules. Biochem Pharmacol. 1987;36:3353–3360. doi: 10.1016/0006-2952(87)90310-8. [DOI] [PubMed] [Google Scholar]
- 40.Foitzik T, Lewandrowski KB, Fernández-del Castillo C, Rattner DW, Klar E, Warshaw AL. Exocrine hyperstimulation but not pancreatic duct obstruction increases the susceptibility to alcohol-related pancreatic injury. Arch Surg. 1994;129:1081–1085. doi: 10.1001/archsurg.1994.01420340095018. [DOI] [PubMed] [Google Scholar]
- 41.Korsten MA, Haber PS, Wilson JS, Lieber CS. The effect of chronic alcohol administration on cerulein-induced pancreatitis. Int J Pancreatol. 1995;18:25–31. doi: 10.1007/BF02825418. [DOI] [PubMed] [Google Scholar]
- 42.Pandol SJ, Periskic S, Gukovsky I, Zaninovic V, Jung Y, Zong Y, Solomon TE, Gukovskaya AS, Tsukamoto H. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117:706–716. doi: 10.1016/s0016-5085(99)70465-8. [DOI] [PubMed] [Google Scholar]
- 43.Ponnappa BC, Marciniak R, Schneider T, Hoek JB, Rubin E. Ethanol consumption and susceptibility of the pancreas to cerulein-induced pancreatitis. Pancreas. 1997;14:150–157. doi: 10.1097/00006676-199703000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Katz M, Carangelo R, Miller LJ, Gorelick F. Effect of ethanol on cholecystokinin-stimulated zymogen conversion in pancreatic acinar cells. Am J Physiol. 1996;270:G171–G175. doi: 10.1152/ajpgi.1996.270.1.G171. [DOI] [PubMed] [Google Scholar]
- 45.Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004;4:567–586. doi: 10.1159/000082182. [DOI] [PubMed] [Google Scholar]
- 46.Pandol SJ. Acute pancreatitis. Curr Opin Gastroenterol. 2006;22:481–486. doi: 10.1097/01.mog.0000239861.89209.5f. [DOI] [PubMed] [Google Scholar]
- 47.Norman JG, Franz MG, Fink GS, Messina J, Fabri PJ, Gower WR, Carey LC. Decreased mortality of severe acute pancreatitis after proximal cytokine blockade. Ann Surg. 1995;221:625–631; discussion 631-634. doi: 10.1097/00000658-199506000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275:G1402–G1414. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 50.Heath DI, Cruickshank A, Gudgeon M, Jehanli A, Shenkin A, Imrie CW. Role of interleukin-6 in mediating the acute phase protein response and potential as an early means of severity assessment in acute pancreatitis. Gut. 1993;34:41–45. doi: 10.1136/gut.34.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Formela LJ, Galloway SW, Kingsnorth AN. Inflammatory mediators in acute pancreatitis. Br J Surg. 1995;82:6–13. doi: 10.1002/bjs.1800820105. [DOI] [PubMed] [Google Scholar]
- 52.Siegmund E, Weber H, Kasper M, Jonas L. Role of PGE2 in the development of pancreatic injury induced by chronic alcohol feeding in rats. Pancreatology. 2003;3:26–35. doi: 10.1159/000069141. [DOI] [PubMed] [Google Scholar]