

Abstract
Originally isolated on the basis of its capacity to stimulate T-cell maturation and proliferation, avian thymic hormone (ATH) is nevertheless a parvalbumin, one of two β-lineage isoforms expressed in birds. We recently learned that addition of Ca2+-free ATH to a solution of 8-anilinonaphthalene-1-sulfonate (ANS) markedly increases ANS emission. This behavior, not observed in the presence of Ca2+, suggests that apolar surface area buried in the Ca2+-bound state becomes solvent-accessible upon Ca2+ removal. In order to elucidate the conformational alterations that accompany Ca2+ binding, we have obtained the solution structure of the Ca2+-free protein using NMR spectroscopy and compared it to the Ca2+-loaded protein, solved by x-ray crystallography. Although the metal ion-binding (CD-EF) domains are largely coincident in the superimposed structures, a major difference is observed in the AB domains. The tight association of helix B with the E and F helices in the Ca2+-bound state is lost upon removal of Ca2+, producing a deep hydrophobic cavity. The B helix also undergoes substantial rotation, exposing the side-chains of F24, Y26, F29, and F30 to solvent. Presumably, the increase in ANS emission observed in the presence of unliganded ATH reflects the interaction of these hydrophobic residues with the fluorescent probe. The increased solvent exposure of apolar surface area in the Ca2+-free protein is consistent with previously collected scanning calorimetry data, which indicated an unusually low change in heat capacity upon thermal denaturation. The Ca2+-free structure also provides added insight into the magnitude of ligation-linked conformational alteration compatible with a high-affinity metal ion-binding signature. The exposure of substantial apolar surface area suggests the intriguing possibility that ATH could function as a reverse Ca2+ sensor.
Keywords: calcium-binding protein, EF-hand protein, parvalbumin, NMR structure, crystal structure
Introduction
The second-messenger role of Ca2+ in eukaryotic signal transduction pathways is largely mediated by EF-hand proteins1–4. The human genome, for example, encodes 242 EF-hand family members5. Some of these, calmodulin being the archetype, have explicit regulatory activity, modulating the activities of effector proteins in a Ca2+-dependent manner. Other EF-hand proteins function as mobile intracellular Ca2+ buffers. Regardless of their precise role, all display the hallmark Ca2+-binding motif – a central ion-binding loop flanked by short amphipathic helical segments. The term “EF-hand” was inspired by the recognition that the spatial arrangement between these structural elements can be mimicked with the fingers of the right hand6.
Despite the general similarity of their metal ion-binding sites, EF-hand proteins exhibit broad variations in divalent ion affinity. We are exploring the physical and structural basis for these differences, using specific parvalbumin (PV) isoforms. Parvalbumins are small (Mr 12,000), vertebrate-specific EF-hand proteins3,7,8. The PV family includes α- and β sub-lineages, distinguished by isoelectric point (pI > 5 for α) and lineage-specific sequence differences9,10. Mammals express two isoforms, one from each lineage11, that exhibit 49% sequence identity. Despite the sequence similarity, in 0.15 M NaCl at pH 7.4, rat α-PV binds Ca2+ with a standard free energy change 3.5 kcal mol-1 more favorable than that of rat β-PV12. Whereas Na+ competes, albeit weakly, for vacant EF-hand sites in both proteins, only the β isoform binds K+. Thus, when K+ replaces Na+ as the major solvent cation, the α isoform experiences an apparent increase in divalent ion affinity, binding Ca2+ a full 5.5 kcal mol−1 more tightly than rat β-PV. Besides improving our understanding of this biologically important class of protein, an explanation for the disparity in binding affinity could furnish insight into protein-ligand interactions in general.
The PDB contains more than 20 high-resolution structures of Ca2+-bound parvalbumins – including carp β (5CPV13), leopard-shark α (5PAL)14, pike β (2PVB)15, rat α (1RWY)16, and rat β (1RRO)17. Their overall structural similarity – average RMSD < 1.0 Å for backbone atoms – suggested that variations in divalent ion-binding affinity might, in fact, reflect differences in the structures of the apo-proteins. Accordingly, solution structures were obtained for the Ca2+-free rat α- and β-parvalbumins18,19. Ca2+ removal from the β isoform evidently provokes a substantial conformational rearrangement, implying that the attenuated divalent ion affinity may reflect the energetic cost associated with isomerizing the apo-protein. If correct, then Ca2+ binding should be accompanied by more muted structural alterations in high-affinity isoforms. That idea is supported by solution structural data for the Ca2+-free rat α isoform, which closely resembles the Ca2+-bound protein. Together, these findings implied a direct correlation between binding affinity and conformational similarity of the Ca2+-free and Ca2+-loaded states. The Ca2+-free parvalbumin structure described herein suggests that the correlation is not quite that straightforward.
Avian thymic hormone (ATH) was originally isolated on the basis of, and named for, its capacity to stimulate avian T-cell maturation and proliferation20,21. Quite unexpectedly, sequence analysis of the purified protein revealed that it was a β-parvalbumin22. In fact, ATH is one of two β-PV isoforms expressed in chicken thymus tissue. The other is known as chicken parvalbumin 323, or CPV3, also believed to serve an endocrine role in the avian immune system24. Both EF-hand sites in ATH qualify as high-affinity, or Ca2+/Mg2+, sites. The Ca2+-binding constants in 0.15 M NaCl, at pH 7.4, are 2.4 × 108 and 1.0 × 108 M−1; the corresponding Mg2+ constants are 2.2 × 104 and 1.2 × 104 M−1 25,26.
We recently learned that addition of Ca2+-free ATH to a solution of the hydrophobic probe 8-anilinonaphthalene-1-sulfonate (ANS) produces a large increase in fluorescence quantum yield and a pronounced blue shift27. Addition of the Ca2+-bound protein is without effect. This behavior, which is not observed with the Ca2+-free forms of either rat α-PV or CPV3, implies that the conformations of the Ca2+-free and Ca2+-bound protein differ substantively. This finding conflicts with the proposed correlation between divalent ion affinity and conformational similarity of the Ca2+-free and Ca2+-loaded states of high-affinity parvalbumin isoforms. In an effort to resolve this issue and to delineate the conformational changes that occur in ATH upon Ca2+ binding, we have determined the solution structure of Ca2+-free ATH. To permit a more meaningful evaluation of the structural changes provoked by Ca2+ removal, we also report the crystal structure of Ca2+-bound ATH.
Results
Ca2+-bound ATH
The structure of Ca2+-bound ATH was solved by X-ray crystallography (Table 1). The crystal morphology (long needles) and space group (P1), its sensitivity to radiation damage, and the unit cell dimensions (containing 8 copies of the protein) made this task challenging. Ultimately, the experimental hurdles were overcome by employing the micro-diffraction beamline at NE-CAT 24-ID-E of the Advanced Photon Source. The structure was solved to a resolution of 1.95 Å, using a wedge-based data collection strategy, featuring a wide oscillation width of 4° per frame, as described in Materials and Methods.
Table 1.
X-ray diffraction data collection and refinement statisticsa
| Wavelength (Å) | 0.9792 |
| Space group | P1 |
| Unit cell parameters (Å, °) |
a = 47.6, b = 51.6, c = 67.6 α = 87.4, β = 85.4, γ = 87.4 |
| No. of molecules in unit cell | 8 |
| Diffraction resolution (Å) | 50 – 1.95 (2.02 – 1.95) |
| No. of observations | 196, 092 |
| No. of unique reflections | 45, 451 |
| Redundancy | 4.3 (4.3) |
| Completeness (%) | 98.0 (96.8) |
| Mean I/σI | 16.6 (3.2) |
| Rmerge | 0.086 (0.416) |
| No. of protein atoms | 6528 |
| No. of water molecules | 449 |
| No. of glycerol molecules | 3 |
| No. of sulfate ions | 4 |
| No. of calcium ions | 16 |
| Rcryst | 0.164 (0.206) |
| Rfreeb | 0.221 (0.283) |
| Maximum-likelihood coordinate error (Å) | 0.27 |
| RMSD bond lengths (Å)c | 0.004 |
| RMSD bond angles (deg.)c | 0.715 |
| Ramachandran plotd | |
| Favored (%) | 99.4 |
| Allowed (%) | 0.6 |
| Outliers (%) | 0.0 |
| Average B-factors (Å2) | |
| Protein | 21 |
| Water | 25 |
| Glycerol molecules | 34 |
| Sulfate ions | 67 |
| Calcium ions | 22 |
| PDB accession code | 3FS7 |
Ca2+-bound ATH displays the characteristic parvalbumin fold, consisting of six α-helices (labeled A-F in Fig. 1A) organized into two domains. The AB domain, spanning residues 1–38, includes the A and B α-helices and an extended loop. The remaining polypeptide chain forms the CD/EF domain, which includes the two EF-hand metal ion-binding motifs. The eight chains in the asymmetric unit of the crystal have very similar structures. Pairwise root mean square differences (RMSDs) for Cα atoms in the eight chains span the range 0.34 – 0.93 Å, with an average of 0.61 Å.
Fig. 1.
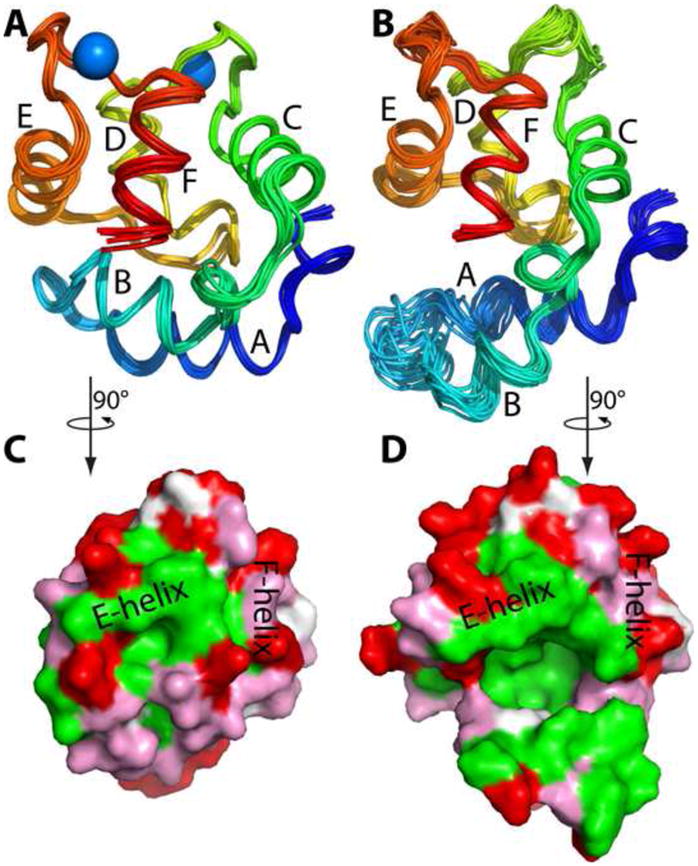
Tertiary structure of ATH. (A) Crystal structure of Ca2+-bound ATH. Superposition of the eight Ca2+-bound molecules in the asymmetric unit. (B) Solution structure of Ca2+-free ATH. An ensemble of the 20 lowest energy structures obtained with CYANA. (C) Surface rendering of the ensemble-average Ca2+-bound ATH structure. (D) Surface rendering of the ensemble-average Ca2+-free ATH structure, emphasizing the hydrophobic cavity produced by the displacement of the B helix. The views depicted in (C) and (D) were obtained by 90° counterclockwise rotation of the views in (A) and (B) about the vertical axis. This figure and Figs. 4 and 6 were produced with PyMOL 63.
Ca2+-free ATH
Resonance assignments
Assignments were made using a series of triple-resonance experiments. CBCACONH and HNCACB spectra yielded tentative assignments for nearly all of the Cα and Cβ nuclei. The CBCACONH experiment provides inter-residue correlations between the amide signal for the ith residue, HN(i), and the Cα and Cβ nuclei in the (i-1)th residue, Cα/Cβ(i-1). The HNCACB spectrum provides the corresponding intra-residue correlation, as well as an inter-residue correlation for the majority of amide signals. The HNCA/HNCOCA and HNCO/HCACOCANH spectral pairs were used to confirm the backbone assignments and to resolve ambiguities. The HNCOCA experiment correlates HN(i) with Cα(i-1). The HNCA experiment correlates HN(i) with Cα(i) and, frequently, HN(i) with Cα(i-1). Due to their narrower spectral windows, these experiments offer greater resolution of the Cα signals. The HNCO experiment correlates HN(i) with C′(i-1), the backbone carbonyl of the preceding residue. The HCACOCANH correlates HN(i) with C′(i) and, often, HN(i) with C′(i-1). Aliphatic carbon assignments beyond Cβ were made with the CCONH experiment, which correlates HN(i) with all of the carbon nuclei in the side-chain of the i–1 residue. The aliphatic side-chain carbon assignments were complete, excluding the 18 carboxylates (Asp, Glu, and C-terminus), the seven carboxamides (5 Gln, 2 Asn), and the R75 guanidinino group.
Aliphatic 1H assignments were made with HBHACONH, HCCONH, 15N-edited TOCSY-HSQC, and HCCH-TOCSY experiments. The HBHACONH spectrum correlates HN(i) with Hα/Hβ(i-1); the HCCONH spectrum correlates correlates HN(i) with all of the carbon-bound protons in the i-1 side-chain; the 15N-TOCSY-HSQC spectrum provides the corresponding intra-residue correlations; and the HCCH-TOCSY spectrum provides intra-residue correlations between each aliphatic carbon nucleus and all of the carbon-linked protons. Assignments for Hδ and Hε in Phe and Tyr were obtained from the HBCBCGCDHD and HBCBCGCDCEHE experiments, which correlate those protons with Cβ. Proton assignments (side-chain and backbone) were 95% complete.
Fig. 2 displays the 1H-15N HSQC spectrum of Ca2+-free ATH at 20 °C. Main-chain amide signals are observed for all residues except I2. The following pairs of resonances exhibit substantial overlap: D41/V33, G48/G98, Y26/L77, K44/D90, D92/D94, T32/I43, and D51/K107. The spectrum of the apo-protein is compared to the spectrum of the Ca2+-loaded protein, collected under identical conditions, in Supplemental Fig. S1.
Fig. 2.
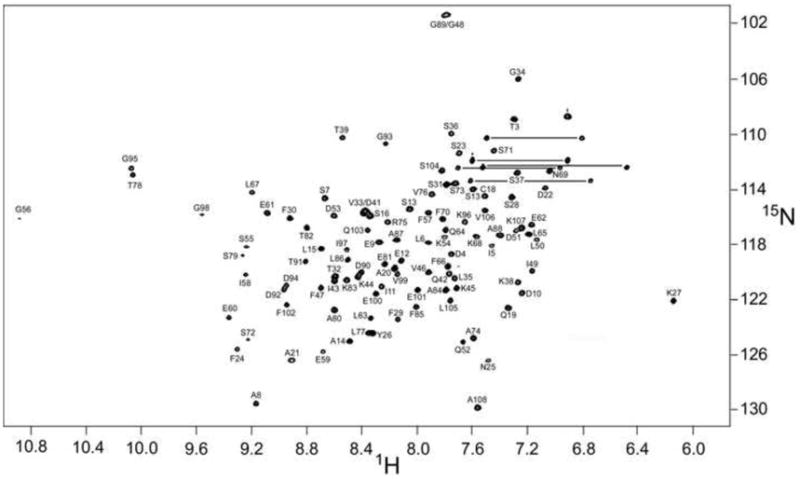
1H-15N HSQC spectrum of Ca2+-free chicken ATH in 0.15 M NaCl, 0.01 M Mes, 0.005 M EDTA, pH 6.0, at 20°C.
Solution structure of Ca2+-free ATH
The tertiary structure of Ca2+-free ATH was calculated with CYANA28, using distance and dihedral angle restraints (Table 2). An ensemble of 20 low-energy conformers is displayed in Fig. 1B. The RMSD, relative to the ensemble average, is 0.75 Å for the backbone atoms (Cβ, Cα, C′, O, and N), 1.12 Å for all heavy atoms. Table 2 lists additional structural quality statistics for the ensemble. 99.9% of the φ,Ψ combinations in the 20 conformers reside in allowed regions of the Ramachandran plot. The average NOE restraint violation is 0.026 Å, and there are no violations exceeding 0.5 Å in six or more of the structures. The average dihedral restraint violation is 0.59°, with no violations exceeding 5° in six or more structures.
Table 2.
List of restraints and statistical analysis for the Ca2+-free ATH solution structure
| Number of experimental restraints | |
| total NOEs | 2355 |
| Intraresidue | 590 |
| Sequential | 588 |
| Medium-range (1 < |i-j| ≤ 4) | 587 |
| Long-range (|i-j| > 4) | 590 |
| TALOS | 74 |
| Residual CYANA target function | 3.47 ± 0.25 |
| Restraint violations | |
| NOE restraints (>0.1Å, 6 or more structures) | 23 |
| NOE restraints (>0.2Å, 6 or more structures) | 12 |
| NOE restraints (>0.3Å, 6 or more structures) | 6 |
| NOE restraints (>0.4Å, 6 or more structures) | 3 |
| Dihedral restraints (>5°, 6 or more structures) | 0 |
| RMSD from experimental restraints | |
| NOE restraints (Å) | 0.026 |
| Dihedral restraints (deg.) | 0.59 |
| RMSD from idealized covalent geometry | |
| bonds (Å) | 0.0035 |
| angles (deg) | 0.57 |
| Dihedral angles (deg) | 21.7 |
| improper angles (deg) | 0.83 |
| Coordinate RMSD from average structure (Å) | |
| backbone (Cβ, Cα, C′, O, N) | 0.75 ± 0.16 |
| all heavy atoms | 1.12 ± 0.16 |
| Ramachandran plot (ensemble averages) | |
| most favored regions (%) | 69.9 |
| allowed regions (%) | 27.7 |
| generously allowed (%) | 2.3 |
| disallowed (%) | 0.1 |
15N relaxation data
Data were collected on Ca2+-free ATH at 20 °C. The R1 and R2 data are well accommodated by a two-parameter exponential decay model. Representative data are presented in Supplemental Fig. S2. R1 and R2 values for 90 of 107 amide vectors are plotted in Fig. 3A and Fig. 3B, respectively. The corresponding numerical values are tabulated in Supplemental Table S1.
Fig. 3.
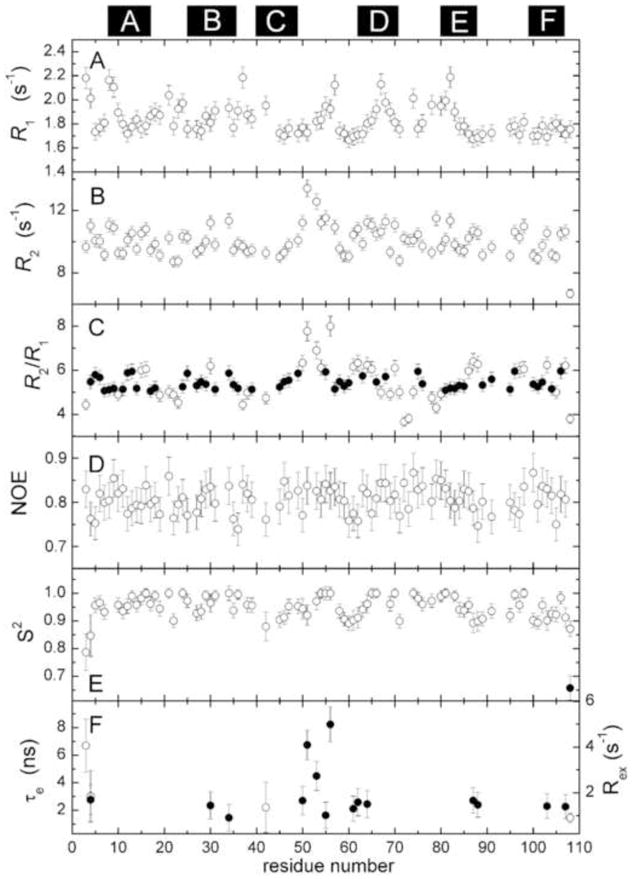
Summary of ATH 15N relaxation data and internal mobility analysis. For reference, the positions of the six helices are indicated at the top of the figure. (A) R1 values. (B) R2 values. (C) Calculated R2/R1 ratios. The filled circles (●) represent amide vectors used in the calculating the mean. (D) {1H}15N NOE values. (E) Order parameter (S2) determined by model-free analysis, as described in the text. Two of the residues required inclusion of a second order parameter (●), corresponding to motion on a slower timescale, to satisfactory model their relaxation behavior. (F) τe (○) and Rex (●) values for residues displaying motion on a timescale exceeding 20 ps.
The rotational correlation time (τc) was estimated from the subset of amide vectors (Fig. 3C, •) exhibiting an R2/R1 ratio within one standard deviation of the mean. The data are well accommodated by a spherically symmetric rotational diffusion model, yielding a τc value of 6.68 ± 0.04 ns. Axially symmetric and fully symmetric models yielded insignificant reductions in χ2.
The {1H}15N NOE values (Fig. 3D) exhibit a high degree of uniformity. The mean NOE value is 0.80 ± 0.04.
Internal mobility and model-free analysis
Main-chain flexibility in Ca2+-free ATH was examined using the Lipari-Szabo model-free treatment29,30. Relaxation data for 79 amide vectors were analyzed, employing a spherically symmetric diffusion model. The results are displayed in Fig. 3E and Fig. 3F, and the model-free parameters are listed in Supplemental Table S2.
The majority of signals (61/79) can be modeled with the overall rotational correlation time (τc) and a generalized order parameter (S2). Three amide vectors require a τe term to describe internal motion on the 20 ps - 10 ns timescale. An additional 14 require an Rex term to describe internal motion on the μs-ms timescale. One amide (D4) can be accommodated only by inclusion of both τe and Rex terms. Finally, residue 108 exhibits behavior consistent with motion on two timescales shorter than the overall rotational correlation time. The average order parameter for the apo-protein is 0.95, comparable to the value of 0.92 obtained for the Ca2+-free forms of rat α-PV and rat β-PV18,19,31.
The data for eleven of the 90 vectors for which relaxation data were collected are not compatible with any of the five standard models, suggesting that those amides are undergoing more complex motions. The residues in question are A8, K9, S23, S37, F57, L67, K68, S72, S73, S79, and T82. Their positions in the Ca2+-free and Ca2+-bound states are displayed in Supplemental Fig. S3.
Comparison of the Ca2+-bound and Ca2+-free ATH structures
The ensemble-averaged Ca2+-free ATH structure (pink) has been superimposed on the crystal structure of the Ca2+-bound protein (green) in Fig. 4. Although the peptide backbones appear largely coincident, a major difference is observed in the region spanning residues 22 through 32. In the Ca2+-loaded structure, helix B packs snugly against the E and F helices. Upon removal of Ca2+, however, the B helix undergoes substantial reorientation, resulting in loss of contact with the CD-EF domain and creation of a deep cavity (Fig. 1C). Analysis with the CASTp server indicates that the hydrophobic cavity has a surface area of 554 Å2 and volume of 817 Å3.
Fig. 4.
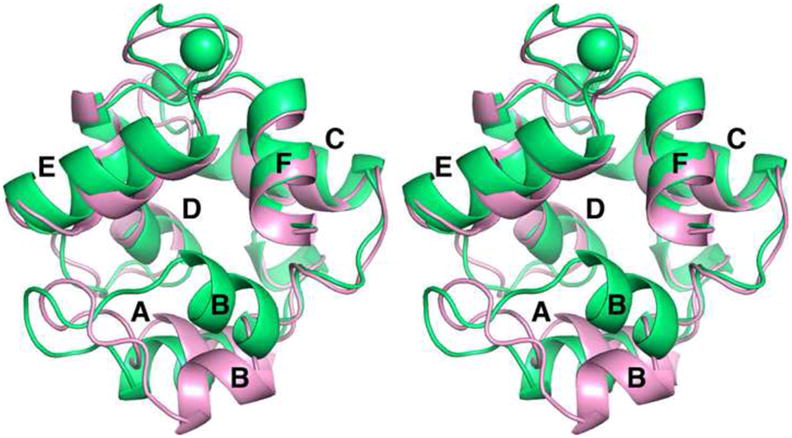
Stereoview of the superimposed Ca2+-bound (green) and Ca2+-free forms (pink) of ATH.
A statistical comparison of the two structures is presented in Fig. 5. This distance difference matrix compares the inter-residue distances, measured between α carbon atoms, in the Ca2+-free and Ca2+-bound forms of ATH. Substantive structural differences are largely confined to residues 20 – 30. The pairwise RMSDs between the eight chains of the crystal structure and the twenty models of the Ca2+-free structure span the range 3.1 – 4.4 Å, with an average of 3.8 ± 0.3 Å (Cα atoms). This range is substantially larger than the RMSDs for the individual chains of either the crystal structure (0.6 Å) or the NMR ensemble (1.1 Å), indicating significant conformational differences between the Ca2+-bound and Ca2+-free structures. In fact, the main structural differences between the two forms of the protein are limited to the AB domain (Fig. 4, Fig. 5, and Supplemental Fig. S4).
Fig. 5.
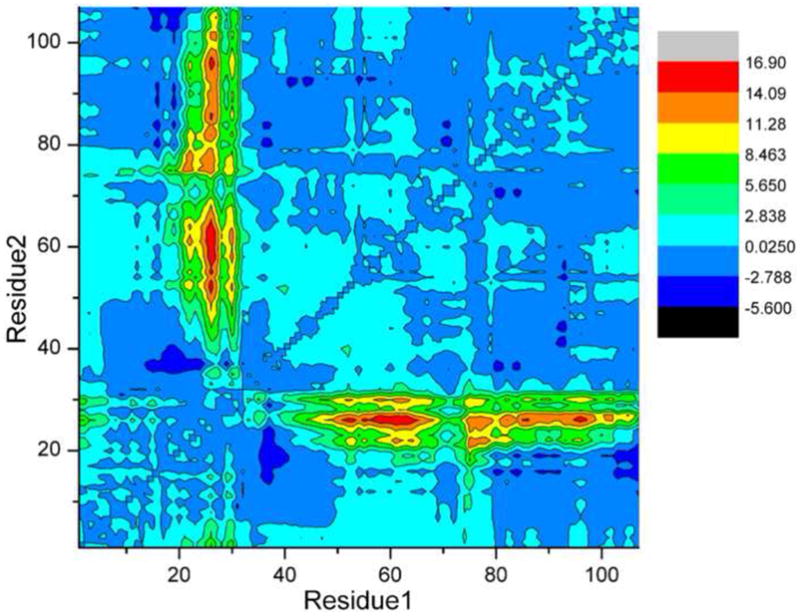
Distance difference matrix. The changes in inter-residue distance (in Å) that accompany Ca2+ removal were calculated with CNS. The quantity rij (Ca2+-free) – rij (Ca2+-bound) is displayed as a contour plot, where rij(Ca2+-free) is the Cα-Cα distance for residues i and j in the Ca2+-free structure, and rij(Ca2+-bound) is the corresponding distance for the Ca2+-bound structure. The differences were averaged over all possible combinations of the eight chains of the X-ray ensemble and the 20 chains of the NMR ensemble.
A detailed stereoview of the Ca2+-free protein in the vicinity of the B helix is displayed in Fig. 6. In panel B, the path of the main-chain in the Ca2+-bound protein is also depicted (in magenta), which serves to emphasize both the magnitude of the structural changes and their highly localized nature. The largest displacement of the main chain (13 Å) in response to Ca2+ removal occurs at Y26, positioned at the N-terminus of helix B. In addition to moving away from the CD-EF domain, the B helix also undergoes a significant rotation. As a consequence, the side-chains of F24, Y26, F29, F30 – normally part of the hydrophobic core – become solvent accessible (Supplemental Fig. S5). In fact, the reorientation of helix B opens a window on the hydrophobic core of the protein. In addition to the residues mentioned above, the following core residues experience some increase in solvent accessibility: I11, L15, F66, L67, F70, F85, and L105.
Fig. 6.
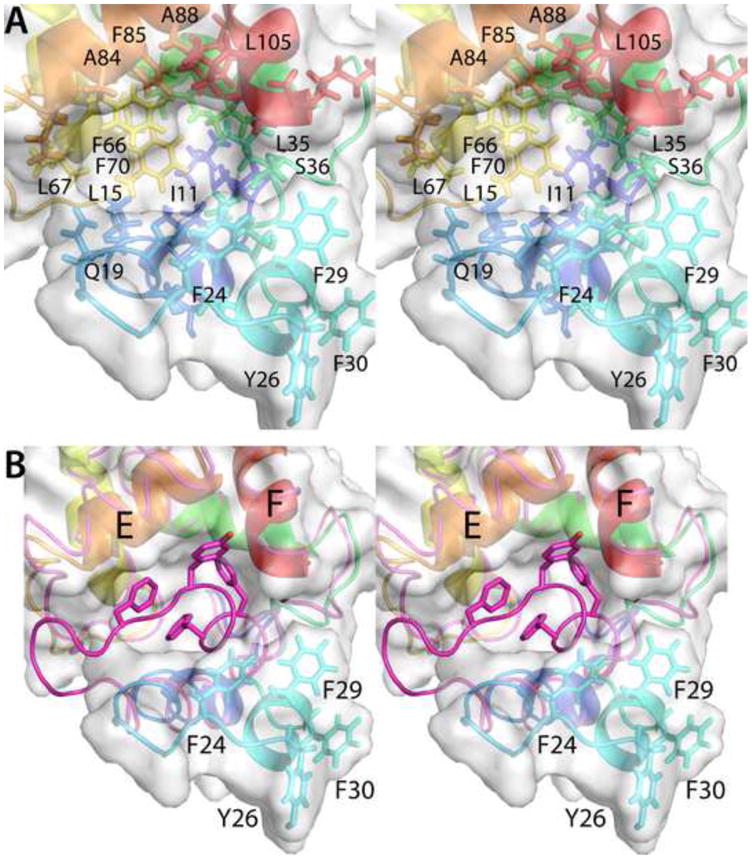
Structural changes accompanying removal of Ca2+ from ATH. (A) Stereoview of the hydrophobic cavity created by the movement of helix B. (B) This stereoview of the region of interest compares the path of the polypeptide backbone in the Ca2+-free and Ca2+-bound (magenta) states. Note that large displacements of the polypeptide chain are confined to the vicinity of the AB loop and the B helix.
A previous analysis of thermal stability indicated that ΔCp, the increase in heat capacity upon denaturation, is lower for Ca2+-free ATH than for Ca2+-free rat β-PV27. Because the magnitude of ΔCp primarily reflects increased exposure of apolar surface area in the unfolded state, the decreased value obtained for ATH suggests that the increase in apolar surface area upon denaturation is atypically small. Exposure of apolar surface area in the native state, as implied by the solution structure, would provide a rationale for the lower ΔCp value.
As noted above, 11 amide vectors exhibit complex dynamics. Their locations are indicated in Supplemental Fig. S3. Residues S23 and S37 bracket the B helix. Thus, it is reasonable that their conformations might sample alternative conformations on a timescale not compatible with the model-free analysis. In the Ca2+-bound state, L67 contacts the side-chains of F24, F29, and F30, helping to anchor the extended D/E loop to the AB domain. The displacement and rotation of the B helix that accompanies Ca2+ removal eliminates those contacts. The anticipated increase in flexibility and mobility of the D/E loop may explain why the dynamics of the amide vectors for L67, K68, S72, and S73 cannot be captured by the model-free treatment. Similarly, S79 and T82 are located at the N-terminal end of the E helix, which associates with the B helix in the Ca2+-loaded protein. The departure of the B helix upon Ca2+ removal and resulting disruption of those contacts could significantly heighten flexibility. A8 and K9 are located at the N-terminal end of the A helix, which is proximal to the C-terminal end of helix B. Their conformations may be sensitive to the pronounced movement of the B helix.
Discussion
Interest in the structure of Ca2+-free ATH was originally sparked by the observation that addition of the Ca2+-free protein to a solution of ANS produced a marked increase in the quantum yield of the fluorophore. This behavior, not observed with the Ca2+-bound protein, suggested that the apo-protein presents a hydrophobic surface with which the fluorescent probe can interact. Presumably, ANS associates with the apolar side-chains that are exposed by the reorientation of the B helix that accompanies Ca2+ removal.
Besides ATH, structural data have previously been acquired for the Ca2+-free states of rat α-PV and rat β-PV18,19. The EF-hand sites in the rat α isoform are typical high-affinity (Ca2+/Mg2+) sites, with average binding constants, in buffered saline at pH 7.4, of 1.2 × 108 and 1 × 104 M−1 for Ca2+ and Mg2+, respectively. The rat β isoform exhibits substantially lower divalent ion affinity12. For example, the total free energy change that accompanies Ca2+ binding at 25 °C is 3.5 kcal/mol less favorable in buffered saline at pH 7.4. Whereas the solution structure obtained for Ca2+-free rat α closely resembles the Ca2+-bound form, the unliganded and bound forms of rat β-PV exhibit some substantive differences. In particular, the hydrophobic core of the protein evidently undergoes significant reorganization when Ca2+ binds. This observation suggested that the attenuation of divalent affinity observed for the rat β isoform may reflect the energetic cost of the conformational change attendant to metal ion binding. More generally, the structural results for the two rat parvalbumin isoforms imply a direct correlation between divalent ion affinity and the similarity of the unliganded and bound states.
The divalent ion affinity measured for ATH eclipses that of rat α-PV. Thus, we would anticipate that the apo- and Ca2+-loaded forms of the protein would be largely indistinguishable. However, as described above, the conformations exhibit a marked difference in the vicinity of the B helix. It would seem that the aforementioned correlation of divalent ion affinity with the conformational similarity of the apo- and Ca2+-bound protein requires amendment.
In ATH, the differences between the apo- and Ca2+-bound forms are confined to the B helix. The conformation of the CD-EF domain is largely independent of the divalent ion-binding status. In the rat β isoform, by contrast, the conformational change associated with Ca2+ binding apparently perturbs the packing of the hydrophobic core in the CD-EF domain as well as the AB/CD-EF interdomain interaction. Thus, the structural rearrangements that accompany Ca2+ binding/removal differ qualitatively in ATH and rat β-PV. In the former, they can be reversed by displacement and rotation of the B helix alone, with minimal involvement of the rest of the molecule. The fact that the conformational difference is localized in ATH may explain why the molecule retains its high affinity for divalent ions, even though binding/dissociation is accompanied by a remarkable structural alteration. In rat β-PV, by contrast, reversal of the conformational alterations linked to metal ion binding involves coordinated movement throughout the molecule.
Under resting state conditions in vivo, assuming intracellular Ca2+ and Mg2+ concentrations of 10−7 and 10−3 M, respectively, ATH would reside predominantly in the Mg2+-bound form. Because the Ca2+-free state studied here is negligibly populated in the cell, one could argue that the structural difference observed between the unliganded and Ca2+-bound forms of ATH has doubtful physiological significance. Interestingly, however, the impact of the Mg2+-bound protein on ANS fluorescence is comparable to that of the apo-protein27. This result suggests that the conformation of the B helix in the Mg2+ bound protein may resemble that observed in the Ca2+-free protein, with comparable exposure of hydrophobic surface area. If correct, then ATH could conceivably function as a reverse Ca2+ sensor. In the Mg2+-bound (resting) state, exposure of apolar surface would permit interaction with a target molecule. Upon exchange of Mg2+ for Ca2+ following a transient increase in cytosolic Ca2+ concentration, the apolar surface would be hidden, and the target molecule would be released. Clearly, structural data for the Mg2+-loaded protein would be highly desirable, and efforts to obtain those data are underway.
Materials and Methods
Protein expression and purification
The chicken ATH coding sequence, optimized for expression in E. coli, was obtained from Genscript (Piscataway, NJ) and cloned between the Nde I and Bam HI sites of pET11a. Bacteria harboring the ATH-pET11 construct were cultured at 37 °C in 15N- or 13C, 15N-labeled Spectra 9 medium (Cambridge Isotope Laboratory, Andover, MA), supplemented with ampicillin (100 μg/mL). IPTG (0.25 mM) was added when the absorbance of the culture at 600 nm reached 0.6. After an additional 20 h, the bacteria were collected by centrifugation. The protein was purified as described previously26. Each liter yielded 20–25 mg of protein, with purity exceeding 98%.
Crystallization
ATH was crystallized from ammonium sulfate at 21 °C by vapor diffusion. Prior to crystallization, the protein was dialyzed extensively against 0.05 M MES, pH 6.0, containing 10 μM Ca2+. Hanging drops were prepared by combining 40 μL of ATH solution (10 mg/mL) with 40 μL of 70% (NH4)2SO4, 0.05 M MES, pH 6.0. Thin rod-like crystals, with a maximum thickness of 20 μm (Fig. 7), formed in 4–8 weeks. The largest of the crystals were prepared for low-temperature data collection by soaking in 15% glycerol. They were then picked up with Hampton loops and plunged into liquid nitrogen.
Fig. 7.
ATH crystal morphology. One of the crystals used for data collection at the NE-CAT 24-ID-E microbeam line of the Advanced Photon Source.
X-ray diffraction data collection and structure determination
Initial X-ray diffraction experiments were performed at beamline 4.2.2 of the Advanced Light Source. The resulting data set, having a high-resolution limit of 2.7 Å, was used for preliminary assessment of diffraction quality and molecular replacement calculations. The space group was P1. The unit cell, which contained eight molecules, had these parameters: a = 48 Å, b = 52 Å, c = 68 Å, α = 87°, β = 85°, γ = 87°. The corresponding Vm is 1.8 Å3/Da, and the solvent content is low, just 31 %.
In an attempt to obtain higher resolution data, additional experiments were performed at the NE-CAT 24-ID-E beamline of the Advanced Photon Source. This beamline is optimized for analysis of diminutive crystals. It employs a highly focused 20 × 100 micron X-ray beam, which may be shaped down to 5 microns, for data collection. Additionally, it is equipped with a Maatel MD-2 micro-diffractometer to facilitate visualization and centering of small crystals.
The crystals analyzed at 24-ID-E had typical dimensions of 300 × 20 × 20 μm. The X-ray beam size was therefore reduced to 20 microns by inserting an aperture. This adjustment substantially reduced background scatter, dramatically increasing the signal-to-noise ratio of the resulting data. Small crystals are susceptible to primary X-ray radiation damage from the highly focused X-rays generated by a third generation synchrotron source32–35. To minimize radiation damage, wedges of data were collected from different parts of the crystal, then merged using HKL200036 to produce complete data sets. Several data collection strategies were investigated by varying beam attenuation, oscillation angle, and wedge size. The distance of the ADSC Q315 detector was set to 250 mm and the detector 2θ angle was zero.
The best data set was obtained by merging the reflections from five non-overlapping wedges, each wedge consisting of twenty frames collected with an oscillation width of 4 ° per frame, exposure time of 2 seconds per frame, and beam attenuation of 43% transmission at 12662 eV. Thus, the merged data set consisted of a continuous 400° of data. The resulting data set is 98 % complete to 1.95 Å resolution, with an average redundancy of 4.3 (Table 1).
Molecular replacement calculations were performed with MOLREP37 using a search model derived from the coordinates of the 1.05 Å resolution structure of rat α-parvalbumin (PDB code 1RWY16). A solution having eight molecules in the asymmetric unit was obtained. The model was improved with iterative rounds of model building in COOT38 and refinement in PHENIX39. Initially, NCS restraints and group B-factors were used, but analysis of Rfree indicated that NCS restraints could be released, and the use of individual B-factors with TLS was appropriate.
NMR sample preparation
Samples were prepared with uniformly labeled 15N-labeled, uniformly 13C,15N-labeled ATH, or uniformly labeled 15N-ATH that was also fractionally labeled (15%) with 13C. In each case,18 mg of protein – sufficient to yield a 0.5 mL sample at 3 mM — was concentrated to 5 mL by ultrafiltration, then dialyzed for 48 h, at 4°C, against 4 L of 0.15 M NaCl, 0.025 M Hepes, 5.0 mM EDTA, pH 7.4. Dialysis was continued for 48 h against 0.15 M NaCl, 0.01 M Mes, 5.0 mM EDTA, pH 6.0. Following the addition of buffer prepared in D2O (0.1 volume) and 10% sodium azide (to a final concentration of 0.1%), the solution was concentrated to 0.5 mL. The resulting sample was loaded into a 5 mm Shigemi microcell (Shigemi, Inc., Allison Park, PA).
NMR spectroscopy
All data were acquired at 20°C on a Varian INOVA 600 MHz spectrometer equipped with a triple-resonance cryoprobe. 1H chemical shifts were referenced relative to DSS; 13C and 15N shifts were referenced indirectly, employing the 1H/X frequency ratios. Data were processed with NMRPipe40 and analyzed with Sparky41.
1H,15N-HSQC spectra were collected using 1H and 15N windows of 8380 Hz (2048 complex points) and 2430 Hz (256 complex points), respectively. 1H,13C-HSQC spectra were collected on the aliphatic carbon region, using 1H and 13C dimensions of 8380 Hz (2048 complex points) and 12060 Hz (256 complex points), respectively. Backbone 13C assignments were made with these pairs of 3D experiments: HNCA42 and HN(CO)CA43; HNCACB44,45 and CBCA(CO)NH46; and HNCO42 and HCACOCANH47. In each case, the 1H and 15N dimensions spanned 8380 Hz (2048 complex points) and 2430 Hz (80 complex points), respectively. The number of complex points and 13C window width were 60 and 2062 Hz (HNCO, HCACOCANH), 160 and 4525 Hz (HNCA, HNCOCA), or 140 and 12065 Hz (HNCACB, CBCACONH). Aliphatic carbon assignments beyond Cβ were made with the CCONH48 experiment, acquired with the 1H and 15N settings just listed and a 13C window of 12065 Hz (160 complex points). Stereospecific assignments for the methyl groups in valine and leucine were obtained by analysis of the splitting patterns observed in the 1H,13C-HSQC spectrum of fractionally labeled protein, as described by Neri et al.52 Those data were acquired with a 1H window of 8380 Hz (2048 complex points) and 13C window of 3015 Hz (1024 complex points).
Aliphatic 1H assignments were made with HBHACONH, HCCONH15N-edited TOCSY-HSQC49, and HCCH-TOCSY50 experiments. The first three were acquired with an 8380 Hz spectral window in both 1H dimensions (256 complex points in t1, 2048 in t3) and an 15N window of 2430 Hz (80 complex points). The HCCH-TOCSY spectrum was acquired with identical 1H settings and a 12065 Hz 13C window (108 complex points). The Hδ and Hε resonances in Phe and Tyr were assigned with the HBCBCGCDHD51 and HBCBCGCDCEHE51 experiments. Those spectra were collected with a 1H window of 8380 Hz (2048 complex points) and a 13C window of 4525 Hz (64 complex points).
Solution structure calculations
For the collection of NOE-based distance restraints, 3D 15N-edited and 13C-edited NOESY-HSQC53 data sets were collected on 13C, 15N-labeled protein, employing mixing times of 125 ms and 100 ms, respectively. Cross peaks were picked manually and integrated in Sparky. φ,Ψ dihedral angle restraints were obtained for 77 residues using TALOS54. Structure calculations were performed with CYANA28. All NOE assignments were made automatically in CYANA. The quality of the final structures was analyzed using PROCHECK55.
15N relaxation data
R1, R2, and {1H}15N NOE data were collected on 15N-labeled protein employing Varian BioPack pulse sequences. R1 data were acquired with these relaxation delays: 50, 100, 150, 250, 350, 450, 600, 800, 1000, and 1200 ms. R2 data were acquired with these delays: 10, 30, 50, 70, 90, 110, 130, 150, 170, and 190 ms. Replicate data were collected at three delay values to evaluate experimental uncertainty. To measure the steady-state heteronuclear {1H}15N-NOE, HSQC spectra were collected, with and without proton saturation (3.0 s), employing a total recycle delay period of 5.0 s. Duplicate experiments provided an estimate of the experimental uncertainty.
Signal intensities were measured automatically in Sparky. R1 and R2 values were estimated by fitting the intensities to a two-parameter single-exponential decay, in Origin, version 7.5 (OriginLab). The NOE values were calculated from the ratio of the signal intensities in the presence and absence of proton saturation.
The relaxation data were analyzed with Tensor256. An overall rotational correlation time (τc) was estimated from the subset of amide vectors having R2/R1 values within one standard deviation of the mean value57. The isotropic rotational diffusion model yielded a τc = 6.68 ± 0.04 ns, which corresponds to a rotational diffusion coefficient of 2.50 × 107 s−1. Axially symmetric and fully anisotropic models did not yield significant improvements in χ2.
Internal mobilities were examined with the Lipari-Szabo model-free formalism29,30. Tensor2 employs the five models suggested by Clore et al.58,59 and the model selection strategy described by Mandel et al.60.
Supplementary Material
Acknowledgments
This work was supported by NSF award MCB0543476 (to M.T.H. and J.J.T.). The authors wish to thank Dr. Wei Wycoff, staff NMR Spectroscopist for the MU NMR Facility, for assistance with NMR data acquisition. The Varian 600MHz spectrometer used for this work was acquired with the assistance of NSF award DBI-0070359, NIH award R01 GM57289 (cryoprobe) and the MU Research Board. We thank Dr. Jay Nix of Advanced Light Source beamline 4.2.2 for help with data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Part of this work is based upon research conducted at the Northeastern Collaborative Access Team beam lines of the Advanced Photon Source, supported by award RR-15301 from the National Center for Research Resources at the National Institute of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract No. W-31-109-ENG-38.
Abbreviations
- ANS
8-anilinonaphthalene-1-sulfonate
- ATH
avian thymic hormone
- CD site
parvalbumin metal ion-binding site flanked by the C and D helices
- CPV3
chicken parvalbumin 3
- DSS
sodium 2,2-dimethyl-2-silapentane-5-sulfonate
- EDTA
ethylenediaminetetraacetic acid
- EF site
parvalbumin metal ion-binding site flanked by the E and F helices
- Hepes
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HSQC
heteronuclear single-quantum coherence
- Mes
2-(N-morpholino)ethanesulfonic acid
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
NOE spectroscopy
- PV
parvalbumin
- R1
longitudinal relaxation rate (1/T1)
- R2
transverse relaxation rate (1/T2)
- RMSD
root-mean-square-difference
- S2
generalized Lipari-Szabo order parameter
- TALOS
torsion angle likelihood obtained from shifts and sequence similarity
- τc
overall rotational correlation time
- τe
internal correlation time
- Rex
rate constant for μs/ms motion resulting from chemical or conformational exchange
Footnotes
The following supplemental data associated with this article can be found in the online version: a comparison of the 1H,15N-HSQC spectra of Ca2+-free and Ca2+-bound ATH (Supplemental Fig. S1); representative R1 and R2 decay data for Ca2+-free ATH (Supplemental Fig. S2); ribbon diagrams of Ca2+-free and Ca2+-bound ATH, depicting the locations of amide resonances not amenable to analysis by the model-free formalism (Supplemental Fig. S3); a plot of RMSD values for the Cα atoms in the superimposed structures of Ca2+-free and Ca2+-bound ATH as a function of residue number (Supplemental Fig. S4); estimated changes in solvent-accessible surface area that accompany Ca2+ removal from ATH as a function of residue number (Supplemental Fig. S5); R1, R2, and NOE values for Ca2+-free ATH (Supplemental Table S1); and Lipari-Szabo model-free parameters for Ca2+-free ATH (Supplemental Table S2).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
Coordinates and structure factors for Ca2+-bound ATH have been deposited in the Protein Data Bank with accession number 3FS7. Coordinates and structural restraints for Ca2+-free ATH have been deposited in the Protein Data Bank with accession number 2KQY. 1H, 15N, and 13C assignments have been deposited in the BioMagnetic Resonance Bank with accession number 16617.
References
- 1.Kretsinger RH. Structure and evolution of calcium-modulated proteins. CRC Crit Rev Biochem. 1980;8:119–174. doi: 10.3109/10409238009105467. [DOI] [PubMed] [Google Scholar]
- 2.Kawasaki H, Kretsinger RH. Calcium-binding proteins 1: EF-hands. Protein Profile. 1995;2:297–490. [PubMed] [Google Scholar]
- 3.Celio MR, Pauls T, Schwaller B. Guidebook to the Calcium-Binding Proteins. Oxford University Press; New York: 1996. [Google Scholar]
- 4.Kawasaki H, Nakayama S, Kretsinger RH. Classification and evolution of EF-hand proteins. BioMetals. 1998;11:277–295. doi: 10.1023/a:1009282307967. [DOI] [PubMed] [Google Scholar]
- 5.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 6.Kretsinger RH, Nockolds CE. Carp muscle calcium-binding protein. II Structure determination and general description. J Biol Chem. 1973;248:3313–3326. [PubMed] [Google Scholar]
- 7.Pauls TL, Cox JA, Berchtold MW. The Ca2+-binding proteins parvalbumin and oncomodulin and their genes: New structural and functional findings. Biochim Biophys Acta. 1996;1306:39–54. doi: 10.1016/0167-4781(95)00221-9. [DOI] [PubMed] [Google Scholar]
- 8.Schwaller B. The continuing disappearance of “pure” Ca2+ buffers. Cell Mol Life Sci. 2009;66:275–300. doi: 10.1007/s00018-008-8564-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodman M, Pechere JF. The evolution of muscular parvalbumins investigated by the maximum parsimony method. J Mol Evol. 1977;9:131–158. doi: 10.1007/BF01732745. [DOI] [PubMed] [Google Scholar]
- 10.Moncrief ND, Kretsinger RH, Goodman M. Evolution of EF-hand calcium-modulated proteins. I Relationships based on amino acid sequences. J Mol Evol. 1990;30:522–562. doi: 10.1007/BF02101108. [DOI] [PubMed] [Google Scholar]
- 11.Fohr UG, Weber BR, Muntener M, Staudenmann W, Hughes GJ, Frutiger S, Banville D, Schafer BW, Heizmann CW. Human α and β parvalbumins. Structure and tissue-specific expression. Eur J Biochem. 1993;215:719–727. doi: 10.1111/j.1432-1033.1993.tb18084.x. [DOI] [PubMed] [Google Scholar]
- 12.Henzl MT, Larson JD, Agah S. Influence of Monovalent Cation Identity on Parvalbumin Divalent Ion-Binding Properties. Biochemistry. 2004;43:2747–2763. doi: 10.1021/bi035890k. [DOI] [PubMed] [Google Scholar]
- 13.Swain AL, Kretsinger RH, Amma EL. Restrained least squares refinement of native (calcium) and cadmium-substituted carp parvalbumin using x-ray crystallographic data at 1.6-Å resolution. J Biol Chem. 1989;264:16620–16628. [PubMed] [Google Scholar]
- 14.Roquet F, Declercq JP, Tinant B, Rambaud J, Parello J. Crystal structure of the unique parvalbumin component from muscle of the leopard shark (Triakis semifasciata). The first X-ray study of an alpha-parvalbumin. J Mol Biol. 1992;223:705–720. doi: 10.1016/0022-2836(92)90985-s. [DOI] [PubMed] [Google Scholar]
- 15.Declercq JP, Evrard C, Lamzin V, Parello J. Crystal structure of the EF-hand parvalbumin at atomic resolution (0.91 Å) and at low temperature (100 K). Evidence for conformational multistates within the hydrophobic core. Protein Sci. 1999;8:2194–2204. doi: 10.1110/ps.8.10.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bottoms CA, Schuermann JP, Agah S, Henzl MT, Tanner JJ. Crystal structure of rat α-parvalbumin at 1.05 Å resolution. Protein Sci. 2004;13:1724–1734. doi: 10.1110/ps.03571004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed FR, Rose DR, Evans SV, Pippy ME, To R. Refinement of recombinant oncomodulin at 1.30 A resolution. J Mol Biol. 1993;230:1216–1224. doi: 10.1006/jmbi.1993.1237. [DOI] [PubMed] [Google Scholar]
- 18.Henzl MT, Tanner JJ. Solution structure of Ca2+-free rat β-parvalbumin (oncomodulin) Protein Sci. 2007;16:1914–1926. doi: 10.1110/ps.072837307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henzl MT, Tanner JJ. Solution structure of the Ca2+-free rat a-parvalbumin. Protein Sci. 2008;17:431–438. doi: 10.1110/ps.073318308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murthy KK, Ragland WL. Immunomodulation by thymic hormones: studies with an avian thymic hormone. Prog Clin Biol Res. 1984;161:481–491. [PubMed] [Google Scholar]
- 21.Barger B, Pace JL, Ragland WL. Purification and partial characterization of an avian thymic hormone. Thymus. 1991;17:181–197. [PubMed] [Google Scholar]
- 22.Brewer JM, Wunderlich JK, Ragland WL. The amino acid sequence of avian thymic hormone, a parvalbumin. Biochimie. 1990;72:653–660. doi: 10.1016/0300-9084(90)90047-k. [DOI] [PubMed] [Google Scholar]
- 23.Hapak RC, Zhao H, Boschi JM, Henzl MT. Novel avian thymic parvalbumin displays high degree of sequence homology to oncomodulin. J Biol Chem. 1994;269:5288–5296. [PubMed] [Google Scholar]
- 24.Novak R, Henzl MT, Ragland WL. Receptor cells for the CPV3 parvalbumin of chicken thymus in spleen and caecal tonsils. J Allergy Clin Immunol. 1997;99:S202. [Google Scholar]
- 25.Serda RE, Henzl MT. Metal ion-binding properties of avian thymic hormone. J Biol Chem. 1991;266:7291–7299. [PubMed] [Google Scholar]
- 26.Henzl MT, Agah S. Divalent ion-binding properties of the two avian β-parvalbumins. Proteins. 2006;62:270–278. doi: 10.1002/prot.20701. [DOI] [PubMed] [Google Scholar]
- 27.Tan A, Henzl MT. Evidence for a Ca2+-Specific Conformational Change in Avian Thymic Hormone, a High-Affinity β-Parvalbumin. Biochemistry. 2009;48:3936–3945. doi: 10.1021/bi900029j. [DOI] [PubMed] [Google Scholar]
- 28.Güntert P. Automated NMR Structure Calculation with CYANA. In: Downing AK, editor. Protein NMR Techniques. Humana Press; Totowa, NJ: 2004. pp. 353–378. [DOI] [PubMed] [Google Scholar]
- 29.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1 Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 30.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2 Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 31.Henzl MT, Wycoff WG, Larson JD, Likos JJ. 15N nuclear magnetic resonance relaxation studies on rat β-parvalbumin and the pentacarboxylate variants, S55D and G98D. Protein Sci. 2002;11:158–173. doi: 10.1110/ps.18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henderson R. Cryoprotection of protein crystals against radiation-damage in electron and X-ray-diffraction. Proc Biol Sci. 1990;241:6–8. [Google Scholar]
- 33.Gonzalez A, Nave C. Radiation-Damage in Protein Crystals at Low-Temperature. Acta Cryst. 1994;D50:874–877. doi: 10.1107/S0907444994006311. [DOI] [PubMed] [Google Scholar]
- 34.Cusack S, Belrhali H, Bram A, Burghammer M, Perrakis A, Riekel C. Small is beautiful: protein micro-crystallography. Nature Struct Biol. 1998;5:634–637. doi: 10.1038/1325. [DOI] [PubMed] [Google Scholar]
- 35.Dimasi N, Flot D, Dupeux F, Marquez JA. Expression, crystallization and X-ray data collection from microcrystals of the extracellular domain of the human inhibitory receptor expressed on myeloid cells IREM-1. Acta Cryst. 2007;F63:204–208. doi: 10.1107/S1744309107004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Cryst. 2000;D56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC. Recent developments in the PHENIX software for automated crystallographic structure determination. J Synchrotron Rad. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 40.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 41.Goddard TD, Kneller DG. Sparky. 2007. p. 3. [Google Scholar]
- 42.Ikura M, Kay LE, Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of larger proteins: Heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 43.Bax A, Ikura M. An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J Biomol NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- 44.Kay LE, Xu GY, Yamazaki T. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J Mag Res. 1994;109:129–133. [Google Scholar]
- 45.Muhandiram DR, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J Mag Res. 1994;103:203–216. [Google Scholar]
- 46.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Amer Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 47.Lohr F, Ruterjans H. A new triple-resonance experiment for the sequential assignment of backbone resonances in proteins. J Biomol NMR. 2005;6:189–197. doi: 10.1007/BF00211783. [DOI] [PubMed] [Google Scholar]
- 48.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of carbon-13 magnetization. J Mag Res. 1993;101:114–119. [Google Scholar]
- 49.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: Application to interleukin 1β. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 50.Kay LE, Xu GY, Singer AU, Muhandiram DR, Forman-Kay JD. A gradient-enhanced HCCH-TOCSY experiment for recording side-chain proton and carbon-13 correlations in water samples of proteins. J Mag Res. 1993;B101:333–337. [Google Scholar]
- 51.Yamazaki T, Forman-Kay JD, Kay LE. Two-dimensional NMR experiments for correlating 13Cβ and 1Hδ/ε chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J Amer Chem Soc. 1993;115:11054–11055. [Google Scholar]
- 52.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 53.Marion D, Kay LE, Sparks SW, Torchia D, Bax A. Three-dimensional heteronuclear NMR of nitrogen-15 labeled proteins. J Amer Chem Soc. 1989;111:1515–1517. [Google Scholar]
- 54.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 55.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 56.Dosset P, Hus JC, Blackledge M, Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J Biomol NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 57.Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J Amer Chem Soc. 1995;117:12562–12566. [Google Scholar]
- 58.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J Amer Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 59.Clore GM, Driscoll PC, Wingfield PT, Gronenborn AM. Analysis of the backbone dynamics of interleukin-1β using two-dimensional inverse detected heteronuclear 15N-1H NMR spectroscopy. Biochemistry. 1990;29:7387–7401. doi: 10.1021/bi00484a006. [DOI] [PubMed] [Google Scholar]
- 60.Mandel AM, Akke M, Palmer AG., III Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 61.Engh RA, Huber R. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Cryst. 1991;A47:392–400. [Google Scholar]
- 62.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: phi, psi, and Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 63.DeLano WL. The PyMOL molecular graphics system. 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.