

Abstract
Replication of herpes simplex virus takes place in the cell nucleus and is carried out by a replisome composed of six viral proteins: the UL30-UL42 DNA polymerase, the UL5-UL8-UL52 helicase-primase, and the UL29 single-stranded DNA-binding protein ICP8. The replisome is loaded on origins of replication by the UL9 initiator origin-binding protein. Virus replication is intimately coupled to recombination and repair, often performed by cellular proteins. Here, we review new significant developments: the three-dimensional structures for the DNA polymerase, the polymerase accessory factor, and the single-stranded DNA-binding protein; the reconstitution of a functional replisome in vitro; the elucidation of the mechanism for activation of origins of DNA replication; the identification of cellular proteins actively involved in or responding to viral DNA replication; and the elucidation of requirements for formation of replication foci in the nucleus and effects on protein localization.
Keywords: DNA Recombination, DNA Repair, DNA Replication, Herpesvirus, Virus Replication, ICP8, UL42, UL5, UL52, UL9
Introduction
Herpesviruses are found in all animals from molluscs to man. During evolution, the viruses have become tightly associated and co-evolved with their hosts. They seem to cross species borders only by accident, and in such rare instances, they may cause unexpected and severe disease. Herpesviruses have an unusual lifestyle; they cause lytic infection in cells, leading to efficient production of new infectious virus particles, and they also establish latent infections in either non-dividing neuronal cells or cycling cells of the immune system. The latent state is characterized by expression of a very limited set of genes to ascertain maintenance of virus chromosomes and to escape recognition by the immune system. The mechanisms for establishing latency appear to differ considerably for different herpesviruses. In contrast, mechanisms for replication of virus DNA during lytic infection and subsequent formation of infectious particles seem to be evolutionarily conserved. There is one notable exception; the mechanism for recognition of origins of DNA replication and initiation of DNA synthesis differs between the herpesvirus families.
Humans can be infected by eight different herpesviruses. Herpes simplex viruses I and II and varicella zoster virus are alphaherpesviruses. Cytomegalovirus and the roseoloviruses, human herpesviruses 6 and 7, are classified as betaherpesviruses. Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus belong to the gammaherpesvirus subfamily.
In this minireview, we discuss recent developments in replication, recombination, and repair of herpes simplex virus DNA. We will take as our starting point a previous minireview in the Journal of Biological Chemistry that provides an insightful and accurate description of basic mechanisms and components of the herpes simplex virus replication machinery (1). Noteworthy new developments have been (i) the presentation of three-dimensional structures for the DNA polymerase, the polymerase accessory factor, and the single-stranded DNA-binding protein (ssDNA)2; (ii) the reconstitution of a functional replisome in vitro; (iii) the elucidation of the mechanism for activation of origins of DNA replication; (iv) the identification of cellular proteins actively involved in or responding to viral DNA replication; and (v) the elucidation of requirements for formation of replication foci in the nucleus and effects on protein localization.
Structure of Replicating DNA
Early in infection, the herpes simplex virus capsid is transported to the nuclear pores and delivers the 150-kb double-stranded linear DNA into the nucleus. Linear DNA, which does not appear to activate the double-strand break response, is rapidly converted to circular genomes (Fig. 1) (2–4). Circularization is not prevented by inhibitors of viral DNA synthesis or protein synthesis, and circular molecules function as templates for DNA synthesis (2). Experiments using mutant cell lines and siRNA-mediated knockdown of cellular DNA ligases demonstrate that DNA ligase IV/XRCC4 is specifically required for formation of end-less genomes and efficient virus replication (Fig. 1) (5).
FIGURE 1.

Herpes simplex virus DNA replication. Upper, the linear genome is circularized by DNA ligase IV/XRCC4 and serves as template for the viral replication machinery. A first phase of theta-type replication, initiated at three redundant origins of replication oriS and oriL, is followed by rolling circle replication. Note that frequent recombination events generate molecules with a more complex structure (see text). The figure is adapted from Ref. 5. UL and US, unique long and short segments, respectively. Middle, the HSV-1 replication fork. Lower, HSV-1 replication causes a series of structural changes in the cell nucleus characterized by co-localization of replication, repair, and recombination proteins (for further details, see Ref. 18).
The linear genome of HSV-1 DNA is composed of distinct sequence elements arranged in the order ab-UL-b′a′c′-US-ca (where UL is the unique long segment and US is the unique short segment). The genome contains three highly similar and functionally redundant origins of DNA replication: two copies of oriS and one copy of oriL. The a sequences serve as cleavage and packaging signals. The presence of direct terminal repeats at the ends still make it possible that homologous recombination can serve as a less efficient pathway for formation of end-less genomes (6).
The commonly accepted model for HSV-1 replication predicts initial bidirectional theta-type replication, resulting in amplification of circular molecules, followed by a switch to rolling circle replication generating concatemers of viral genomes (Fig. 1). In reality, replicating HSV-1 DNA appears to exist in a complex non-linear branched form from which only small amounts of monomer DNA can be released by restriction enzyme cleavage (7). In contrast, human herpesvirus 6 has a simpler genome structure, a-U-a, and contains only a single origin for lytic DNA replication. The replication intermediates also have a simpler structure. They are not highly branched and consist of head-to-tail concatemers as well as circular monomers or oligomers (8). These observations argue in favor of the traditional model for lytic replication of herpesviruses, but they also demonstrate that the presence of multiple origins of replication and repeated sequence elements generates replication intermediates that may require extensive editing before infectious genomes are produced.
The UL12 alkaline exonuclease seems to be involved in processing of replicated DNA because a knock-out mutant shows severely impaired growth and generates capsids containing abnormal genomes (9). The observation that large numbers of non-infectious yet DNA-containing virus particles are produced in cells infected by a virus lacking the alkaline exonuclease demonstrates the need for proper editing of primary replication products.
Replication Machinery
DNA replication is initiated at the redundant origins of replication oriL and oriS. The origins of replication are recognized and activated by the origin-binding protein (OBP), encoded by the UL9 gene (10, 11).
DNA synthesis is carried out by a replisome composed of a heterodimeric DNA polymerase encoded by the UL30 and UL42 genes; a trimeric helicase-primase encoded by the UL5, UL8, and UL52 genes; and a ssDNA-binding protein commonly referred to as ICP8 (infected cell protein 8), encoded by the UL29 gene (Fig. 1) (1). A functional replisome can be reconstituted from purified proteins (12, 13). The replisome can use a minicircle template for rolling circle replication and generate leading and lagging strand products. Rolling circle replication has also been visualized by electron microscopy (14). The approximate rate of unwinding, 60–65 bp/s, approaches the rate of fork movement in vivo, and it is not further stimulated by the HSV-1 DNA polymerase (15). OBP helicase activity is not required for fork movement, and it cannot replace helicase-primase at the fork (12). A coupling between leading and lagging strand synthesis is suggested by the observation that assembled replisomes are able to carry out synthesis of double-stranded DNA even in the presence of a competing template-primer, but evidence for a direct coupling is still lacking (12). Synthesis of leading and lagging strand products using the minicircle template can be achieved by the UL30-UL42 DNA polymerase and the UL5-UL52 helicase-primase subassembly in the absence of ICP8 and UL8 (12, 13). However, DNA synthesis is stimulated by ICP8, and at least in vitro, UL8 has to be present in seemingly equimolar amounts for stimulation to occur (12). The lagging strand products, ranging between 0.2 and 4 kb in vitro, are likely to be further processed by Fen-1 and DNA ligase I (12, 13, 16). Additional proteins such as the viral uracil glycosylase UL2 may associate with the replisome and contribute to replication fidelity (17).
HSV-1 DNA replication takes place in nuclear foci, which later turn into larger replication compartments (Fig. 1) (18). The formation of replication compartments requires active DNA synthesis. A large number of cellular proteins localize to the replication compartments or co-immunoprecipitate with viral replication proteins such as ICP8 (19). The functional role of such proteins during virus replication remains to be elucidated.
Activation of Origins of DNA Replication
The HSV-1 replicon consists of the replicator sequences oriS and oriL and the initiator protein OBP assisted by ICP8. OBP is a superfamily II DNA helicase with a C-terminal domain for sequence-specific binding to origins of replication. The replicon has evolved from an ancestor common to the roseoloviruses, human herpesviruses 6 and 7, and the alphaherpesviruses (20). oriS from HSV-1 has two binding sites, boxes I and II, for the initiator protein OBP and an additional weak site, box III. Boxes I and III together form a stable hairpin (20). The binding sites are always separated by an AT-rich spacer sequence, and at least for HSV-1, the efficiency of initiation of DNA replication varies periodically with the length of spacer sequence (21, 22). The recognition sequence for OBP, TTCGCAC, is highly conserved among alphaherpesviruses but differs from its counterpart in roseoloviruses (20). OBP binds cooperatively to oriS, and the assembly of a stable complex containing two OBP dimers on oriS as well as ICP8 has been visualized by electron microscopy (23). OBP alone can destabilize the AT-rich spacer sequence (24). In the presence of ATP, the preinitiation complex will proceed to unwind DNA and generate stretches of ∼300-bp single-stranded DNA (ssDNA) covered with ICP8 within 15 min on linear DNA (23).
Biochemical investigations have revealed that linear oriS is converted to ssDNA containing a box I/box III hairpin by OBP and ICP8 in the presence of ATP (25). The hairpin is stably bound, in a sequence-specific manner, by the C-terminal domain of OBP, and the single-stranded AT-rich spacer is contacted by the N-terminal helicase domains (Fig. 2) (26). A dimer of OBP can simultaneously bind two double-stranded DNA ligands but only one copy of a box I/box III hairpin with a 10-nucleotide single-stranded tail (26). It has been argued that this finding reflects a conformational change in OBP, from an origin-binding conformation to a helicase conformation, which can be facilitated by the interaction between ICP8 and the WPXXXGAXXFXXL motif found in the extreme C terminus of OBP (20, 26). When ICP8 is in contact with ssDNA, this interaction can no longer be sustained (27). Thus, OBP seems to be able to attract and deliver ICP8 in an orderly manner to activated oriS (Fig. 2).
FIGURE 2.
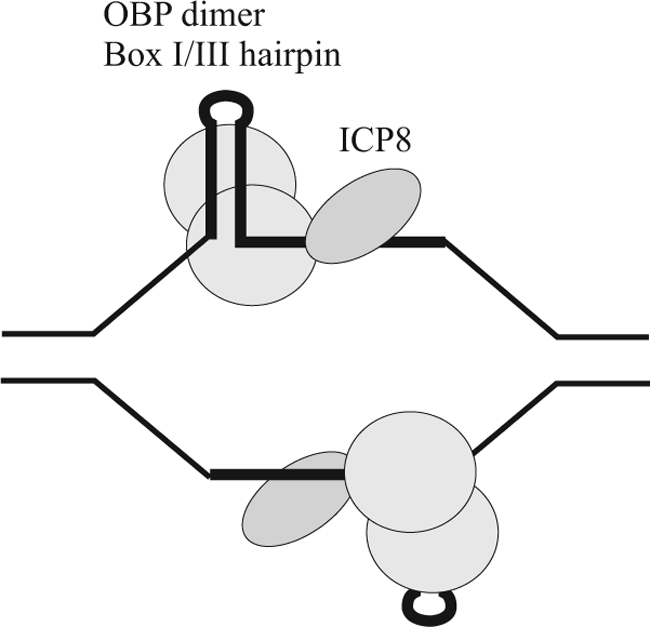
HSV-1 origins of replication activated by OBP and ICP8 in the presence of ATP. A dimer of OBP binds to a hairpin with helicase domains covering 3′-ssDNA and delivers ICP8 to ssDNA (26).
The C-terminal domain of OBP can, as a monomer, bind sequence-specifically to the major groove of DNA (28). However, a second monomer can, at least in vitro, be rapidly recruited form a stable 2:1 complex (29). A ternary 2:1:1 complex between the C-terminal domain of OBP, ICP8, and double-stranded DNA has been observed (29). These observations may suggest that activation of oriS is accompanied by major conformational changes in the C-terminal DNA-binding domain of OBP. It remains to be seen if these features can be recapitulated with a single dimer of OBP or a complex of two dimers on oriS.
It has not yet been possible to establish in vitro a reconstituted system for origin-dependent DNA synthesis, and we therefore lack information about recruitment of helicase-primase to activated oriS and synthesis of the first primers. It is in fact possible that additional factors such as DNA polymerase α-primase may have specific roles during HSV-1 DNA replication (30).
Properties of Replisome Proteins
UL30-UL42 DNA Polymerase
The HSV-1 DNA polymerase consists of a catalytic subunit, the product of the UL30 gene, and an accessory subunit, the UL42 protein, which enhances the processivity of the enzyme. The UL30 polymerase is a family B DNA polymerase that is 25% identical to DNA polymerase δ from Saccharomyces cerevisiae (31, 32). The three-dimensional structure resembles not only that of DNA polymerase δ but also that of phage RB69 DNA polymerase (Fig. 3) (31, 32). A pre-N-terminal domain (amino acids 1–140) precedes the N-terminal domain, which shares three motifs with S. cerevisiae DNA polymerase δ (31). The first motif has a topology resembling that of the OB-fold; motif II has similarity to the RNA-binding motif found in ribonucleoproteins; and the third motif consists of two α-helices connected by a short helix or a loop. The 3′–5′ exonuclease, the palm, the fingers, and the thumb subdomains are arranged as for other family B members. The HSV-1 UL30-UL42 polymerase complex is reported to misincorporate dNTPs as frequently as 1 in 300 incorporation events, and it exhibits an average elongation rate of 44 nucleotides/s (33). Mutational inactivation of the 3′–5′ exonuclease causes decreased fidelity and modest strand displacement activity (16, 34). The antiviral compound acyclovir is incorporated less efficiently than dGTP but causes chain termination, and it is removed with a half-life of 1 h (36). Interestingly, the polymerase can cleave an apurinic/apyrimidinic site in duplex DNA as well as remove the product 5′-deoxyribose phosphate by a lyase activity (37). The presence of a separate UL2 uracil glycosylase as well as apurinic/apyrimidinic and 5′-deoxyribose phosphate lyase activities in the virus polymerase indicates important roles in enhancing replication fidelity (17).
FIGURE 3.

Structure of HSV-1 replication proteins. Shown are the UL30 DNA polymerase (31), the UL42 processivity factor with a C-terminal peptide from UL30 DNA polymerase (42), and a filament composed of ICP8 and ssDNA (65).
The UL42 subunit serves as a processivity factor for the UL30 DNA polymerase (39, 40). The extreme C terminus of UL30 binds tightly to UL42, and a peptide corresponding to the last 18 amino acids of UL30 can in fact inhibit long-chain DNA synthesis (41). A crystal structure of UL42 truncated at its C terminus in complex with a 36-amino acid peptide corresponding to the C terminus of UL30 reveals a remarkable resemblance to a proliferating cell nuclear antigen protomer despite a lack of sequence similarity (Fig. 3) (42). HSV-1 UL42 is active as a monomer and binds tightly to DNA via a positively charged surface opposite the UL30-binding site (43). It is nevertheless capable of translocating efficiently along DNA (43, 44). UL42 is reported to be a phosphoprotein, but the functional consequences of phosphorylation or any other post-translational modification remain to be investigated (45).
UL5-UL8-UL52 Helicase-Primase
The HSV-1 helicase-primase was first identified as a DNA-dependent ATPase and subsequently shown to be composed of three subunits: the UL5 helicase, the UL52 primase, and the accessory protein UL8 (46, 47). A UL5-UL52 subassembly retains all enzymatic activities, but it fails to localize to the nucleus in the absence of UL8 (48). UL8 is also required for efficient unwinding of long duplex DNA substrates in the presence of the ssDNA-binding protein ICP8, and surface resonance experiments reveal that the UL5-UL8-UL52 complex, but not the UL5-UL52 subassembly, interacts physically with ICP8 (49). The same features are recapitulated in experiments using the herpesvirus replisome on a minicircle template (see above). It is commonly assumed that helicase-primase operates as a trimer at the replication fork; however, a recent study suggests that it may also exist in higher order complexes (50).
The UL5 subunit can be identified as a superfamily I DNA helicase, and it operates on the lagging strand template, moving in a 5′–3′ direction. The six helicase motifs have been identified, and functional roles have been established by site-directed mutagenesis (51). The UL52 primase has a catalytic DXD motif and an evolutionarily conserved C-terminal zinc finger, and both are required for function (52–54). The UL8 protein has no apparent cellular homolog that could be identified by sequence analysis. It is reported to interact not only with the UL5-UL52 complex but also with OBP (55), ICP8 (49), and UL30 DNA polymerase (56). The properties of UL52 primase in complex with UL5 have been examined in some detail. It synthesizes 2–13-nucleotide-long products, but only products longer than 8 nucleotides are used by polymerase (30). Efficient priming occurs only at 3′-G-Pyr-Pyr sequences (57); it is an inaccurate enzyme and misincorporates natural NTPs as well as synthetic substrates at high frequency (58, 59). It is also affected by template and primer slippage (58). Mutations in the UL52 zinc finger also abolish the helicase activity of UL5, illustrating the functional interdependence of the two enzymes in a single complex (60).
Helicase-primase is also the target for a novel series of highly efficient non-nucleoside antiviral compounds (61–63). Acquired resistance to these drugs is associated with mutations in the helicase and the primase subunits, thereby emphasizing the interdependence of the two enzymes (63, 64).
ssDNA-binding Protein ICP8
A high-resolution crystal structure of a truncated version of ICP8 lacking only the last 60 amino acids has been reported (Fig. 3) (65). Truncated ICP8 still binds ssDNA but with loss of cooperativity (66). The crystal structure reveals a large N-terminal domain (residues 9–1038) and a small C-terminal domain (residues 1049–1129). The front side of the neck region resembles the OB-fold, and a zinc finger is likely to provide structural integrity. An attractive model suggests how the helical C-terminal domain and the last 60 amino acids act together to account for the formation of a flexible stretched ssDNA molecule (Fig. 3) (64). In the model, 14 nucleotides are covered by one ICP8 molecule, which is in good agreement with experimental results (27).
The functional roles of ICP8 during activation of oriS and propagation of the replication fork have been experimentally addressed in vivo and in vitro (see above), but ICP8 has also properties of a recombinase; it can promote strand annealing as well as helix destabilization. The process has been visualized demonstrating how two coiled ICP8-ssDNA nucleoprotein filaments generate intertwined coiled-coil structures in which strand annealing occurs (66). ICP8 has also been demonstrated to promote strand invasion in vitro, and together with helicase-primase, it can promote strand exchange (67, 68). A possible role could be to promote recombination-dependent DNA synthesis during repair of double-strand breaks (69, 70). It has, however, not yet been possible to assess the significance of the recombinase properties of ICP8 during the course of a virus infection.
Regulation of DNA Synthesis
Herpes simplex virus DNA synthesis requires ICP4-dependent expression of early genes encoding the seven replication proteins discussed above. However, it is also possible that activation of the origins may be controlled during an infectious cycle as well as during reactivation from latency. In addition, coordination of replication with recombination and repair might require modification of replication proteins. It has been noted that the initiator protein OBP is required only during the early part of the lytic infection and that expression of truncated versions composed of the C-terminal DNA-binding domain might serve as an inhibitor of initiation (72, 73). The activity of OBP may be controlled by phosphorylation and subsequent ubiquitin-dependent degradation mediated by the F-box protein NFB42, a mechanism suitable for establishing and maintaining latency in neuronal cells (74, 75).
Repair and Recombination of HSV-1 DNA
Herpes simplex virus replication is intimately coupled to homologous recombination. It is known that homologous recombination is responsible for the inversion of the unique long and short segments of the genome, and it is possible that the frequency of recombination is enhanced by strand breaks produced during replication or by cellular nucleases (76, 77). The enzymes and regulatory molecules required for homologous recombination have only been partially characterized. Knockdown experiments with Rad51, Rad52, and Rad54 may result in a small reduction in virus replication (78, 79). However, if the same experiment is performed using UV light-treated virus particles, the virus yield is reduced by ∼50–150-fold, indicating that homologous recombination serves as an efficient pathway for repair of damaged DNA (79). These observations do not exclude that viral proteins such as ICP8 can perform similar functions. An additional way to promote homologous recombination may involve the alkaline exonuclease UL12 and ICP8; together, they may promote strand exchange in vitro (80). In addition, UL12 interacts with the Mre11-Rad50-Nbs1 complex, providing a connection with cellular pathways for double-strand break repair (81). Active virus replication also activates ATM and downstream signaling (3, 4), but ATR signaling is disabled (82). Because activation of ATR depends on binding of replication protein A to stretches of ssDNA (83), it is tempting to speculate that competition between ICP8 and replication protein A for binding to ssDNA may affect the response to DNA damage.
A tight association between virus replication and DNA repair is suggested by the presence of a gene for uracil glycosylase in the HSV-1 genome. The enzyme forms a complex with the viral DNA polymerase and collaborates to perform DNA repair (37). The presence of a dUTPase and a ribonucleotide reductase lacking allosteric control mechanisms may indicate an enhanced risk for misincorporation of dNTPs during replication (35). It is also of interest to note that components of the mismatch repair system, together with proliferating cell nuclear antigen and replication factor C, can be found at sites of Epstein-Barr virus DNA synthesis (38).
Clearly, there are close connections between virus replication, repair, and recombination. A recent study demonstrates the requirement for homologous recombination as well as nucleotide excision repair and the translesion synthesis DNA polymerase η for replication of UV light-damaged HSV-1 (79). The virus replisome appears to be more sensitive to DNA damage than the transcription apparatus because expression from immediate-early and early genes is largely unaffected under conditions in which replication-dependent expression from late genes is severely reduced (79). The immediate effects of DNA damage on the virus replisome have not been studied.
Concluding Remarks
High-resolution structural information and extensive functional characterization of the replication proteins form the foundation for future research. Detailed studies of replication processes in vivo and in vitro can be combined with an understanding of the cellular contributions as well as response to virus replication. Such studies will shed light on the biology of herpesviruses and also serve to elucidate fundamental cellular processes.
This work was supported by grants from the Swedish Cancer Foundation, the Swedish Research Council, and the Sahlgrenska University Hospital Läkarutbildningsavtal. This minireview will be reprinted in the 2011 Minireview Compendium, which will be available in January, 2012.
- ssDNA
- single-stranded DNA
- OBP
- origin-binding protein.
REFERENCES
- 1. Lehman I. R., Boehmer P. E. (1999) J. Biol. Chem. 274, 28059–28062 [DOI] [PubMed] [Google Scholar]
- 2. Strang B. L., Stow N. D. (2005) J. Virol. 79, 12487–12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lilley C. E., Carson C. T., Muotri A. R., Gage F. H., Weitzman M. D. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5844–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shirata N., Kudoh A., Daikoku T., Tatsumi Y., Fujita M., Kiyono T., Sugaya Y., Isomura H., Ishizaki K., Tsurumi T. (2005) J. Biol. Chem. 280, 30336–30341 [DOI] [PubMed] [Google Scholar]
- 5. Muylaert I., Elias P. (2007) J. Biol. Chem. 282, 10865–10872 [DOI] [PubMed] [Google Scholar]
- 6. Yao X. D., Elias P. (2001) J. Biol. Chem. 276, 2905–2913 [DOI] [PubMed] [Google Scholar]
- 7. Martinez R., Sarisky R. T., Weber P. C., Weller S. K. (1996) J. Virol. 70, 2075–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Severini A., Sevenhuysen C., Garbutt M., Tipples G. A. (2003) Virology 314, 443–450 [DOI] [PubMed] [Google Scholar]
- 9. Porter I. M., Stow N. D. (2004) J. Gen. Virol. 85, 583–591 [DOI] [PubMed] [Google Scholar]
- 10. Elias P., O'Donnell M. E., Mocarski E. S., Lehman I. R. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 6322–6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olivo P. D., Nelson N. J., Challberg M. D. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 5414–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falkenberg M., Lehman I. R., Elias P. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 3896–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stengel G., Kuchta R. D. (2011) J. Virol. 85, 957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skaliter R., Makhov A. M., Griffith J. D., Lehman I. R. (1996) J. Virol. 70, 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Falkenberg M., Elias P., Lehman I. R. (1998) J. Biol. Chem. 273, 32154–32157 [DOI] [PubMed] [Google Scholar]
- 16. Zhu Y., Wu Z., Cardoso M. C., Parris D. S. (2010) J. Virol. 84, 7459–7472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bogani F., Corredeira I., Fernandez V., Sattler U., Rutvisuttinunt W., Defais M., Boehmer P. E. (2010) J. Biol. Chem. 285, 27664–27672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Livingston C. M., DeLuca N. A., Wilkinson D. E., Weller S. K. (2008) J. Virol. 82, 6324–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor T. J., Knipe D. M. (2004) J. Virol. 78, 5856–5866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Olsson M., Tang K. W., Persson C., Wilhelmsson L. M., Billeter M., Elias P. (2009) J. Biol. Chem. 284, 16246–16255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lockshon D., Galloway D. A. (1988) Mol. Cell. Biol. 8, 4018–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gustafsson C. M., Hammarsten O., Falkenberg M., Elias P. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 4629–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Makhov A. M., Lee S. S., Lehman I. R., Griffith J. D. (2003) Proc. Natl. Acad. U.S.A. 100, 898–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He X., Lehman I. R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3024–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aslani A., Olsson M., Elias P. (2002) J. Biol. Chem. 277, 41204–41212 [DOI] [PubMed] [Google Scholar]
- 26. Macao B., Olsson M., Elias P. (2004) J. Biol. Chem. 279, 29211–29217 [DOI] [PubMed] [Google Scholar]
- 27. Gustafsson C. M., Falkenberg M., Simonsson S., Valadi H., Elias P. (1995) J. Biol. Chem. 270, 19028–19034 [DOI] [PubMed] [Google Scholar]
- 28. Simonsson S., Samuelsson T., Elias P. (1998) J. Biol. Chem. 273, 24633–24639 [DOI] [PubMed] [Google Scholar]
- 29. Manolaridis I., Mumtsidu E., Konarev P., Makhov A. M., Fullerton S. W., Sinz A., Kalkhof S., McGeehan J. E., Cary P. D., Griffith J. D., Svergun D., Kneale G. G., Tucker P. A. (2009) J. Biol. Chem. 284, 16343–16353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cavanaugh N. A., Kuchta R. D. (2009) J. Biol. Chem. 284, 1523–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu S., Knafels J. D., Chang J. S., Waszak G. A., Baldwin E. T., Deibel M. R., Jr., Thomsen D. R., Homa F. L., Wells P. A., Tory M. C., Poorman R. A., Gao H., Qiu X., Seddon A. P. (2006) J. Biol. Chem. 281, 18193–18200 [DOI] [PubMed] [Google Scholar]
- 32. Swan M. K., Johnson R. E., Prakash L., Prakash S., Aggarwal A. K. (2009) Nat. Struct. Mol. Biol. 16, 979–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaudhuri M., Song L., Parris D. S. (2003) J. Biol. Chem. 278, 8996–9004 [DOI] [PubMed] [Google Scholar]
- 34. Song L., Chaudhuri M., Knopf C. W., Parris D. S. (2004) J. Biol. Chem. 279, 18535–18543 [DOI] [PubMed] [Google Scholar]
- 35. Averett D. R., Lubbers C., Elion G. B., Spector T. (1983) J. Biol. Chem. 258, 9831–9838 [PubMed] [Google Scholar]
- 36. Hanes J. W., Zhu Y., Parris D. S., Johnson K. A. (2007) J. Biol. Chem. 282, 25159–25167 [DOI] [PubMed] [Google Scholar]
- 37. Bogani F., Boehmer P. E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11709–11714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Daikoku T., Kudoh A., Sugaya Y., Iwahori S., Shirata N., Isomura H., Tsurumi T. (2006) J. Biol. Chem. 281, 11422–11430 [DOI] [PubMed] [Google Scholar]
- 39. Hernandez T. R., Lehman I. R. (1990) J. Biol. Chem. 265, 11227–11232 [PubMed] [Google Scholar]
- 40. Gottlieb J., Marcy A. I., Coen D. M., Challberg M. D. (1990) J. Virol. 64, 5976–5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bridges K. G., Hua Q., Brigham-Burke M. R., Martin J. D., Hensley P., Dahl C. E., Digard P., Weiss M. A., Coen D. M. (2000) J. Biol. Chem. 275, 472–478 [DOI] [PubMed] [Google Scholar]
- 42. Zuccola H. J., Filman D. J., Coen D. M., Hogle J. M. (2000) Mol. Cell 5, 267–278 [DOI] [PubMed] [Google Scholar]
- 43. Randell J. C., Coen D. M. (2001) Mol. Cell 8, 911–920 [DOI] [PubMed] [Google Scholar]
- 44. Komazin-Meredith G., Santos W. L., Filman D. J., Hogle J. M., Verdine G. L., Coen D. M. (2008) J. Biol. Chem. 283, 6154–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marsden H. S., Campbell M. E., Haarr L., Frame M. C., Parris D. S., Murphy M., Hope R. G., Muller M. T., Preston C. M. (1987) J. Virol. 61, 2428–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Crute J. J., Mocarski E. S., Lehman I. R. (1988) Nucleic Acids Res. 16, 6585–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crute J. J., Tsurumi T., Zhu L. A., Weller S. K., Olivo P. D., Challberg M. D., Mocarski E. S., Lehman I. R. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 2186–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barnard E. C., Brown G., Stow N. D. (1997) Virology 237, 97–106 [DOI] [PubMed] [Google Scholar]
- 49. Falkenberg M., Bushnell D. A., Elias P., Lehman I. R. (1997) J. Biol. Chem. 272, 22766–22770 [DOI] [PubMed] [Google Scholar]
- 50. Chen Y., Bai P., Mackay S., Korza G., Carson J. H., Kuchta R. D., Weller S. K. (2011) J. Virol. 85, 968–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Graves-Woodward K. L., Gottlieb J., Challberg M. D., Weller S. K. (1997) J. Biol. Chem. 272, 4623–4630 [DOI] [PubMed] [Google Scholar]
- 52. Dracheva S., Koonin E. V., Crute J. J. (1995) J. Biol. Chem. 270, 14148–14153 [DOI] [PubMed] [Google Scholar]
- 53. Biswas N., Weller S. K. (1999) J. Biol. Chem. 274, 8068–8076 [DOI] [PubMed] [Google Scholar]
- 54. Chen Y., Livingston C. M., Carrington-Lawrence S. D., Bai P., Weller S. K. (2007) J. Virol. 81, 8742–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McLean G. W., Abbotts A. P., Parry M. E., Marsden H. S., Stow N. D. (1994) J. Gen. Virol. 75, 2699–2706 [DOI] [PubMed] [Google Scholar]
- 56. Marsden H. S., McLean G. W., Barnard E. C., Francis G. J., MacEachran K., Murphy M., McVey G., Cross A., Abbotts A. P., Stow N. D. (1997) J. Virol. 71, 6390–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ramirez-Aguilar K. A., Low-Nam N. A., Kuchta R. D. (2002) Biochemistry 41, 14569–14579 [DOI] [PubMed] [Google Scholar]
- 58. Ramirez-Aguilar K. A., Kuchta R. D. (2004) Biochemistry 43, 9084–9091 [DOI] [PubMed] [Google Scholar]
- 59. Urban M., Joubert N., Hocek M., Alexander R. E., Kuchta R. D. (2009) Biochemistry 48, 10866–10881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen Y., Carrington-Lawrence S. D., Bai P., Weller S. K. (2005) J. Virol. 79, 9088–9096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Crute J. J., Grygon C. A., Hargrave K. D., Simoneau B., Faucher A. M., Bolger G., Kibler P., Liuzzi M., Cordingley M. G. (2002) Nat. Med. 8, 386–391 [DOI] [PubMed] [Google Scholar]
- 62. Kleymann G., Fischer R., Betz U. A., Hendrix M., Bender W., Schneider U., Handke G., Eckenberg P., Hewlett G., Pevzner V., Baumeister J., Weber O., Henninger K., Keldenich J., Jensen A., Kolb J., Bach U., Popp A., Mäben J., Frappa I., Haebich D., Lockhoff O., Rübsamen-Waigmann H. (2002) Nat. Med. 8, 392–398 [DOI] [PubMed] [Google Scholar]
- 63. Chono K., Katsumata K., Kontani T., Kobayashi M., Sudo K., Yokota T., Konno K., Shimizu Y., Suzuki H. (2010) J. Antimicrob. Chemother. 65, 1733–1741 [DOI] [PubMed] [Google Scholar]
- 64. Biswas S., Kleymann G., Swift M., Tiley L. S., Lyall J., Aguirre-Hernández J., Field H. J. (2008) J. Antimicrob. Chemother. 61, 1044–1047 [DOI] [PubMed] [Google Scholar]
- 65. Mapelli M., Panjikar S., Tucker P. A. (2005) J. Biol. Chem. 280, 2990–2997 [DOI] [PubMed] [Google Scholar]
- 66. Mapelli M., Mühleisen M., Persico G., van Der Zandt H., Tucker P. A. (2000) J. Virol. 74, 8812–8822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Makhov A. M., Griffith J. D. (2006) J. Mol. Biol. 355, 911–922 [DOI] [PubMed] [Google Scholar]
- 68. Nimonkar A. V., Boehmer P. E. (2003) J. Biol. Chem. 278, 9678–9682 [DOI] [PubMed] [Google Scholar]
- 69. Nimonkar A. V., Boehmer P. E. (2002) J. Biol. Chem. 277, 15182–15189 [DOI] [PubMed] [Google Scholar]
- 70. Nimonkar A. V., Boehmer P. E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10201–10206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Deleted in proof.
- 72. Schildgen O., Gräper S., Blümel J., Matz B. (2005) J. Virol. 79, 7273–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Link M. A., Schaffer P. A. (2007) J. Virol. 81, 10699–106711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Eom C. Y., Lehman I. R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9803–9807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Eom C. Y., Heo W. D., Craske M. L., Meyer T., Lehman I. R. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 4036–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weber P. C., Challberg M. D., Nelson N. J., Levine M., Glorioso J. C. (1988) Cell 54, 369–381 [DOI] [PubMed] [Google Scholar]
- 77. Huang K. J., Ku C. C., Lehman I. R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 8995–9000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kudoh A., Iwahori S., Sato Y., Nakayama S., Isomura H., Murata T., Tsurumi T. (2009) J. Virol. 83, 6641–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Muylaert I., Elias P. (2010) J. Biol. Chem. 285, 13761–13768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Reuven N. B., Staire A. E., Myers R. S., Weller S. K. (2003) J. Virol. 77, 7425–7433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Balasubramanian N., Bai P., Buchek G., Korza G., Weller S. K. (2010) J. Virol. 84, 12504–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mohni K. N., Livingston C. M., Cortez D., Weller S. K. (2010) J. Virol. 84, 12152–12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Choi J. H., Lindsey-Boltz L. A., Kemp M., Mason A. C., Wold M. S., Sancar A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 13660–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]