

Abstract
The spindle assembly checkpoint (SAC) ensures accurate segregation of chromosomes by monitoring kinetochore attachment of spindles during mitosis. Proper progression of mitosis depends on orderly ubiquitination and subsequent degradation of various mitotic inhibitors. At the molecular level, upon removal of SAC, Cdc20 activates E3 ubiquitin ligase anaphase-promoting complex/cyclosome that, along with E2 ubiquitin-conjugating enzyme UbcH10, executes this function. Both Cdc20 and UbcH10 are overexpressed in many cancer types and are associated with defective SAC function leading to chromosomal instability. The precise mechanism of correlated overexpression of these two proteins remains elusive. We show that Cdc20 transcriptionally up-regulates UbcH10 expression. The WD40 domain of Cdc20 is required for this activity. Physical interaction between Cdc20 and anaphase-promoting complex/cyclosome-CBP/p300 complex and its subsequent recruitment to the UBCH10 promoter are involved in this transactivation process. This transcriptional regulatory function of Cdc20 was observed to be cell cycle-specific. We hypothesize that this co-regulated overexpression of both proteins contributes to chromosomal instability.
Keywords: CBP, Cell Cycle, p300, Transcription, Ubiquitin-conjugating Enzyme (Ubc), APC/C, Cdc20, Spindle Assembly Checkpoint, UbcH10
Introduction
Chromosomal instability has been found to be a prominent cause for aneuploidy and consequently the onset of cancer. Proper chromosomal segregation during the mitotic stage of cell division prevents the occurrence of chromosomal instability and thus rules out the generation of aneuploid cells (1, 2). The bipolar segregation of duplicated chromosomes during metaphase to anaphase transition is monitored by spindle assembly checkpoint (SAC).4 Cells are allowed to proceed toward chromosomal segregation after all the kinetochores of sister chromatids are attached to bipolar spindles. But in case any defect occurs at kinetochore-spindle attachment or in the generation of tension across the bipolar spindle arrangement, the cells remain arrested at metaphase by SAC until all the defects are corrected (3). At the biochemical level, this mitotic progression of cells is mediated by sequential ubiquitination and proteasomal degradation of different substrates. The anaphase-promoting complex/cyclosome (APC/C) is the mitotic E3 ubiquitin ligase that, along with E2 ubiquitin carrier protein UbcH10, mediates this ubiquitination activity (4–6). For proper functional activity of APC/C, the presence of one of two WD40 domain-containing adapter proteins, Cdc20 and Cdh1, is required (7). Cdc20 activates APC/C at metaphase, and APC/CCdc20 ubiquitinates securin, thus releasing the endopeptidase separase. Free separase then cleaves the SCC1 subunit of the cohesin complex, which holds together the sister chromatids at metaphase plate. After separation, chromatids move to the opposite poles, and the cells enter the anaphase. Cdc20 is the direct target of SAC. When SAC is on, Cdc20 remains sequestered by the mitotic checkpoint complex (MCC) comprising Mad2, BubR1, and Bub3, and APC/CCdc20 remains inactive (3, 8). Recently, it has been suggested that UbcH10 terminates SAC-mediated mitotic arrest by ubiquitinating Cdc20 and thereby releasing inhibitory MCC from Cdc20 (5). Concordant with that, p31comet blocks Mad2 to sequester Cdc20 further (9). On the contrary, the deubiquitinating enzyme USP44 deubiquitinates Cdc20 so that MCC can sequester it from activating APC/C before the kinetochore attachment to spindle poles (10). Consistent with their mitotic role, both Cdc20 and UbcH10 accumulate gradually during G2 phase with a peak at mitosis and then sharply decrease as the cells exit from mitosis (11–13). Down-regulation of Cdc20 as well as UbcH10 occurs by APC/CCdh1-mediated ubiquitination assisted by UbcH10 itself after ensuring the proper mitotic exit upon degradation of all mitotic substrates (12, 13).
Defects in the functioning of SAC may lead to chromosomal missegregation thereby resulting in the generation of aneuploid cells. Mutations and/or deregulated expression of various SAC genes have been found in a number of cancer tissues (14–16). Cdc20 overexpression has been observed in various cancer tissues (17) and reported to cause premature anaphase onset resulting in aneuploidy in cancer cells (18). Expression of the Mad2-binding deficient and thereby SAC-defective mutant Cdc20 promotes tumor formation in mice (19). These results suggest that proper functioning of Cdc20 is crucial for orderly execution of cell division. However, the downstream effects of Cdc20 overexpression in relation to SAC inactivation and aneuploidization largely remain unknown. In this regard, it is also noteworthy that UbcH10 overexpression may override SAC-mediated cell cycle arrest thereby resulting in defective chromosomal segregation and subsequent onset of aneuploidy (5, 20). Therefore, regulation of UbcH10 expression might also be an essential step for proper function of the checkpoint. Indeed, UbcH10 expression is found to be up-regulated in a number of cancer tissues of different origins, and this up-regulation is related with poor prognosis in some cancer types (21–25). The UBCH10 gene encoding locus 20q13.1 is also reported to be amplified in several tumors (26). Dominant negative UbcH10 blocks the APC/C-mediated ubiquitination activity, thus preventing the degradation of mitotic substrates causing cells to accumulate at mitosis (27). Moreover, small interfering RNA (siRNA)-mediated down-regulation of UbcH10 expression causes a lower rate of proliferation in both normal and cancer cells (25, 26, 28). However, the mechanism of UbcH10 up-regulation in oncogenic condition is still largely unresolved.
Thus, maintenance of levels of SAC proteins is an important regulatory mechanism to check aneuploidy and subsequent tumorigenesis. Besides degradation of cell cycle regulators via the ubiquitin proteasome pathway, another emerging mechanism for cell cycle control is transcriptional regulation of its components. Transcriptional deregulation of the SAC gene MAD2 was correlated with mitotic abnormality in human cancers (29). Promoter methylation-associated differential BubR1 expression is considered as a key determinant of checkpoint control in cancer cells (30). Cdc20 expression is transcriptionally regulated by tumor suppressor protein p53 (31, 32). p53 also regulates transcription of other SAC genes such as MAD1 and BUB1B (33, 34). It has been reported that WD repeat-containing mitotic checkpoint proteins can act as transcriptional repressors during interphase (35). In a previous study, it was shown that CBP/p300 and APC/C cooperate to regulate transcription (36). APC/C-mediated activation of CBP/p300 was found to regulate acetyltransferase activity of CBP/p300. In this study, we show that WD40 repeat containing protein Cdc20 modulates the APC/C-CBP/p300 complex to regulate the transcription of UBCH10 gene.
EXPERIMENTAL PROCEDURES
Cell Culture, Synchronization, Drug Treatment, and Transfection
Human cell lines HeLa and HCT116 were purchased from the American Type Culture collection (Manassas, VA). UPCI:SCC084 and UPCI:SCC104 cells were kind gifts from Dr. Susanne M. Gollin (University of Pittsburgh). HepG2 cells were kindly provided by Dr. S. Adhya (Indian Institute of Chemical Biology, India). All the cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum and antibiotics (1% PenStrep glutamine and 0.006% gentamicin, Invitrogen) in a 37 °C incubator under a 5% CO2 atmosphere. For cell synchronization, cells were treated with cold thymidine (2.5 mm; United States Biochemical, Cleveland, OH) for 16 h followed by an 8-h release. The thymidine treatment was repeated for another 22 h. Following this double thymidine block, cells were released in thymidine-free complete medium and harvested at various time points. For mitotic arrest, cells were treated with nocodazole (100 ng/ml; Sigma) for 16 h before harvesting. The proteasomal inhibitor MG115 (Sigma) was added 5 h before harvesting at a final concentration of 25 μm. Transient transfections were done with various plasmids and siRNA constructs in different cell lines using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. Except in case of synchronization experiments, all transient transfections were performed either for 48 h (expression analysis) or for 72 h (siRNA analyses). The siRNA transfection of synchronized cells was done 4 h before the first thymidine addition.
Plasmids and siRNA Constructs
UBCH10 promoter regions were amplified from human genomic DNA with the primers listed in supplemental Table S1. The amplified regions were cloned into the linearized pTZ57R/T (Fermentas, Lithuania) by the T/A cloning method. The fragments were then subcloned into luciferase reporter vector pGL3 basic (Promega, Madison, WI) using restriction enzymes SacI and HindIII (New England Biolabs, Beverly, MA). The full-length FLAG-tagged Cdc20 expression plasmid pCDNA5/FRT/TO FLAG-CDC20 was a kind gift from Dr. Jonathon Pines (Gurdon Institute, Cambridge, UK). Another full-length Cdc20 expression plasmid pJS55/hCDC20 was a kind gift from Dr. Joan V. Ruderman (Harvard Medical School, Boston). Del N and Del C mutants of Cdc20 in pEGFP-N3 were a gift from Dr. J. Weinstein (Amgen, Thousand Oaks, CA). hTERT-luciferase expression plasmid was a kind gift from Dr. Riccardo Dalla-Favera (Columbia University, New York). Various siRNA constructs directed against Cdc20 (sc-36160, Santa Cruz Biotechnology, used in Figs. 1, C and D, 2, D and E, 4F, and 5, A and C, and supplemental Fig. S6; catalog no. AM 16706, Ambion (Austin, TX) used in Figs. 1, E and G, 2G, and Fig. 6, B and C, and supplemental Fig. S7), UbcH10 (Santa Cruz Biotechnology), Cdc27 and Mad2 (Santa Cruz Biotechnology), and scrambled control (Ambion) were used at a final concentration of 80 nm.
FIGURE 1.

Cdc20 positively regulates UbcH10 expression. A, ectopic expression of Cdc20 up-regulates endogenous UbcH10 mRNA. HepG2 cells were transiently transfected with 0, 500, and 1000 ng of pCDNA5-FLAG-CDC20 expression plasmids. Total RNA was isolated and reverse-transcribed. cDNAs were subjected to quantitative real time PCR (qPCR) using primers for CDC20, UBCH10, and BUB3. Relative expression values were normalized to the G3PDH transcripts levels. The data represent three independent determinations (average ± S.E.). B, ectopic expression of Cdc20 up-regulates endogenous UbcH10 protein. HepG2 cells were transiently transfected with 0, 250, 500, and 1000 ng of pCDNA5-FLAG-CDC20 expression plasmids, and cell extracts were prepared followed by Western blot analysis with antibodies against FLAG, UbcH10, Mad2, and β-actin. C, knockdown of Cdc20 down-regulates endogenous UbcH10 mRNA. HepG2 cells were transiently transfected with either siRNA oligos against Cdc20 mRNA or scrambled siRNA oligo as control. Total RNA was isolated and reverse-transcribed. cDNAs were subjected to quantitative real time (RT)-PCR using primers for CDC20, UBCH10, and MAD2. Values were normalized to the values of G3PDH transcripts. The data represent three independent determinations (average ± S.E.). D, knockdown of Cdc20 down-regulates endogenous UbcH10 protein. HepG2 cell extracts from Cdc20 siRNA-transfected cells were prepared followed by Western blot analysis with antibodies against Cdc20, UbcH10, BubR1, Bub3, and Mad2 and β-actin. E, rescue of UbcH10 down-regulation in Cdc20 knockdown cells. HepG2 cells were transiently transfected with either siRNA oligos to Cdc20 mRNA or scrambled siRNA oligo as control. One set of Cdc20 siRNA-treated cells was transfected with pCDNA5-FLAG-CDC20. Cell extracts were prepared followed by Western blot analysis with antibodies against Cdc20, UbcH10, and β-actin. F, decreased UbcH10 expression in HepG2 cells does not alter Cdc20 expression. HepG2 cells were transiently transfected with siRNA oligos to UbcH10 mRNA or scrambled siRNA oligo as control. Cell extracts were prepared followed by Western blot analysis with antibodies against UbcH10, Cdc20, and β-actin. G, alteration of UbcH10 expression by Cdc20 is not due to proteasomal degradation. HepG2 cells were transiently transfected with siRNA oligos to Cdc20 mRNA or scrambled siRNA oligo as control and treated with proteasomal inhibitor drug MG115. Cell extracts were prepared followed by Western blot analysis with antibodies against Cdc20, UbcH10, and β-actin.
FIGURE 2.

Cdc20 up-regulates transcription of UBCH10. A, map of UBCH10 promoter regions used for luciferase reporter assay. pSN1 construct contains the −694 to +39 region of UBCH10 gene cloned into pGL3 basic vector. Two deletion constructs, pSN2 and pSN3, contain the −322 to +39 and −142 to +39 regions of the UBCH10 gene, respectively. B, ectopic expression of Cdc20 up-regulates UBCH10 promoter activity. HepG2 cells were transiently co-transfected with pSN1 (100 ng) along with 0, 250, and 500 ng of pCDNA5-FLAG-CDC20. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). Lysates were also subjected to Western blot analysis with antibodies against FLAG and β-actin. C, mitotic arrest activates the UBCH10 promoter and increases in Cdc20 protein. HepG2 cells were transiently transfected with pSN1 (100 ng) and treated with the spindle-disrupting drug nocodazole (100 ng/ml). Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). Lysates were also subjected to Western blot analysis with antibodies against Cdc20 and β-actin. D, knockdown of Cdc20 suppresses activity of UBCH10 but not of hTERT promoter. HepG2 cells were transiently co-transfected with pSN1 (100 ng) or hTERT promoter luciferase plasmid (100 ng) and siRNA oligos directed against Cdc20 mRNA or scrambled siRNA as control. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). Lysates were also subjected to Western blot analysis with antibodies against Cdc20 and β-actin. E, nocodazole treatment does not have any effect on UBCH10 promoter activity upon Cdc20 knockdown. HepG2 cells were transiently co-transfected with pSN1 (100 ng) and siRNA oligos directed against Cdc20 mRNA or scrambled siRNA oligo as control and treated with or without nocodazole. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). F, Mad2 knockdown does not alter UBCH10 promoter activity. HepG2 cells were transiently co-transfected with pSN1 (100 ng) and siRNA oligos directed against Mad2 mRNA or scrambled siRNA oligo as control. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). Lysates were also subjected to Western blot analysis with antibodies against Mad2 and β-actin. G, SAC does not influence Cdc20-mediated regulation of the UBCH10 promoter activity. HepG2 cells were transiently co-transfected with pSN1 (100 ng) and siRNA oligos directed against Cdc20 mRNA or Mad2 mRNA or scrambled siRNA oligo as control. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.).
FIGURE 4.

Cdc20, APC/C, and CBP/p300 occupy UBCH10 promoter and cause chromatin remodeling. A, Cdc20 occupies the UBCH10 promoter in UPCI:SCC104 cells. ChIP assay was done using antibodies specific for RNA pol II and Cdc20 and no antibody as control. Precipitated DNA was PCR-amplified using primers encompassing −322- to +39-nt region of UBCH10 promoter relative to the transcription start site. B, Cdc20 and CBP occupy the UBCH10 promoter in HepG2 cells. ChIP assay was performed using antibodies specific for RNA pol II, Cdc20, and CBP and normal IgG as control. Precipitated DNA was PCR-amplified as indicated in A. C, APC/C is present on UBCH10 promoter in HepG2 cells. ChIP assay was performed using antibodies specific for RNA pol II, Cdc20, CBP, p300, Cdc27, and normal IgG as control. Precipitated DNA was PCR-amplified as indicated in A. D, MCC is not present on UBCH10 promoter. ChIP assay was performed using antibodies specific for RNA pol II, Cdc20, CBP, Mad2, and normal IgG as control. Precipitated DNA was PCR-amplified as indicated in A. E, ectopic expression of Cdc20 enhances histone acetylation of the UBCH10 promoter. HepG2 cells were transiently transfected with pCDNA5-FLAG-CDC20 or empty vector as control. Chromatin immunoprecipitation was done using antibodies specific for RNA pol II, Cdc20-acetylated histone 3 (Lys-9/14), and normal IgG as control. Precipitated chromatin was estimated by quantitative real time PCR. The results are expressed as percent of input. Bars represent mean ± S.E. of two independent determinations from two separate chromatin preparations. F, knockdown of Cdc20 reduces histone acetylation of UBCH10 promoter. HepG2 cells were transiently transfected with siRNA oligos to Cdc20 mRNA or scrambled siRNA oligo as control. Precipitated DNA was PCR-amplified as indicated in A.
FIGURE 5.

Cdc20 regulates APC/C-CBP/p300 interaction and influences their recruitment to the UBCH10 promoter. A, APC/C-CBP interaction is reduced in HepG2 cells upon Cdc20 knockdown. HepG2 cells were transiently transfected with siRNA oligos to Cdc20 mRNA or scrambled siRNA oligo as control. Whole cell extracts were immunoprecipitated (IP) with antibodies specific for Cdc27 or CBP or normal IgG. Immunocomplexes and input (20% of the whole cell extracts) were probed with antibodies to the indicated proteins. WB, Western blot. B, APC/C and CBP recruitment to the UBCH10 promoter is reduced upon Cdc20 knockdown. HepG2 cells were transiently transfected with siRNA oligos to Cdc20 mRNA or scrambled siRNA as control. ChIP assay was done using antibodies specific for Cdc20, Cdc27, and CBP and normal IgG as control. Precipitated DNA was PCR-amplified using primers encompassing −322- to +39-nt region of the UBCH10 promoter relative to the transcription start site. C, Cdc20 and CBP recruitment to the UBCH10 promoter is reduced upon Cdc27 knockdown. HepG2 cells were transiently transfected with siRNA oligos to Cdc27 mRNA or scrambled control. ChIP assay was performed using antibodies specific for Cdc20, Cdc27, and CBP and IgG control. Precipitated DNA was PCR-amplified using primers as indicated in B. D, knockdown of Cdc27 suppresses UBCH10 promoter activity. HepG2 cells were transiently co-transfected with pSN1 (100 ng) and siRNA oligos directed against Cdc27 mRNA or scrambled siRNA as control. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). Lysates were also subjected to Western blot analysis with antibodies against Cdc27 and β-actin. E, ectopic expression of Cdc20 does not activate UBCH10 promoter in Cdc27 knockdown cells. HepG2 cells were transiently co-transfected with pSN1 (100 ng) and pCDNA5-FLAG-CDC20 and/or siRNA oligos directed against Cdc27 mRNA or scrambled control. Protein lysates were isolated for luciferase assay. The data represent three independent determinations (average ± S.E.). Lysates were also subjected to Western blot analysis with antibodies against FLAG, Cdc27, and β-actin.
FIGURE 6.

Cdc20 regulates UbcH10 expression in a cell cycle-specific manner. A, schematic diagram of cell synchronization procedure. Transfection, thymidine addition, and different incubation times are shown. B, cell cycle-specific correlation between Cdc20 and UbcH10 mRNA expression. HepG2 cells were transiently transfected with siRNA oligos to Cdc20 mRNA or scrambled control and synchronized by double thymidine block as shown in A. Total RNA was isolated from cells at every 2-h interval from the time of the second thymidine release up to 14 h and reverse-transcribed. cDNAs were subjected to quantitative real time PCR (qPCR) using primers for CDC20 and UBCH10. Relative expression values were normalized to the G3PDH transcript levels. The data represent three independent determinations (average ± S.E.). C, cell cycle-specific correlation between Cdc20 and UbcH10 expression at the protein level. HepG2 cells were transiently transfected with siRNA oligos to Cdc20 mRNA or scrambled control and synchronized by double thymidine block as shown in the schematic (A). Protein lysates were prepared with cells at every 2-h interval from the time of the second thymidine release up to 14 h and subjected to Western blot analysis with antibodies against Cdc20, UbcH10, and β-actin, respectively. D, Cdc20-APC/C interaction is cell cycle-specific. HepG2 whole cell extracts were immunoprecipitated (IP) with antibodies specific for Cdc27 or normal IgG. Immunocomplexes and inputs (20% of the whole cell extracts) were probed with antibodies to the indicated proteins by Western blot (WB) analysis. E, Cdc20 recruitment to UBCH10 promoter is cell cycle-specific. HepG2 cells were synchronized by double thymidine block, and cells were harvested at 2, 8, and 14 h. from the time of second thymidine release. ChIP analysis was performed by qPCR using primers designed to amplify sequences between the −322- and +39-nt region relative to the transcription start site. The results shown are the percent of input where immunoprecipitations were performed using either control IgG and antibody against Cdc20. Bars represent mean ± S.E. of two independent determinations from two separate chromatin preparations. F, co-regulated expression of Cdc20 and UbcH10 influences metaphase to anaphase transition. HepG2 cells were synchronized by double thymidine block, transiently transfected with pCDNA5-FLAG-CDC20, and treated with either siRNA oligos to UbcH10 mRNA or scrambled siRNA as control. After the second thymidine release, these cells were treated with nocodazole and harvested at 10, 12, and 14 h after release (left panel). Cell extracts were prepared followed by Western blot analysis with antibodies against cyclin B1, FLAG, UbcH10, and β-actin. G, Cdc20-mediated regulation of UbcH10 influences mitotic progression. HepG2 cells were synchronized by double thymidine block, transiently transfected with pCDNA5-FLAG-CDC20, and treated with either siRNA oligos to UbcH10 mRNA or scrambled siRNA as control. After the second thymidine release, these cells were treated with nocodazole and fixed at 10, 12, and 14 h after release, stained with DAPI, and visualized under a fluorescence microscope. Frequencies of mitotic cells were calculated as mitotic index. The data represent three independent determinations (average ± S.D.).
Quantitative Real Time PCR
Total RNA from cell lines was isolated using TRIzol (Invitrogen) according to manufacturer's protocol. Five micrograms of isolated RNA was treated with DNase (Promega, Madison) in a total volume of 10 μl, and 2 μl of this mixture was used for cDNA preparation using random hexamer and MMLV-RT (Promega, Madison). Real time PCR was performed on the 7500 Fast Real Time PCR system (Applied Biosystems, Foster City) using power SYBR Green PCR master mix (Applied Biosystems, Foster City). Primer sets for CDC20, UBCH10, BUB3, and GAPDH are listed in supplemental Table S2. The comparative threshold cycle method (ΔΔCt) was used to quantify relative amounts of product transcripts with GAPDH as endogenous reference control.
Western Blotting and Antibodies
The whole cell lysate or the immunocomplexes were resolved by SDS-PAGE (6–12% gel) and transferred onto a PVDF membrane (Millipore, Billerica, MA). Various primary antibodies used are mouse monoclonal Cdc20 (E-7, Santa Cruz Biotechnology), mouse monoclonal FLAG (Sigma), rabbit polyclonal UbcH10 (Upstate, Billerica, MA), mouse monoclonal Mad2, goat polyclonal BubR1, Bub3, and Cdc27 (Santa Cruz Biotechnology), mouse monoclonal CBP (Santa Cruz Biotechnology), mouse monoclonal cyclin B1 (Cell Signaling Technology, Beverly, MA), and mouse monoclonal α-tubulin and β-actin antibody (Sigma). Bands were detected using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL) after treating with HRP-conjugated secondary antibody (Sigma).
Luciferase Assay
After transfection and/or treatment, cells were washed with phosphate-buffered saline (PBS) and subsequently lysed with luciferase cell culture lysis buffer supplied with the luciferase assay kit (Promega, Madison, WI). After a short vortex, whole cell lysates were centrifuged at 4 °C at 13,000 rpm for 2 min, and 15–30 μl of supernatants was mixed with 30–60 μl of luciferase assay substrate. Luminescence was measured as relative luciferase unit in a GLOMAX luminometer (Promega). Total protein concentration in each lysate was measured by protein assay reagent (Bio-Rad) and then used to normalize the luciferase activity of each lysate. Each assay was performed in duplicate and repeated three times. Fold activation values were calculated as mean of three separate experiments.
Chromatin Immunoprecipitation (ChIP) Assay
Sonicated genomic DNA (Bioruptor, Diagenode, NJ) was subjected to ChIP assay using Quick ChIP kit from Imgenex Corp. (San Diego). Immunoprecipitation was carried out with 10 μg each of antibodies specific for RNA pol II, Cdc20, CBP, p300, Cdc27, and Mad2 (Santa Cruz Biotechnology), acetyl-histone H3 (Lys-9/Lys-14) (Cell Signaling Technology), and normal IgG control (Sigma). PCR amplification of immunoprecipitated chromatin was done using primers indicated in the Table S3.
Co-immunoprecipitation Assay
HepG2 cells, transiently transfected with Cdc20/negative control siRNA, were washed with PBS and resuspended in 50 mm Tris (pH 7.5), 15 mm EDTA, 150 mm NaCl, 0.1% Triton X-100, 0.01% SDS buffer containing protease inhibitor mixture (Sigma). The cells were lysed by freeze-thaw cycle, and the supernatants were incubated overnight with antibodies specific for Cdc20, Cdc27, and CBP (Santa Cruz Biotechnology). Normal IgG (Sigma) was taken as a control for immunoprecipitation. The antibody-protein complex was precipitated with protein-G-Sepharose beads (Bangalore Genei, Bangalore) and washed, and subsequently the protein complex was eluted by SDS-lysis buffer. The eluted samples were then processed for Western blot analysis with antibodies specific for either Cdc20 or Cdc27 or CBP (Santa Cruz Biotechnology).
Determination of Mitotic Index
Synchronized HepG2 cells were treated with nocodazole (100 ng/ml) for various time periods (as mentioned in respective cases). Cells were fixed with ice-cold acetomethanol (1:1) and stained with 4,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Frequencies of mitotic cells were counted under a fluorescence microscope (Leica DM 3000, Leica Microsystems, Heerbrugg, Switzerland) among 200–300 cells each time. Mitotic index was calculated as mean of three separate experiments.
FACS Analysis
Cells were synchronized by thymidine treatment, and after release, ∼106 cells were harvested at respective time points, washed twice with PBS, and resuspended in 0.25 ml of cold PBS. Cells were fixed by adding 2 ml of cold 70% ethanol dropwise into the samples while vortexing gently and then incubated at 4 °C for a minimum of 24 h. After fixation, cells were washed twice with PBS and resuspended in 1 ml of PBS containing 100 μg/ml propidium iodide (Sigma) and 20 μg/ml RNase A (Invitrogen). Fixed cells were kept at room temperature for 40 min and then analyzed by FACS (FACSARIATM III, BD Biosciences).
Bioinformatic Analysis
The dataset of overexpression of Cdc20 and UbcH10 in primary tumors was obtained from Oncomine 4.3 research edition database. Cancer versus normal datasets of Cdc20 overexpression with fold change between ≥2 and ≤50 (p value ≤0.05) were selected. Corresponding values of fold change of UbcH10 overexpression were taken from these datasets. Correlation between Cdc20 and UbcH10 expression was calculated using Wessa.Net-Free statistics and forecasting software (Calculators) version 1.1.23-r6.
RESULTS
UbcH10 Expression Is Correlated with Cdc20 Expression
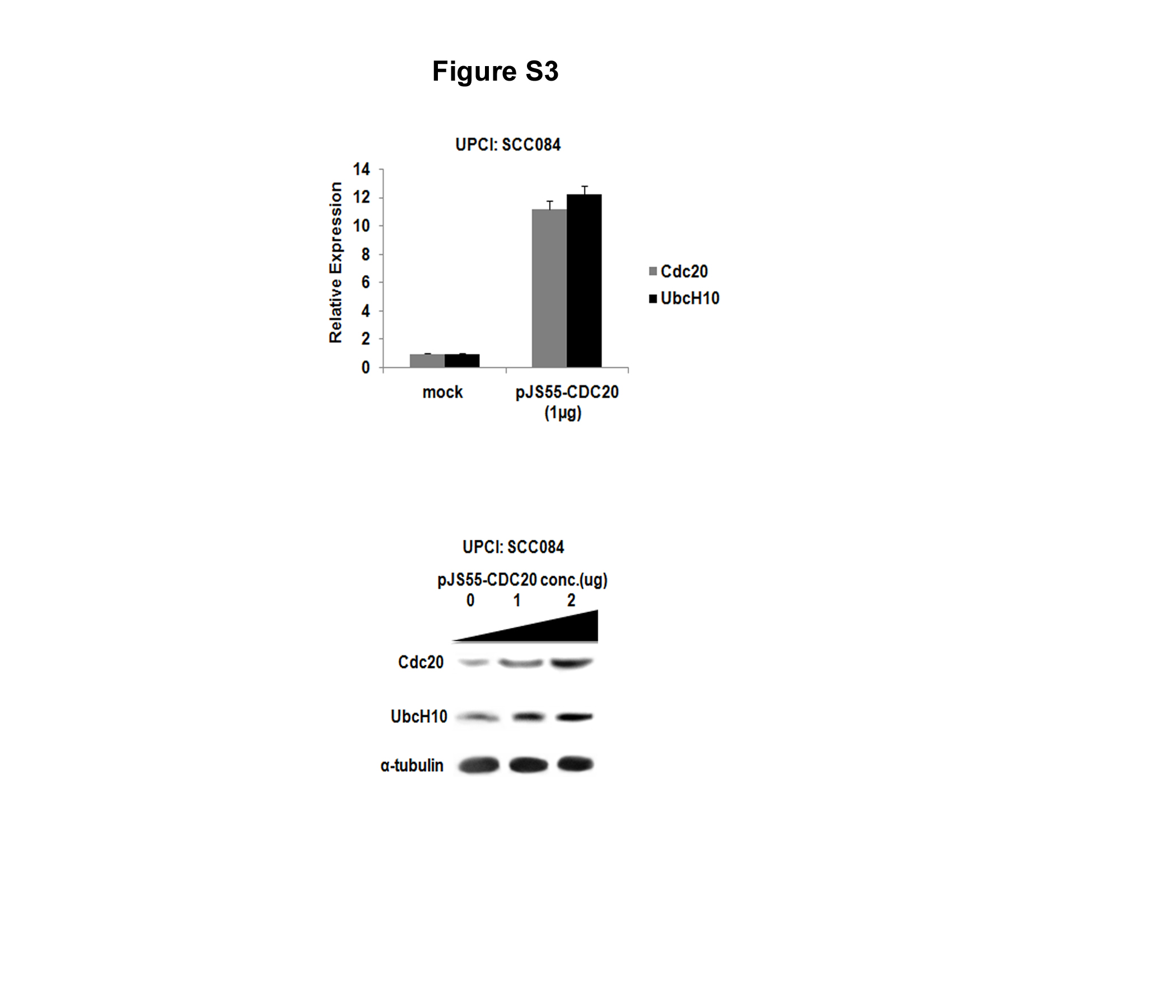
An initial clue to the fact that Cdc20 might regulate the expression of UbcH10 came from the observation that both primary tumors and cancer cells expressing high levels of Cdc20 also had higher levels of UbcH10 (supplemental Figs. S1 and S2, A and B). This led us to examine the effect of ectopic expression of Cdc20 on UbcH10 expression. Up-regulation of UbcH10 was noticed both at the mRNA and protein levels upon increasing Cdc20 expression in HepG2 and UPCI:SCC084 cells (Fig. 1, A and B; supplemental Fig. S3, A and B). Conversely, Cdc20 knockdown significantly lowered the expression of UbcH10 in HepG2 cells (Fig. 1, C and D). Under both conditions, Mad2, Bub3, and BubR1 and other important SAC components remained unaltered (see Fig. 1, A–D). To rule out the possibility of the off-target effect of siRNA sequence, we performed the rescue experiment using another siRNA from a different source (see under “Experimental Procedures”). As shown in Fig. 1E, the ectopic expression of Cdc20 rescued the level of UbcH10 even in Cdc20 knockdown conditions. The specificity of the effect of Cdc20 on UbcH10 expression was also indicated from the observation that knockdown of UbcH10 did not alter Cdc20 expression (Fig. 1F). To eliminate the possibility of the decrease in UbcH10 level due to protein degradation, Cdc20 siRNA transfected HepG2 cells were treated with or without the proteasomal inhibitor MG115. As shown in Fig. 1G, treatment with MG115 failed to restore the UbcH10 protein level. In summary, these results suggest that Cdc20 positively regulates the expression of UbcH10.
Cdc20 Positively Regulates the Transcription of UBCH10
To address the possibility that Cdc20 regulates UbcH10 expression at the transcription level, we cloned a 694-bp upstream region of the UBCH10 gene (NM_007019.2, −694 to +39 nt), including the transcription start site into a luciferase assay vector pGL3 basic (pSN1, Fig. 2A). A dose-dependent increase in luciferase activity was observed upon co-transfection of pSN1 with Cdc20 expression vector pCDNA5-FLAG-CDC20 in HepG2 cells (Fig. 2B). However, a similar effect of Cdc20 was not observed with the UBCH10 promoter construct that contains the same sequence in reverse orientation (pSN4, supplemental Fig. S4). The up-regulation of UBCH10 promoter-driven luciferase (pSN1) activity was also observed in nocodazole-treated HepG2 cells with the concomitant increase in Cdc20 level (Fig. 2C).
The specificity of the Cdc20-mediated activation of UBCH10 promoter was established from the following observations: (a) knockdown of Cdc20 down-regulated UBCH10 promoter activity in HepG2 cells (Fig. 2D). However, under this condition, the activity of hTERT promoter remained unaltered (Fig. 2D). (b) Up-regulation of the UBCH10 promoter activity was not observed in nocodazole-treated HepG2 cells upon Cdc20 knockdown (Fig. 2E). It is to be noted that Cdc20 knockdown led to the mitotic arrest of HepG2 cells similar to that of nocodazole treatment as revealed by the FACS analysis (supplemental Fig S7B) as well as stabilization of cyclin B1 (supplemental Fig S7C). (c) Finally, knockdown of Mad2 did not affect the UBCH10 promoter activity (Fig. 2F). Interestingly, the apparent down-regulation of UbcH10 in Cdc20-depleted cells was unaffected by eliminating SAC via co-depletion of Mad2 (Fig. 2G).
Deletion analysis of the UBCH10 promoter revealed that the sequence between the −322 to −142 nt of the transcription start site was required to confer Cdc20 response of the UBCH10 promoter (Fig. 3A). The WD40 repeat region of Cdc20 (255–355 amino acids) is required for various protein-protein interactions (37). We investigated the possibility of this WD40 region playing any role in the transcriptional regulatory function of Cdc20. Toward that end, we used both the N-terminal deletion (Δ1–167 amino acids) and C-terminal deletion (Δ168–499 amino acids) constructs of the CDC20 gene to test for their ability to induce UBCH10 promoter activity (Fig. 3B) (38). Co-transfection of either of these deletion constructs with pSN1 showed that the WD40 region containing Del N construct (Del N-CDC20) resulted in up-regulation of promoter activity, although there was no change in luciferase activity upon transfection of the Del C construct (Del C-CDC20) that lacks the WD40 domain (Fig. 3C). These results suggest that Cdc20 regulates transcription of UBCH10, a hitherto unknown function of Cdc20, and that the WD40 region of Cdc20 is required in this transcription regulation function.
FIGURE 3.

Characterization of Cdc20-responsive region on the UBCH10 promoter and involvement of WD40 repeats of Cdc20 in UBCH10 transcription regulation. A, −322- to −142-nt region of the UBCH10 promoter is required for Cdc20 response. HepG2 cells were transiently co-transfected with 100 ng of each of two deletion constructs pSN2 or pSN3 and 0 or 250 ng of pCDNA5-FLAG-CDC20 plasmid. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.). B, map of Cdc20 deletion constructs (DelC-CDC20 and DelN-CDC20) are shown along with full-length CDC20 (GM1). WD40 repeat region is marked. aa, amino acids. C, WD40 repeat region of Cdc20 is required for UBCH10 transcription regulation. HepG2 cells were transiently co-transfected with pSN1 (100 ng) and 0 or 250 ng of GM1 or DelC-CDC20 or DelN-CDC20 construct. Protein lysates were prepared for luciferase assay. The data represent three independent determinations (average ± S.E.).
Cdc20 Interacts with UBCH10 Promoter in Vivo and Promotes Chromatin Remodeling
We next determined whether Cdc20 was present in the transcription complex on the UBCH10 promoter by chromatin immunoprecipitation (ChIP) assay. ChIP analysis using primers between −322- and +39-nt region showed that Cdc20 was physically present on the UBCH10 promoter in both HepG2 and UPCI:SCC104 cell lines (Fig. 4, A and B). The specificity of Cdc20 recruitment to the UBCH10 promoter was established by the following experiments: (a) there was no amplification of the immunoprecipitated DNA using primers from the flanking regions of the UBCH10 promoter (supplemental Fig. S5A); (b) no amplification from the immunoprecipitated DNA was observed using primers from the promoter of an unrelated gene TYR (NM_000372.3) (supplemental Fig. S5B), and (c) quantitative ChIP PCR analysis revealed a dose-dependent increase in Cdc20 recruitment to the UBCH10 promoter (supplemental Fig S5C).
We also observed recruitment of CBP/p300 to the UBCH10 promoter by ChIP assay (Fig. 4, B and C). Interestingly, there is a report that the APC/C and CBP/p300 cooperate to regulate transcription (36, 39). Therefore, we examined the recruitment of APC/C to UBCH10 promoter in HepG2 cells. ChIP assay using antibody against Cdc27, a subunit of APC/C (40), revealed the presence of the same on the UBCH10 promoter (Fig. 4C). We also performed ChIP assay using anti-Mad2 antibody to exclude the presence of MCC on the UBCH10 promoter (Fig. 4D). Finally, we showed that there was increased acetylation of histone H3 lysine 9/14 of the UBCH10 promoter upon ectopic expression of Cdc20 (Fig. 4E). On the contrary, Cdc20 down-regulation resulted in decreased acetylation of the same on the UBCH10 promoter (Fig. 4F). All these observations suggest that recruitment of Cdc20 together with APC/C and p300/CBP causes chromatin remodeling to activate the UBCH10 promoter.
Cdc20 Activates UBCH10 by Modulating Transcriptional Activity of APC/C-CBP/p300 Complex
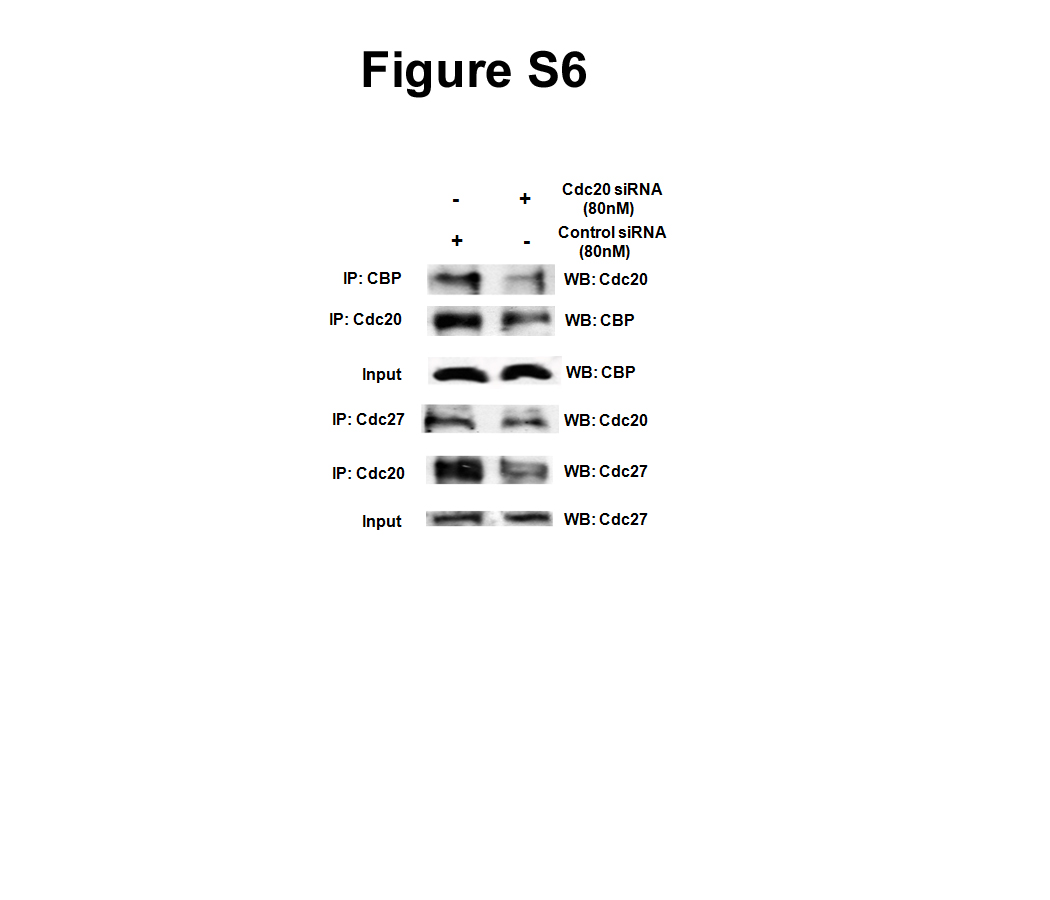
It has been reported earlier that APC/C-CBP/p300 complex can regulate the transcription of a gene (36). To investigate the possibility that transcription activity of the APC/C-CBP/p300 complex might be regulated by Cdc20, co-immunoprecipitation experiment was done to assess the APC/C-CBP/p300 interaction upon knockdown of Cdc20. As expected, Cdc20 knockdown resulted in the lowering of its interaction with both APC/C and CBP (supplemental Fig. S6). Intriguingly, Cdc20 knockdown also led to a decrease in Cdc27-CBP interaction (Fig. 5A). Furthermore, ChIP data revealed reduced recruitment of CBP and Cdc27 on the UBCH10 promoter upon knockdown of Cdc20 (Fig. 5B). Our previous observation that Cdc27 is present on the UBCH10 promoter (Fig. 4C) led us to examine the recruitment of Cdc20 and CBP on the UBCH10 promoter upon knockdown of Cdc27. ChIP data showed reduced recruitment of Cdc20 and CBP on the same promoter under Cdc27 knockdown conditions (Fig. 5C). Furthermore, analysis of the UBCH10 promoter-driven luciferase activity in Cdc27 siRNA-treated HepG2 cells showed lowering of UBCH10 promoter activity (Fig. 5D). This down-regulation of the UBCH10 promoter activity in Cdc27 siRNA-treated cells could not be restored by ectopic expression of Cdc20 (Fig. 5E). Taken together, these data suggested that Cdc20 modulates the association between APC/C and CBP/p300 and Cdc20-APC/C-CBP/p300 complex positively regulates UBCH10 gene expression.
Cdc20 Regulates UbcH10 Expression in a Cell Cycle-specific Manner
Finally, we investigated the cell cycle-specific regulation of UbcH10 expression by Cdc20 in HepG2 cells. We followed the expression of UbcH10 and Cdc20 in synchronized cells after release from the G1 block (Fig. 6A). FACS analysis showed cells were at G1 phase at 2 h and mostly at S and G2 phase at 8 h, which after completing cell division again returned to G1 phase at 14 h after release from thymidine block (supplemental Fig. S7A). Quantitative RT-PCR as well as Western blot data showed that the expression of both the genes increased up to 10 h from time of release and subsequently decreased with time (Fig. 6, B and C, left panels). Interestingly, in Cdc20 siRNA-treated synchronized cells there was a concomitant reduction of endogenous UbcH10 expression (Fig. 6, B and C, right panels). We next examined the formation of the Cdc20-APC/C complex in these synchronized cells. Time point co-immunoprecipitation assay showed the association between APC/C and Cdc20 increased as the cells proceeded toward mitotic phase and decreased as it exits this phase (Fig. 6D). Concordant with that, time point ChIP-qPCR assay showed highest recruitment of Cdc20 on UBCH10 promoter when APC/C-Cdc20 interaction was also high (Fig. 6E). This result also shows the specificity of the Cdc20-UBCH10 promoter interaction that follows the pattern of UbcH10 expression during cell cycle. Together, these data revealed correlation between Cdc20 and UbcH10 expression and cell cycle-specific regulation of physical interaction between Cdc20-APC/C with UBCH10 promoter.
Finally, we investigated the effect of Cdc20-mediated regulation of UbcH10 expression on mitotic progression. Toward that end, first we followed cyclin B1 degradation in synchronized HepG2 cells overexpressing Cdc20 under nocodazole-treated conditions (Fig. 6F, left and middle panels). Second, we checked the effect of Cdc20 overexpression on cyclin B1 degradation under UbcH10 knockdown conditions in nocodazole-treated HepG2 cells (Fig. 6F, middle and right panels). Western blot analysis showed degradation of cyclin B1 upon ectopic expression of Cdc20 even in nocodazole-arrested HepG2 cells indicating overriding of mitotic arrest (Fig. 6F, left and middle panels). However, UbcH10 knockdown blocked cyclin B1 degradation even in Cdc20-overexpressing HepG2 cells (Fig. 6F, middle and right panels). To examine the cellular effect of this Cdc20-mediated regulation of UbcH10 expression, we measured mitotic index of synchronized HepG2 cells upon nocodazole treatment. The data showed that the lowering of mitotic index by Cdc20 overproduction was rescued by knockdown of UbcH10 (Fig. 6G; supplemental Fig. S8). Thus, these results suggest that Cdc20 overexpression can drive mitotic slippage in a UbcH10-dependent manner.
DISCUSSION
The Fizzy/Cdc20 family of conserved proteins are essential for the activation of APC/C (41). The role of Cdc20 in recruiting substrates to APC/C through their C-terminal WD40 domain is well established (13). In addition to substrate recruitment, the N-terminal C box of Cdc20 was also found to trigger substrate ubiquitination by APC/C (42, 43). Recently, in a genome-wide siRNA screen, Cdc20 knockdown resulted in mitotic defects in the cell (44). Thus, all the functions of Cdc20 are attributed to proper mitotic progression during the cell cycle. Here, for the first time we report a new function of Cdc20 as a novel transcription regulator. We showed that a unique function of Cdc20 is its ability to positively regulate the expression of mitotic ubiquitin-conjugating enzyme, UbcH10, through its interaction with APC/C and CBP/p300.
Initially, we observed a correlation between Cdc20 and UbcH10 levels in several cancer cell lines and primary cancer tissues. Cdc20 protein has a WD40 repeat domain in its C-terminal region (13). It is known that WD repeats are involved in various protein-protein interactions regulating an array of functions, including chromatin remodeling and transcription (45). Indeed, in a previous report, WD repeats containing mitotic checkpoint proteins were implicated in transcriptional repression during interphase (35). We used several strategies to investigate whether Cdc20 has any role on regulation of UbcH10 expression. First, we showed that both ectopic expression of Cdc20 and knockdown of endogenous Cdc20 specifically altered the endogenous UbcH10 levels in various cell lines. Second, using a promoter-reporter assay we established that Cdc20 transcriptionally activates the UBCH10 promoter. Finally, through ChIP assay we provided evidence that Cdc20 was physically interacting with the UBCH10 promoter. Moreover, we found that the Cdc20-responsive element resides between −322- and −142-nt region of UBCH10 promoter. We also found that the WD repeat region of Cdc20 is required for this UbcH10 transactivation. Chromatin remodeling is widely associated with transcriptional regulation of genes (46). Here, the ChIP data revealed acetylation-mediated chromatin remodeling during the activation of UbcH10 expression by Cdc20.
To investigate the possibility of co-activators being associated with Cdc20-mediated transcriptional regulation of UBCH10, we found the recruitment of CBP/p300 and APC/C in the same upstream region of the UBCH10 promoter. The CBP/p300 proteins act as transcriptional co-activators and regulate transcription of a number of genes through a variety of ways (47). First, they act as a bridge connecting different transcription factors to the transcription apparatus. Second, they nucleate the association of diverse cofactor proteins into multicomponent transcription complexes. Finally, histone acetyltransferase activity of CBP/p300 influences transcription by modulating nucleosomal chromatinization. In a previous report, APC/C and CBP/p300 were shown to regulate transcription cooperatively (36, 39). It was shown that APC/C interacts with and promotes the acetyltransferase activity of CBP/p300 and activates transcription. APC/C is well known to regulate cell cycle progression through its E3 ubiquitin ligase activity (4). Besides the ubiquitin ligase activity of APC/C was also found to regulate neuronal morphogenesis and connectivity (48). Thus, cell cycle-dependent and -independent functions of APC/C are now emerging (49, 50). Co-immunoprecipitation data showed decreased CBP/p300-APC/C interaction under Cdc20 knockdown conditions. Also, ChIP data revealed lower recruitment of CBP/p300 and APC/C on the UBCH10 promoter under Cdc20 knockdown conditions. Collectively, these data indicate that Cdc20 up-regulates UBCH10 transcription through APC/C-CBP/p300 transcription complex, and its presence actually promotes the formation of the transcription complex and their subsequent recruitment to the UBCH10 promoter (Fig. 7). However, as neither Cdc20 nor APC/C has any known DNA binding domain, it remains to be seen how APC/CCdc20 is directed to a specific promoter.
FIGURE 7.

Proposed model of Cdc20-mediated regulation of UbcH10 expression. Respective proteins are shown in different shapes. Ac stands for acetylation of histones. The black line indicates UBCH10 promoter. UBCH10 gene is shown as rectangular box, and transcription start site is indicated at “+1”. The thick black up arrow indicates transcriptional up-regulation from UBCH10 promoter.
Cell cycle-regulated expression of both Cdc20 and UbcH10 has been documented with the highest expressions at mitotic phase, which is consistent with their role during mitosis (11–13). Interestingly, our results suggest that this correlated expression of UbcH10 during the progression of the cell cycle is regulated by the expression of Cdc20. We found knockdown of Cdc20 affected endogenous UbcH10 expression as the cells proceeded through G2 to mitotic phase. Time course co-immunoprecipitation data revealed increased association between APC/C and Cdc20 at these phases of the cell cycle. Consistent with this observation, ChIP qPCR data suggest that recruitment of Cdc20 on the UBCH10 promoter was dictated by its physiological level. We propose that cell cycle-specific regulation of UbcH10 expression by Cdc20 is required to maintain a threshold level of both proteins, which in turn would ensure proper execution of mitotic progression. To validate such a notion, we examined the degradation pattern of cyclin B1, which is a protein marker for studying the mitotic progression. As reported earlier, Cdc20 overproduction showed mitotic slippage even in nocodazole-arrested cells (18). Here, we report this mitotic slippage to be UbcH10-dependent as UbcH10 down-regulation resulted in stabilization of cyclin B1 degradation even in Cdc20-overexpressing cells. In light of our findings, there are two nonexclusive interpretations as follows: 1) where the UbcH10 dependence reflects coordinated function of these enzymes in proteolysis; 2) the possibility that Cdc20-mediated up-regulation of UbcH10 also contributes efficient degradation of metaphase to anaphase inhibitors.
The results presented in this study have an important implication in proper mitotic checkpoint function. Ubiquitination-mediated proteolysis of mitotic inhibitors and thereby cell cycle progression largely depends on UbcH10. It is known to act as E2 ubiquitin carrier protein together with E3 ubiquitin ligase APC/C (27). Recently, another E2 molecule, UBE2S, has been reported to elongate ubiquitin chain preinitiated by UbcH10 (51–53). Overexpression of UbcH10 leads to chromosome missegregation and tumor formation (54), whereas deactivation blocks the cells at metaphase (27). Therefore, UbcH10 is a critical player of the ubiquitination pathway associated with mitotic progression. Similarly, deregulated expression of Cdc20 has been observed in many cancer types. Cdc20 overexpression was found to cause aneuploidization (18), and its expression was reported to be regulated by tumor suppressor protein p53 (31). We hypothesized that oncogenic function of overexpressed Cdc20 also requires a higher level of UbcH10. Indeed the bioinformatic analysis of the Oncomine data base strongly suggests correlated overexpression of both the proteins in many tumor types. Thus, the observation that Cdc20 regulates transcription of UbcH10 in a cell cycle-dependent manner provides a molecular basis of these findings. It could be speculated that Cdc20 overexpression and thus UbcH10 up-regulation synergistically cause the onset of aneuploidy and oncogenicity.
Supplementary Material
Acknowledgments
We thank Dr. Santu Bandyopadhyay, Dr. Saikat Chakrabarti, and Dr. Keya Chaudhuri (Indian Institute of Chemical Biology, India) for FACS, statistical analysis, and microscopic analysis, respectively. We also thank Dr. Kunal Ray (Indian Institute of Chemical Biology, India) for critically reading the manuscript.
This work was supported in part by Department of Biotechnology Grants BT/PR/5524/Med/14/649/2004 and BT/01/COE/05/04 and Council of Scientific and Industrial Research Grant IAP 001 (to S. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8, Tables S1–S3, and Oncomine dataset.
- SAC
- spindle assembly checkpoint
- nt
- nucleotide
- APC/C
- anaphase promoting complex
- oligo
- oligonucleotide
- pol
- polymerase
- qPCR
- quantitative PCR
- MCC
- mitotic checkpoint complex.
REFERENCES
- 1. Bharadwaj R., Yu H. (2004) Oncogene 23, 2016–2027 [DOI] [PubMed] [Google Scholar]
- 2. Chi Y. H., Jeang K. T. (2007) J. Cell. Biochem. 102, 531–538 [DOI] [PubMed] [Google Scholar]
- 3. Musacchio A., Salmon E. D. (2007) Nat. Rev. Mol. Cell Biol. 8, 379–393 [DOI] [PubMed] [Google Scholar]
- 4. Peters J. M. (2006) Nat. Rev. Mol. Cell Biol. 7, 644–656 [DOI] [PubMed] [Google Scholar]
- 5. Reddy S. K., Rape M., Margansky W. A., Kirschner M. W. (2007) Nature 446, 921–925 [DOI] [PubMed] [Google Scholar]
- 6. Thornton B. R., Toczyski D. P. (2006) Genes Dev. 20, 3069–3078 [DOI] [PubMed] [Google Scholar]
- 7. Visintin R., Prinz S., Amon A. (1997) Science 278, 460–463 [DOI] [PubMed] [Google Scholar]
- 8. Baker D. J., Dawlaty M. M., Galardy P., van Deursen J. M. (2007) Cell. Mol. Life Sci. 64, 589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mapelli M., Filipp F. V., Rancati G., Massimiliano L., Nezi L., Stier G., Hagan R. S., Confalonieri S., Piatti S., Sattler M., Musacchio A. (2006) EMBO J. 25, 1273–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stegmeier F., Rape M., Draviam V. M., Nalepa G., Sowa M. E., Ang X. L., McDonald E. R., 3rd, Li M. Z., Hannon G. J., Sorger P. K., Kirschner M. W., Harper J. W., Elledge S. J. (2007) Nature 446, 876–881 [DOI] [PubMed] [Google Scholar]
- 11. Cho R. J., Huang M., Campbell M. J., Dong H., Steinmetz L., Sapinoso L., Hampton G., Elledge S. J., Davis R. W., Lockhart D. J. (2001) Nat. Genet. 27, 48–54 [DOI] [PubMed] [Google Scholar]
- 12. Rape M., Kirschner M. W. (2004) Nature 432, 588–595 [DOI] [PubMed] [Google Scholar]
- 13. Yu H. (2007) Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- 14. Weaver B. A., Cleveland D. W. (2006) Curr. Opin. Cell Biol. 18, 658–667 [DOI] [PubMed] [Google Scholar]
- 15. Yuan B., Xu Y., Woo J. H., Wang Y., Bae Y. K., Yoon D. S., Wersto R. P., Tully E., Wilsbach K., Gabrielson E. (2006) Clin. Cancer Res. 12, 405–410 [DOI] [PubMed] [Google Scholar]
- 16. Yuen K. W., Montpetit B., Hieter P. (2005) Curr. Opin. Cell Biol. 17, 576–582 [DOI] [PubMed] [Google Scholar]
- 17. Rhodes D. R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9309–9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mondal G., Sengupta S., Panda C. K., Gollin S. M., Saunders W. S., Roychoudhury S. (2007) Carcinogenesis 28, 81–92 [DOI] [PubMed] [Google Scholar]
- 19. Li M., Fang X., Wei Z., York J. P., Zhang P. (2009) J. Cell Biol. 185, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rape M., Reddy S. K., Kirschner M. W. (2006) Cell 124, 89–103 [DOI] [PubMed] [Google Scholar]
- 21. Berlingieri M. T., Pallante P., Guida M., Nappi C., Masciullo V., Scambia G., Ferraro A., Leone V., Sboner A., Barbareschi M., Ferro A., Troncone G., Fusco A. (2007) Oncogene 26, 2136–2140 [DOI] [PubMed] [Google Scholar]
- 22. Berlingieri M. T., Pallante P., Sboner A., Barbareschi M., Bianco M., Ferraro A., Mansueto G., Borbone E., Guerriero E., Troncone G., Fusco A. (2007) Eur. J. Cancer 43, 2729–2735 [DOI] [PubMed] [Google Scholar]
- 23. Fujita T., Ikeda H., Taira N., Hatoh S., Naito M., Doihara H. (2009) BMC Cancer 9, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okamoto Y., Ozaki T., Miyazaki K., Aoyama M., Miyazaki M., Nakagawara A. (2003) Cancer Res. 63, 4167–4173 [PubMed] [Google Scholar]
- 25. Pallante P., Berlingieri M. T., Troncone G., Kruhoffer M., Orntoft T. F., Viglietto G., Caleo A., Migliaccio I., Decaussin-Petrucci M., Santoro M., Palombini L., Fusco A. (2005) Br. J. Cancer 93, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wagner K. W., Sapinoso L. M., El-Rifai W., Frierson H. F., Butz N., Mestan J., Hofmann F., Deveraux Q. L., Hampton G. M. (2004) Oncogene 23, 6621–6629 [DOI] [PubMed] [Google Scholar]
- 27. Townsley F. M., Aristarkhov A., Beck S., Hershko A., Ruderman J. V. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 2362–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang L., Bao Y., Luo C., Hu G., Huang C., Ding X., Sun K., Lu Y. (2010) J. Cancer Res. Clin. Oncol. 136, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeong S. J., Shin H. J., Kim S. J., Ha G. H., Cho B. I., Baek K. H., Kim C. M., Lee C. W. (2004) Cancer Res. 64, 8666–8673 [DOI] [PubMed] [Google Scholar]
- 30. Park H. Y., Jeon Y. K., Shin H. J., Kim I. J., Kang H. C., Jeong S. J., Chung D. H., Lee C. W. (2007) Exp. Mol. Med. 39, 195–204 [DOI] [PubMed] [Google Scholar]
- 31. Banerjee T., Nath S., Roychoudhury S. (2009) Nucleic Acids Res. 37, 2688–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kidokoro T., Tanikawa C., Furukawa Y., Katagiri T., Nakamura Y., Matsuda K. (2008) Oncogene 27, 1562–1571 [DOI] [PubMed] [Google Scholar]
- 33. Chun A. C., Jin D. Y. (2003) J. Biol. Chem. 278, 37439–37450 [DOI] [PubMed] [Google Scholar]
- 34. Oikawa T., Okuda M., Ma Z., Goorha R., Tsujimoto H., Inokuma H., Fukasawa K. (2005) Mol. Cell. Biol. 25, 4046–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoon Y. M., Baek K. H., Jeong S. J., Shin H. J., Ha G. H., Jeon A. H., Hwang S. G., Chun J. S., Lee C. W. (2004) FEBS Lett. 575, 23–29 [DOI] [PubMed] [Google Scholar]
- 36. Turnell A. S., Stewart G. S., Grand R. J., Rookes S. M., Martin A., Yamano H., Elledge S. J., Gallimore P. H. (2005) Nature 438, 690–695 [DOI] [PubMed] [Google Scholar]
- 37. Fraschini R., Beretta A., Sironi L., Musacchio A., Lucchini G., Piatti S. (2001) EMBO J. 20, 6648–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mondal G., Baral R. N., Roychoudhury S. (2006) Biochem. J. 396, 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu J., Fuchs S. Y. (2006) Cancer Biol. Ther. 5, 760–762 [DOI] [PubMed] [Google Scholar]
- 40. Pines J. (2009) Mol. Cell 34, 135–136 [DOI] [PubMed] [Google Scholar]
- 41. Pesin J. A., Orr-Weaver T. L. (2008) Annu. Rev. Cell Dev. Biol. 24, 475–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benanti J. A., Toczyski D. P. (2008) Mol. Cell 32, 460–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kimata Y., Baxter J. E., Fry A. M., Yamano H. (2008) Mol. Cell 32, 576–583 [DOI] [PubMed] [Google Scholar]
- 44. Neumann B., Walter T., Hériché J. K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U., Cetin C., Sieckmann F., Pau G., Kabbe R., Wünsche A., Satagopam V., Schmitz M. H., Chapuis C., Gerlich D. W., Schneider R., Eils R., Huber W., Peters J. M., Hyman A. A., Durbin R., Pepperkok R., Ellenberg J. (2010) Nature 464, 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith T. F., Gaitatzes C., Saxena K., Neer E. J. (1999) Trends Biochem. Sci. 24, 181–185 [DOI] [PubMed] [Google Scholar]
- 46. Li B., Carey M., Workman J. L. (2007) Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 47. Chan H. M., La Thangue N. B. (2001) J. Cell Sci. 114, 2363–2373 [DOI] [PubMed] [Google Scholar]
- 48. Yang Y., Kim A. H., Bonni A. (2010) Curr. Opin. Neurobiol. 20, 92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harper J. W., Burton J. L., Solomon M. J. (2002) Genes Dev. 16, 2179–2206 [DOI] [PubMed] [Google Scholar]
- 50. Manchado E., Eguren M., Malumbres M. (2010) Biochem. Soc. Trans. 38, 65–71 [DOI] [PubMed] [Google Scholar]
- 51. Garnett M. J., Mansfeld J., Godwin C., Matsusaka T., Wu J., Russell P., Pines J., Venkitaraman A. R. (2009) Nat. Cell Biol. 11, 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Williamson A., Wickliffe K. E., Mellone B. G., Song L., Karpen G. H., Rape M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 18213–18218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu T., Merbl Y., Huo Y., Gallop J. L., Tzur A., Kirschner M. W. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Ree J. H., Jeganathan K. B., Malureanu L., van Deursen J. M. (2010) J. Cell Biol. 188, 83–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}