

Abstract
The major L-type voltage-gated calcium channels in heart consist of an α1C (CaV1.2) subunit usually associated with an auxiliary β subunit (CaVβ2). In embryonic cardiomyocytes, both the complete and the cardiac myocyte-specific null mutant of CaVβ2 resulted in reduction of L-type calcium currents by up to 75%, compromising heart function and causing defective remodeling of intra- and extra-embryonic blood vessels followed by embryonic death. Here we conditionally excised the CaVβ2 gene (cacnb2) specifically in cardiac myocytes of adult mice (KO). Upon gene deletion, CaVβ2 protein expression declined by >96% in isolated cardiac myocytes and by >74% in protein fractions from heart. These latter protein fractions include CaVβ2 proteins expressed in cardiac fibroblasts. Surprisingly, mice did not show any obvious impairment, although cacnb2 excision was not compensated by expression of other CaVβ proteins or changes of CaV1.2 protein levels. Calcium currents were still dihydropyridine-sensitive, but current density at 0 mV was reduced by <29%. The voltage for half-maximal activation was slightly shifted to more depolarized potentials in KO cardiomyocytes when compared with control cells, but the difference was not significant. In summary, CaVβ2 appears to be a much stronger modulator of L-type calcium currents in embryonic than in adult cardiomyocytes. Although essential for embryonic survival, CaVβ2 down-regulation in cardiomyocytes is well tolerated by the adult mice.
Keywords: Calcium, Calcium Channels, Cardiac Muscle, Gene Knockout, Heart
Introduction
The L-type calcium channels in heart are high voltage-activated, and their minimal composition includes the pore-forming CaV1.2α1 subunit and the auxiliary CaVβ2 subunit (1). CaVβ subunits are believed to enhance the trafficking of the channels to the plasma membrane and to produce shifts in the voltage dependence of channel activation. From the four mammalian genes coding for CaVβ subunits, the cacnb2 gene is predominantly expressed in the heart (2). Overexpressing the rat neuronal CaVβ2a transgene in cardiomyocytes showed increased Ca2+ entry and progressive cell necrosis that led to pump dysfunction and premature death (3). Generation of a ubiquitous and a cardiomyocyte-specific null mutant of cacnb2 (4) causes early embryonic death because of a morphologically and functionally compromised heart. In contrast, targeted deletion of the cacnb1 or cacnb3 genes and the functional inactivation of cacnb4 caused by the spontaneous lethargic (lh) mutation appear not to effect cardiac function (5). In CaVβ2 null cardiomyocytes at the embryonic days preceding death, the calcium current was reduced to approximately one-fourth to one-third of the current in wild type cells (4). The remaining calcium current was ineffective to support cardiomyocyte contraction and cardiac pump function; as a consequence, embryonic heart failure associated with pericardial effusion and, finally, embryonic death after embryonic day 10.5 occurred (4). Although it was still sensitive to dihydropyridines (4), the remaining current appears not to depend on the presence of the CaVβ2 subunit.
To study the impact of CaVβ2 deletion in the heart of adult animals, we induced excision of the cacnb2 gene in cardiomyocytes of adult mice. We show that CaVβ2 protein expression declined by >96% in isolated cardiomyocytes following gene deletion and, surprisingly, we observed only moderately impaired L-type calcium currents with a <29% reduction in current density. Different from the embryo, CaVβ2 protein expression is not essential for L-type calcium currents in adult cardiomyocytes and survival of the adult mouse.
EXPERIMENTAL PROCEDURES
Animal Care and Generation of Inducible and Cardiomyocyte-specific Cavβ2 deletion in Mice
The CaVβ2flox/flox mouse (4) (mixed background of C57BL/6 × 129/SvJ) was crossed with the CaVβ2+/− mouse (4) to obtain the CaVβ2flox/− mouse. The latter was crossed with an α-MHC-MerCreMer mouse (6) expressing a Tamoxifen-inducible Cre recombinase protein fused to two mutant estrogen receptor ligand-binding domains (MerCreMer) under the control of the α-myosin heavy chain promoter to obtain the CaVβ2flox/−/MerCreMertg/0 mouse, which allows for inducible, cardiomyocyte-specific disruption of the Cacnb2 gene in adult mice. Cacnb2 gene excision in 10–12-week-old mice to obtain CaVβ2flox/−/MerCreMertg/0 knock-out (KO) mice was accomplished by i.p. administration of Tamoxifen (60 mg/kg) for 5 consecutive days in week 1 and week 3. Tamoxifen was freshly dissolved in Mygliol 812 oil (Caelo) at a concentration of 20 mg/ml. Wild type mice, CaVβ2flox/− mice, and CaVβ2flox/−/MerCreMertg/0 mice injected with Mygliol 812 oil only (mock) served as controls. Hearts were harvested at weeks 6, 9, and 12 following Tamoxifen administration. All animal procedures were performed in accordance with German legislation on the protection of animals and approved by the Saarland's Institutional Animal Care and Use Committee.
Organs of Tamoxifen-injected mice were obtained under terminal Avertin (2,2,2-tribromoethanol, Fluka) anesthesia. Briefly, the abdominal and thoracic cavities were exposed, and the renal artery was punctured to reduce the blood flow going to the heart. The heart was quickly excised and placed into ice-cold saline solution, where the excess of fat tissue and surrounding vessels was removed. Immediately, the lungs and liver were dissected from the mouse and placed into a Petri dish. Before weighing the organs, the excess of blood was gently removed by placing the organs into a paper tissue. Each organ was weighed (wet weight), the hearts were saved for Western blots, and the lungs and liver were dried at 70 °C over 48 h to get the dry weight. The tibia was removed, carefully cleaned from skin and muscle, and measured out.
Antibodies and Western Blots
Microsomal membrane protein fractions from heart and brain were solubilized with Laemmli buffer, denatured, and subjected to SDS-PAGE. Proteins were transferred onto a nitrocellulose membrane (Hybond-C Extra, GE Healthcare) and probed with antibodies for CaVβ (in-house generated antibodies 424 and 425 (anti-mouse CaVβ2 (4)), 237 (anti-mouse CaVβ1 (7)), 828 and MM2 (anti-mouse CaVβ3 (8)), and 830 and 1051 (anti-mouse CaVβ4 (9)), CaV1.2 (kindly provided by Dr. Franz Hofmann, München, Germany), GAPDH (Santa Cruz Biotechnology), P4HB (Acris 11245-1-AP), α-actinin (Sigma A 7811), and ryanodine receptor (Thermo Scientific). Specificity of CaVβ antibodies was confirmed by using microsomal membrane protein fractions from wild type mice and mice deficient in CaVβ2, CaVβ3, and CaVβ4 (the lh mouse, respectively). Proteins were detected using horseradish peroxidase-coupled secondary antibodies and the Western Lightning chemiluminescence reagent Plus (PerkinElmer Life Sciences). Original scans were saved as TIFF files from LAS 3000 (Fujifilm), which were further processed in Adobe Photoshop. Images were cropped, resized proportionally, and brought to the resolution required for publication.
Isolation of Mouse Cardiac Cells and Primary Culture of Cardiac Fibroblasts
Ventricular cells were obtained from dissociated hearts of female adult wild type mice (F1 between C57Bl/6N × 129/SvJ, 10 weeks). Mice were anesthetized by intraperitoneal injection of Avertin (0.5 mg/g of body weight) including heparin (20 units/g of body weight) to avoid blood clots; the hearts were quickly removed via thoracotomy and transferred to an ice-cold modified Tyrode's solution (TS),3 containing (in mm): 134 NaCl; 4 KCl; 1.2 MgSO4; 1.2 Na2HPO4; 10 HEPES; 11 glucose; pH 7.36. After that, the hearts were cannulated via the aorta, attached to a Langendorff apparatus, and perfused retrogradely with carbogen (5% CO2, 95% O2)-saturated TS containing 200 μm EGTA and 10 mm 2,3-butanedione-monoxime for 7 min. The perfusion solution was then changed to constantly gassed (carbogen) TS containing a collagenase/protease mixture (Liberase TM, Roche Applied Science) at a final concentration of 133 μg/ml for 5–6 min. The ventricles were isolated, dissected, and transferred to TS supplemented with 4% bovine serum albumin (BSA), 12.5 μm Ca2Cl, and 1.4 ng/ml DNase I (Sigma). The ventricles were cut into small pieces followed by homogenization of the tissue by gently pipetting. The suspension was filtered through a nylon filter (pore size 150 μm) and maintained for 10 min in the same solution for sedimentation; cardiomyocytes were mainly concentrated in the pellet. Fibroblasts remaining in the supernatant were collected by centrifugation (250 × g, 10 min) and washed once in M199 medium (Invitrogen). They were seeded on a 25-cm2 culture flask (one heart per flask), and the medium was changed every 24 h until the cells were confluent; the cells were split by trypsin (Invitrogen) treatment and harvested between passages 1 and 2 for RNA isolation or Western blot analysis. The cardiomyocytes contained in the pellet were resuspended in TS solution containing BSA, Ca2Cl, and DNase and were transferred into a polystyrene culture dish; then, they were incubated (37 °C in 5% CO2) for 2 h to remove adherent cells, and the non-attaching cardiomyocytes were saved. The cardiomyocytes were collected by centrifugation (40 × g, 1.5 min) and subsequently resuspended in ice-cold Ca2+/Mg2+-free Dulbecco's PBS (Invitrogen) and washed twice, the first time by centrifuging at 40 × g (1.5 min) and the second time at 40 × g (1 min) to remove debris and possible remaining blood cells. The final pellet was snap-frozen in liquid nitrogen and maintained at −80 °C until protein isolation for Western blots.
Cardiomyocytes for laser capture microdissection (LCM) were obtained as described before. The cardiomyocytes were washed and resuspended in sterile ice-cold Ca2+/Mg2+ free Dulbecco's PBS, and then they were cytospun on LCM glass slides covered by 1-mm PEN membrane (Zeiss) and directly fixed by immersion in absolute methanol for 1 min. The samples were kept on ice inside 50-ml polypropylene tubes to avoid complete drying until cardiomyocyte isolation. The collection of the cells was performed using a Zeiss P.A.L.M. LCM microscope (Zeiss/Microlaser Technologies) with a 20× objective (Plan-NEOFLUAR 0.5 ∞/0.17). Cardiomyocytes were collected in 0.5-ml LCM tubes (AdhesiveCap 500 opaque, Zeiss). Cardiomyocytes were identified by their morphology; only isolated and well preserved and rod-shaped cells were chosen for LCM. About 60 cardiomyocytes were collected per cap; cells were lysed in Buffer RLT (Qiagen), frozen in dry ice, and kept at −80 °C until RNA extraction. For RT-PCR, RNA from four caps (227 cells) was pooled.
RT-PCR
Total RNA was extracted using the RNeasy micro kit (Qiagen). We used 40 ng of total RNA from cardiomyocytes and complete heart and 80 ng of total RNA from cardiac fibroblasts for one-step RT-PCR (Invitrogen). The following intron-spanning primers were used for amplification of CaVβ2 fragments: mb2_32 (5′-GTT CGG CAG ACT CCT ACA CC-3′) and mb2_10 (5′-CTC TAG TTT GAC TGG GCT TGG-3′), resulting in a CaVβ2-specific 326-bp fragment. For amplification of the 225-bp fragment containing part of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1) as control, we used total RNA from complete heart and the following oligonucleotides: UW_637 (5′-GCT CGA GAT GTC ATG AAGG-3′) and UW_638 (5′-AGT TGA GAG AGA TCA TCT CCA CC-3′). PCR fragments were analyzed on a 2% agarose gel.
Pulse-Chase and Immunoprecipitation Experiments
COS-7 cells were irradiated with 30 grays to prevent continuous proliferation. 24 h later, cells were transfected with CaVβ2 cDNA or CaVβ2 plus CaV1.2 cDNAs, respectively, using FuGENE 6 (Roche Applied Science) according to the manufacturer's specifications. 24 h after transfection, cells were starved for 2 h in Met- and Cys-free DMEM-medium followed by a 2-h labeling in medium supplemented with 100 μCi of l-[35S]Met and l-[35S]Cys). Radioactive media were eventually washed out with PBS (time 0) and replaced with DMEM medium. Cells were harvested at time 0 and 12, 24, 48, 72, 96, and 120 h thereafter and lysed in the presence of 0.05 m Tris, 0.15 m NaCl, 0.005 M EDTA, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, pH 8, and CaVβ2 proteins were immunoprecipitated by antibody 425. The precipitated proteins were run on SDS-PAGE and transferred to nitrocellulose filters, which were analyzed by Western blotting and by exposure to PhosphorImager screens followed by quantitative analysis using the AIDA Image Analyzer software.
Immunofluorescence and Confocal Imaging
Isolated myocytes were plated onto glass coverslips precoated with extracellular matrix proteins (R&D Systems) and rinsed with PBS before fixation (4% formaldehyde in PBS, 10 min, room temperature). Thereafter cells were washed once for 10 min in PBS and permeabilized with 0.4% Triton X-100 for 10 min. After an additional 10-min washing step, they were incubated in blocking solution (PBS containing 5% bovine serum albumin) at 21 °C for 10 min. Cells were incubated with the primary antibodies for the ryanodine receptor (1:200) and CaVβ2 (antibody 425, 1:100) for 1 h at 21 °C. After three washes with PBS containing 0.1% Tween 20 at 21 °C, cells were incubated with secondary antibodies (DyLight 649 AffiniPure goat anti-rabbit, Jackson ImmunoResearch Europe, and ATTO 550-conjugated goat anti-mouse, Sigma) at 21 °C for 30 min. Cells were washed again under the same conditions and rinsed once with water. The coverslips were mounted on a drop of mounting medium (ProLong® Gold antifade reagent, Invitrogen) and stored at 4 °C until further analysis.
Coverslips with the labeled myocytes were transferred onto the stage of a Leica SP5-II laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany), and imaging was performed through a 63× oil immersion objective (HCX PL APO, 1.4, Leica). We used a 561 nm laser (CYA-015, Melles-Griot, Bensheim, Germany) and a 633 nm laser (He-Ne, Lasos, Jena, Germany) to excite ATTO550 and DyLight649, respectively. For detection of the fluorescence, the following two spectral bandwidths were used: 565–628 nm (for ATTO550) and 640–800 nm (for DyLight649). Settings for laser excitation as well as emission settings (spectral bandwidth, photomultiplier tube gain, offset) were kept constant between measurements. Z-stacks were acquired as series of confocal sections (each 1024 × 1024 pixels in size) interspaced by 0.15-μm steps. Resulting data were stored onto an OMERO database system (the Open Microscopy Environment) for archiving and later post processing. For analysis and composition of the figures, z-stacks were deconvolved in AutoQuant software (AutoDeblur X, MediaCybernetics), stored as 16-bit grayscale images, and imported into Adobe Illustrator CS5 for composition of final figures. Two sets of figures are provided, one optimized for screen display and another one for printing, taking into account the different results of presentation. For this, all grayscale images were treated the same, i.e. their look-up tables were adapted together to ensure appropriate presentation of grayscale data on a printer or the computer display and to preserve intensity differences between the images.
Electrophysiology
Mouse ventricular myocytes were isolated by using a Langendorff perfusion technique as described previously (10) but adapted for mice. In short, Langendorff hearts were perfused 6 min with a Ca2+-free solution and afterward for 15 min with an enzyme solution (Ca2+-free and 1 mg/ml collagenase A (Roche Applied Science) supplemented with 0.1 mg/ml protease XIV (Sigma)). After cutting the heart into small pieces of ∼2 mm3, these remainders were washed with a Krebs solution (see below) but with 0.18 mm CaCl2 to obtain isolated single cells, which were used for the experiments.
Whole cell voltage clamp recordings were obtained using a ruptured patch clamp with an EPC9 patch clamp amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany). For control of voltage-clamp protocols and data acquisition, we used the pCLAMP 9 software (Molecular Devices, Foster City, CA) run on an IBM-compatible PC, which was connected to the amplifier via a TL-1 DMA interface (Molecular Devices). Patch pipettes (1–2.5 megaohms) were pulled from Vitrex capillary tubes (Modulohm) using a DMZ universal puller (Zeitz Instruments). Series resistance was compensated to the maximum extent possible (40–60%). An Ag-AgCl wire was used as reference electrode. Membrane capacitive transients were electronically compensated, and the linear background components were digitally subtracted when possible before data analysis. In the case of tail current measurements, linear background components and capacitive transients were subtracted. Current traces were filtered at 1 kHz and digitized at 5 kHz. Potentiation of L-type Ca2+ currents was achieved by the application of 1 μm BayK-8644, and inhibition was achieved by 2 μm nifedipine (both from Bayer Leverkusen). All experiments were performed at 22–25 °C.
In all experiments, a bath (Krebs) solution was used (in mm): 156 CsCl; 1.5 CaCl2; 1 MgCl2; 10 HEPES; 10 glucose; adjusted with CsOH to pH 7.4. This solution also nulled the membrane potential of the myocytes. The following pipette solution was used (in mm): 130 CsCl; 20 tetraethylammonium chloride; 1 MgCl2; 1 EGTA; 5 Na2ATP; 0.1 GTP; 5 Hepes adjusted with CsOH to pH 7.2.
Voltage step protocols were applied from a holding potential of −80 mV for 200 ms to a prepulse of −40 mV followed by a 300-ms test pulse between −50 and +80 mV. For inactivation, from a holding potential of −80 mV, prepulses of 300 ms were applied from −90 to 0 mV with an increment of +10 mV. Currents after the prepulse were measured at 0 mV. In all protocols, the interval between the pulse protocols was 2 s.
The data were analyzed using WinASCD (24). IV curves and inactivation curves were fitted by
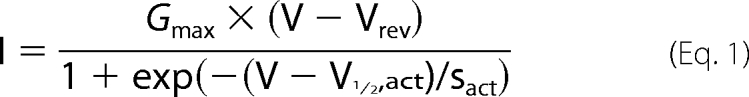 |
where I (pA/pF) is the measured current at the test potential V (in mV), and the fitted parameters are the maximal conductance Gmax (nS/pF), the potential of half-maximal activation V½,act (in mV), the slope of activation sact (in mV), and the reversal potential Vrev (in mV). Inactivation was fitted by
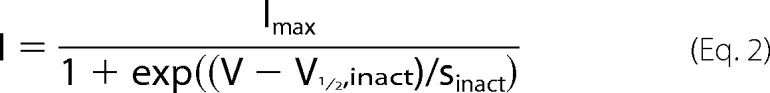 |
where I is the measured current after the prepotential at 0 mV, Imax is the maximal current at 0 mV and at the prepotential of −90 mV, V is the prepotential, V½,inact is the potential for 50% inactivation, and sinact is the slope factor for inactivation. For the statistical analysis, the two-side t test was applied (<0.05 considered significant), and one-way analysis of variance was followed by multiple comparisons-Holm-Sidak t test.
RESULTS AND DISCUSSION
Different from the CaVβ2−/− mice and mice with a cardiomyocyte-restricted excision of the Cacnb2 gene, which died in utero (4), CaVβ2+/− (4), CaVβ2flox/flox (4), CaVβ2−/flox, and CaVβ2−/flox/MerCreMertg/0 mice were born healthy and did not exhibit any obvious abnormalities during adulthood. KO mice (CaVβ2−/flox/MerCreMertg/0 at 6, 9, or 12 weeks after induction for cacnb2 gene deletion by Tamoxifen) also appeared healthy and did not exhibit any obvious abnormalities or symptoms of cardiac decompensation (Fig. 1). The heart rate (Fig. 1A) and heart weight (Fig. 1, B and C), the lung and liver weights (Fig. 1, D, E, G, and H), as well as the water content of lung and liver (Fig. 1, F and I) were not different between KO mice or the controls (control 1, CaVβ2+/flox/MerCreMertg/0; control 2, CaVβ2−/flox).
FIGURE 1.

KO mice (Cavβ2−/flox/MerCreMertg/0 mice after induction for cacnb2 gene deletion by Tamoxifen), like controls receiving identical Tamoxifen treatment, did not exhibit any signs of cardiac decompensation. A–I, heart rate (A), heart weight (B and C), lung weight (D and E), liver weight (G and H), and lung (F) and liver water content (I) (given as the percentage of organ wet weight) were not significantly different among the three genotypes. Control 1, CaVβ2+/flox/MerCreMertg/0; control 2, CaVβ2−/flox. Values are shown as mean ± S.E.
In brain, all four CaVβ subunits are expressed (Fig. 2A), whereas in adult heart, only the CaVβ2 protein is detectable (Fig. 2B). CaVβ2 expression levels in heart were not different among the various genotypes (Fig. 2, C and D) but was different in heart from KO mice, where CaVβ2 transcript expression was hardly detectable (Fig. 2C) and protein expression was significantly reduced (Fig. 2D, lane 4), indicating that the MerCreMer transgene is tightly regulated within the heart in the absence of Tamoxifen and that CaVβ2 expression in the heart can be effectively controlled by Tamoxifen-induced cacnb2 gene excision, as expected (6, 11). CaVβ1, CaVβ3, and CaVβ4 proteins were not detectable, and the expression level of CaV1.2 was not changed (Fig. 2D). All CaV proteins were still being detectable in brain (Fig. 2D, lanes 5 and 6), indicating that there was no apparent compensatory expression of these CaV subunits in the heart of KO mice.
FIGURE 2.

Cavβ-expression. A and B, CaVβ1(β1), CaVβ2(β2), CaVβ3(β3), and CaVβ4(β4) in brain (A) and expression of Cavβ2 in heart (B) (100 μg/lane). C, CaVβ2 transcript expression in cardiac poly(A+)-RNA (10 μg/lane) from WT (lane 1), CaVβ2−/flox/MerCreMertg/0 (mock, lane 2), Tamoxifen-treated CaVβ2−/flox/MerCreMertg/0 (KO, lane 3), CaVβ2+/flox/MerCreMertg/0 (lane 4), and CaVβ2+/flox/MerCreMer0/0 (lane 5). Loading controls are as follows: GAPDH expression (middle) and ethidium bromide stain (bottom). D, CaVβ2 protein expression in heart (45 μg) from WT (lane 1), CaVβ2+/− (lane 2), CaVβ2−/flox/MerCreMertg/0 (mock, lane 3), and Tamoxifen-treated CaVβ2−/flox/MerCreMertg/0 (KO) (lane 4). Lane 5, 40 μg; lane 6, 80 μg. Protein expression in brain is shown as control (exposed for 15 min when compared with 1 min (lanes 1–4)). GAPDH was used as a loading control. E, estimation of the CaVβ2 protein expression levels in hearts from controls (lanes 1, 3, and 5) and KO (lanes 2, 4, and 6) using the CaVβ2 antibodies 425 (upper) and 424 (lower). Coomassie Brilliant Blue stain was used as a loading control.
Tamoxifen treatment resulted in a significant reduction of the CaVβ2 protein immunoreactivity in heart, and to estimate the amount of remaining protein, we performed experiments like the one shown in Fig. 2E, employing two independent antibodies for CaVβ2. The amount of CaVβ2 protein remaining in hearts from KO mice (n = 46) was 21.8 ± 5.7% (antibody 425) and 25.9 ± 7.5% (antibody 424) when compared with controls (n = 46). These values are in agreement with the Tamoxifen-induced Cre-mediated recombination frequency of >70% in heart revealed by Southern blot analysis (6). Part of the remaining CaVβ2 proteins may therefore be expressed in non-myocytes, which are present in heart at a fair number (6, 16).
CaVβ2 immunoreactivity (Fig. 3A, right column) in isolated cardiomyocytes from control mice shares the typical cross-striation pattern with the ryanodine receptor type 2 (RyR2) immunoreactivity (Fig. 3, A and B, left columns), reflecting the spatial juxtaposition of the voltage-gated calcium channels and the ryanodine receptors. In myocytes from KO mice, the CaVβ2 immunoreactivity is considerably reduced (Fig. 3B, right column), whereas the RyR2 immunoreactivity is unchanged (Fig. 3B, left column). To estimate the amount of the remaining CaVβ2 protein in isolated KO cardiomyocytes, we performed Western blots using lysates from these cells (Fig. 3, C and D). The amount of CaVβ2 protein left in isolated myocytes from KO mice was 3.7 ± 0.9% (three independent blots) of the amount present in control cells (Fig. 3, C and D) and much less than the 21.8–25.9% detectable in protein fractions from KO hearts (Fig. 2E). Apparently, CaVβ2 protein is also expressed in non-myocyte cellular components of the heart. By using RT-PCR, we could identify CaVβ2 transcripts (Fig. 3E, lane 4) and proteins in three independent preparations of early passage cardiac fibroblasts (Fig. 3F, lanes 3–5).
FIGURE 3.

Cavβ2 (β2) expression in isolated cardiomyocytes and cardiac fibroblasts. Isolated ventricular myocytes of control (A) and KO mice (B) were probed for ryanodine receptors (RyR, left columns) and β2 (right columns). During confocal imaging, the operator was blinded to the genotype of the cardiomyocytes. A, myocytes labeled for RyR (panels a1 and a2) display the typical cross-striation pattern. Probing for β2 (panels b1 and b2) in the same cell depicts a high degree of similarity in its pattern when compared with the RyR distribution. Panels a2 and b2 have been generated by optically zooming into the cell and rescanning the lower left part of the myocyte at a different z-position. Panels c1, c2, d1, and d2, representative overviews of RyR (panels c1 and c2) and β2 (panels d1 and d2) immunoreactivity of additional myocytes. B, the β2 immunoreactivity is almost absent in ventricular myocytes from β2-KO mice. Ventricular myocytes were treated similarly to the cells in A. Scale bars represent 10 μm. C, β2 protein expression in isolated cardiomyocytes (20 μg of protein/lane) from KO and from controls. Expression of α-actinin was used as a loading control. D, estimation of the β2 protein expression levels in isolated cardiac myocytes from controls and KO. The amount of protein applied is as indicated. Coomassie Brilliant Blue stain was used as a loading control. M, marker proteins. E, RT-PCR amplification of a specific β2 326-bp fragment from single cardiomyocytes obtained as chippings by laser microdissection (lane 1, 40 ng of RNA), mouse heart (lane 2, 40 ng of RNA), cardiac fibroblasts (lane 4, 80 ng of RNA). Amplification of hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1, from total RNA obtained from mouse heart, lane 3) and PCR performed in the absence of template (H2O, lane 5) served as controls. F, the β2 protein expression in isolated cardiomyocytes (lane 1, 30 μg of protein, lane 2, 10 μg) and in three independent cardiac fibroblast preparations of passage 2 (lanes 3–5). Expression of the proline 4-hydroxylase gene (P4HB) gene (∼57 kDa) was used as fibroblast-specific loading control.
The hallmarks of CaVβ function are an increase in current density and a shift in voltage dependence of activation toward more hyperpolarized potentials (1). In isolated adult cardiomyocytes, L-type calcium currents are readily detectable (Fig. 4). Whole cell current density (ICa,0mV) and maximal whole cell conductance (Gmax) were reduced from −9.1 ± 0.64 pA/pF (control, n = 9) to 6.5 ± 0.44 pA/pF (KO, n = 8), i.e. by 28.6% (ICa,0mV) (Fig. 4E) and from 0.19 ± 0.02 nS/pF (control, n = 9) to 0.15 ± 0.02 nS/pF (KO, n = 8), i.e. by 21.1% (Gmax). Although the potential of half-maximal activation was slightly shifted to more depolarized potentials in KO cardiomyocytes (V½,act, −10.8 ± 0.9 mV; n = 7) when compared with control cells (V½,act, −12.1 ± 1.1 mV, n = 9), the difference was not significant (Fig. 4E). Similarly, the kinetics of inactivation is not significantly different between control and KO for all potentials (Fig. 4, F, G, and H). Currents of either group were sensitive to dihydropyridines (Fig. 5) with Bay-K8644 increasing current densities of ICa,0mV in KO cells to −8.0 ± 0.55 pA/pF (n = 7) (Fig. 4D), i.e. by 23.1% when compared with the current density in the absence of Bay-K8644, and to −11.3 ± 0.72 pA/pF, i.e. by 24.2% in control cells (Fig. 5C, n = 7), and with nifedipine inhibiting ICa,0mV to −3.5 ± 0.47 pA/pF, i.e. by 46.2% (KO cells, n = 7) and to −3.4 ± 0.31 pA/pF, i.e. by 62.6% (control cells). The channel agonist Bay-K8644 induced in cells of both genotypes a significant shift in the potential of half-maximal activation (V½,act) toward more negative potentials, from −12.1 ± 1.1 to −16.2 ± 1.4 mV (controls) and from −10.8 ± 0.9 to −15.3 ± 1.2 mV (KO); no differences in shift were obtained by comparing both groups.
FIGURE 4.

Reduced ICa in adult cardiomyocytes from KO mice. A–G, representative ICa and IV relations (A and C) and inactivation (B and D) in a control (A and B) and KO cardiomyocyte (C and D). E, summary. *, p < 0.05; **, p < 0.005. V indicates test potential (in A and C) and prepotential (in B and D), respectively. F and G, current families (F, controls; G, KO). Holding potential was −100 mV followed by a prestep of 100 ms to −40 mV and the test potentials after the prestep to the indicated voltages. Note that that the first part of all traces is the current at the −40 mV prepotential. H, summary of time constants of inactivation at the indicated potentials obtained from mono-exponential fits from F and G. No significant differences were measured between control and KO mice for all potentials (p > 0.05).
FIGURE 5.

Dihydropyridine sensitivity of ICa in cardiomyocytes. A and B, representative ICa in the absence (1) and presence of Bay-K8644 (2) or nifedipine (3). C and D, current density and V½,act (mV) in the absence and presence of Bay-K8644 (Bay-K) or nifedipine (Nife) in control cells (C) and KO cells (D). *, p < 0.05.
In another set of experiments, we compared wild type (WT) and CaVβ2+/− mice (het). The ICa,0mV and the Gmax were not significantly different (ICa,0mV, WT, −8.2 ± 0.7 pA/pF, n = 27; het, −8.8 ± 0.9 pA/pF, n = 7; Gmax, WT, 0.19 ± 0.11 nS/pF, n = 26; het, 0.21 ± 0.16 nS/pF, n = 7). Parameters of activation (V½,act, WT, −10.8 ± 0.6 mV, n = 27; het, −10.4 ± 1.4 mV, n = 7; s, WT, 6.4 ± 0.31 mV; het, 5.8 ± 0.40 mV) and inactivation (V½,inact, WT, −29.2 ± 0.7 mV, n = 26; het, −27.4 ± 1.2 mV, n = 7; s, WT, 6.9 ± 0.34 mV; het, 7.4 ± 0.45 mV) were also not statistically different.
Considering that CaVβ subunits may exist in pools that dynamically interact with the CaV1.2 protein (12, 13), the reduced current density could be caused by the loss of that pool responsible for trafficking and surface expression. CaVβ2 proteins already in complex with the plasma membrane CaV1.2 and modulating channel activation might be more stable and not require de novo protein synthesis. Berrow et al. (14) estimated a 55–60-h half-life of CaVβ-subunits upon application of a CaVβ antisense oligonucleotide, and 50% of the CaV1.2 protein is degraded 25 h after the chase (15). We also performed pulse-chase experiments and could not detect a difference of the half-life of 9.5–10 h of the mouse CaVβ2 protein when expressed in the absence or presence of CaV1.2 in COS cells (two experiments), arguing against the longevity of a pool of CaVβ2 proteins not affected by cacnb2 gene excision.
Current understanding of CaVβ function draws almost exclusively from heterologous expression of recombinant subunits in model systems, which may differ from cardiomyocytes. Studies in cardiomyocytes, in most cases, made use of overexpressing the neuronal rat CaVβ2a variant (3, 17), which is characterized by its intrinsic palmitoylation, although it is very unlikely the main CaVβ subunit associated with native cardiac calcium channels (2, 18, 19). This neuronal rat CaVβ2a (20) corresponds to CaVβ2-N3, according to the nomenclature for human (19), mouse (2), and canine CaVβ2 subunits (19), whereas the CaVβ2 variant predominantly expressed in mouse heart is CaVβ2-N4 (2, 19), which corresponds to human CaVβ2-N4 (19) or CaVβ2b (21) and rabbit CaVβ2a (22).
Other strategies pursued to knock down endogenous cardiac CaVβ proteins were difficult to control because appropriate antibodies were not available in that study (23). Our results now indicate that in the adult mouse heart, CaVβ2 is the most abundant CaVβ, that CaVβ2 is also expressed in cardiac fibroblasts, and that CaVβ2 has an effect on calcium channel density similar to its effect on cardiomyocytes of mouse embryos. However, although it is essential for embryo survival, adult mice tolerate quite well the decrease of CaVβ2.
Acknowledgments
We thank Christine Wesely, Stefanie Buchholz, Kerstin Fischer, and Christin Matka for expert technical assistance and Sabine Pelvay, Elisabeth Ludes, and Martin Jung for immunizing and bleeding rabbits.
This work was supported by Interuniversity Attraction Poles Programme – Belgian State – Belgian Science Policy, P6/28, the Research Council of the KU Leuven (Grant GOA 2004/07), the Flemish Government (Excellentiefinanciering, Grant EF/95/010) (to B. N. and J. P.), the Deutsche Forschungsgemeinschaft (Grants SFB530, KFO196, and GK 1326 (to P. L., M. F., and V. F.), Grant SFB894 (to P. W., M. F., and V. F.), the Homburger Forschungsförderungsprogramm HOMFOR (P. L., M. M., P. W., M. F., and V. F.), and the Forschungskommission der Universitaet des Saarlandes (P. L., M. F., and V. F.).
- TS
- Tyrode's solution
- LCM
- laser capture microdissection
- pF
- picofarads
- nS
- nanosiemens
- het
- heterozygous
- RyR
- ryanodine receptor.
REFERENCES
- 1. Dolphin A. C. (2009) Curr. Opin. Neurobiol. 19, 237–244 [DOI] [PubMed] [Google Scholar]
- 2. Link S., Meissner M., Held B., Beck A., Weissgerber P., Freichel M., Flockerzi V. (2009) J. Biol. Chem. 284, 30129–30137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakayama H., Chen X., Baines C. P., Klevitsky R., Zhang X., Zhang H., Jaleel N., Chua B. H., Hewett T. E., Robbins J., Houser S. R., Molkentin J. D. (2007) J. Clin. Invest. 117, 2431–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weissgerber P., Held B., Bloch W., Kaestner L., Chien K. R., Fleischmann B. K., Lipp P., Flockerzi V., Freichel M. (2006) Circ. Res. 99, 749–757 [DOI] [PubMed] [Google Scholar]
- 5. Striessnig J., Koschak A. (2008) Channels 2, 233–251 [DOI] [PubMed] [Google Scholar]
- 6. Sohal D. S., Nghiem M., Crackower M. A., Witt S. A., Kimball T. R., Tymitz K. M., Penninger J. M., Molkentin J. D. (2001) Circ. Res. 89, 20–25 [DOI] [PubMed] [Google Scholar]
- 7. Murakami M., Fleischmann B., De Felipe C., Freichel M., Trost C., Ludwig A., Wissenbach U., Schwegler H., Hofmann F., Hescheler J., Flockerzi V., Cavalié A. (2002) J. Biol. Chem. 277, 40342–40351 [DOI] [PubMed] [Google Scholar]
- 8. Berggren P. O., Yang S. N., Murakami M., Efanov A. M., Uhles S., Köhler M., Moede T., Fernström A., Appelskog I. B., Aspinwall C. A., Zaitsev S. V., Larsson O., de Vargas L. M., Fecher-Trost C., Weissgerber P., Ludwig A., Leibiger B., Juntti-Berggren L., Barker C. J., Gromada J., Freichel M., Leibiger I. B., Flockerzi V. (2004) Cell 119, 273–284 [DOI] [PubMed] [Google Scholar]
- 9. Badou A., Jha M. K., Matza D., Mehal W. Z., Freichel M., Flockerzi V., Flavell R. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15529–15534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Droogmans G., Nilius B. (1989) J. Physiol. 419, 627–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verrou C., Zhang Y., Zürn C., Schamel W. W., Reth M. (1999) Biol. Chem. 380, 1435–1438 [DOI] [PubMed] [Google Scholar]
- 12. Obermair G. J., Tuluc P., Flucher B. E. (2008) Curr. Opin. Pharmacol. 8, 311–318 [DOI] [PubMed] [Google Scholar]
- 13. Cantí C., Davies A., Berrow N. S., Butcher A. J., Page K. M., Dolphin A. C. (2001) Biophys. J. 81, 1439–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berrow N. S., Campbell V., Fitzgerald E. M., Brickley K., Dolphin A. C. (1995) J. Physiol. 482, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Catalucci D., Zhang D. H., DeSantiago J., Aimond F., Barbara G., Chemin J., Bonci D., Picht E., Rusconi F., Dalton N. D., Peterson K. L., Richard S., Bers D. M., Brown J. H., Condorelli G. (2009) J. Cell Biol. 184, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Banerjee I., Fuseler J. W., Price R. L., Borg T. K., Baudino T. A. (2007) Am. J. Physiol. Heart Circ. Physiol. 293, H1883–H1891 [DOI] [PubMed] [Google Scholar]
- 17. Jaleel N., Nakayama H., Chen X., Kubo H., MacDonnell S., Zhang H., Berretta R., Robbins J., Cribbs L., Molkentin J. D., Houser S. R. (2008) Circ. Res. 103, 1109–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei S. K., Colecraft H. M., DeMaria C. D., Peterson B. Z., Zhang R., Kohout T. A., Rogers T. B., Yue D. T. (2000) Circ. Res. 86, 175–184 [DOI] [PubMed] [Google Scholar]
- 19. Foell J. D., Balijepalli R. C., Delisle B. P., Yunker A. M., Robia S. L., Walker J. W., McEnery M. W., January C. T., Kamp T. J. (2004) Physiol. Genomics 17, 183–200 [DOI] [PubMed] [Google Scholar]
- 20. Perez-Reyes E., Castellano A., Kim H. S., Bertrand P., Baggstrom E., Lacerda A. E., Wei X. Y., Birnbaumer L. (1992) J. Biol. Chem. 267, 1792–1797 [PubMed] [Google Scholar]
- 21. Takahashi S. X., Mittman S., Colecraft H. M. (2003) Biophys. J. 84, 3007–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hullin R., Singer-Lahat D., Freichel M., Biel M., Dascal N., Hofmann F., Flockerzi V. (1992) EMBO J. 11, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cingolani E., Ramirez Correa G. A., Kizana E., Murata M., Cho H. C., Marbán E. (2007) Circ. Res. 101, 166–175 [DOI] [PubMed] [Google Scholar]
- 24. Droogmans G. (1997) WinASCD, Katholieke Universiteit Leuven, The Netherlands [Google Scholar]