

Abstract
proBDNF, a precursor of brain-derived neurotrophic factor (BDNF), is anterogradely transported and released from nerve terminals, but the mechanism underlying this process remains unclear. In this study, we report that proBDNF forms a complex with Huntingtin associated protein-1 (HAP1) and sortilin, which plays an important role in proBDNF intracellular trafficking and stabilization. The interaction of proBDNF with both HAP1A and sortilin in co-transfected HEK293 cells is confirmed by both fluorescence resonance energy transfer and co-immunoprecipitation. The frequent co-localization (>90%) of endogenous HAP1, sortilin, and proBDNF is also found in cultured cortical neurons. Mapping studies using GST pulldown and competition assays has defined the interacting region of HAP1 with proBDNF within amino acids 371–445 and the binding sequences of proBDNF to HAP1 between amino acids 65 and 90. Fluorescence recovery after photobleaching confirms the defective movement of proBDNF-containing vesicles in neurites of HAP1−/− neurons, which can be partially restored by reintroducing HAP1 cDNA into the neurons. However, the effect is significantly increased by simultaneously reintroducing both HAP1 and sortilin. proBDNF and HAP1 are highly co-localized with organelle markers for the Golgi network, microtubules, molecular motor, or endosomes in normal neurons, but this co-localization is reduced in HAP1−/− neurons. Co-immunoprecipitation and Western blot showed that sortilin stabilizes the proBDNF·HAP1 complex in co-transfected HEK293 cells, helping to prevent proBDNF degradation. Furthermore, the complex facilitates furin cleavage to release mature BDNF.
Keywords: Brain, Dynein, Golgi, Neuron, Trafficking, BDNF, Furin, Huntingtin-associated Protein 1, Neurons, Sortilin
Introduction
BDNF2 is a member of the neurotrophin family that plays a role in neuronal development and plasticity (1–6). Like all neurotrophins, BDNF is synthesized as a precursor (proBDNF) encompassing two domains, the prodomain and the BDNF domain (7). The proBDNF is cleaved by prohormone convertases such as furin and plasminogen/plasmin (8–10) to release the mature form (11). The newly synthesized proBDNF is transported anterogradely in neurons and released from the nerve terminal in an activity-dependent manner (9, 12–16). The released BDNF regulates neuronal survival, differentiation, dendritic morphology, and synaptic plasticity, which are important for memory and learning (14, 17–19).
The neurotrophic receptor-mediated retrograde transport of BDNF is well recognized (20–22), but the mechanism of its anterograde transport remains unclear. A recent study has suggested that Huntingtin protein (Htt), the p150Glued component of the dynactin complex (p150Glued), and microtubules are involved in the anterograde transport of BDNF (23). Kinesin (24–26) and dynein (24, 27, 28) motor proteins move along microtubules and participate in axonal anterograde and retrograde transport, respectively. It has been found that dynactin binding of dynein mediates vesicle endoplasmic reticulum-Golgi transport (29). Huntingtin-associated protein 1 (HAP1) interacts with p150glued subunit of dynactin (30) and regulates trafficking and membrane targeting of vesicles. HAP1 interacts with a number of proteins including the GABAA receptor (31, 32) and neurotrophic factor receptors (33, 34). HAP1 and sortilin co-exist in stigmoid-like bodies, a type of protein aggregation structure of unknown function found in neurons of healthy animals, in vivo (35). More emerging evidence suggests that both sortilin and carboxypeptidase E play significant roles in post-translational Golgi sorting of BDNF to the regulated secretory pathway (36, 37).
Sortilin is highly expressed in neuronal cells (38) and mostly distributed in the trans-Golgi network (TGN) (39–42). Its extracellular domain contains a homologous sequence to yeast vacuolar protein sorting 10 protein (VPS10p), which acts as a multi-ligand receptor, and binds to many proteins including proneurotrophins and p75 neurotrophin receptor (p75NTR) (43). The short intracellular domain is involved in Golgi-endosome sorting (42, 44).
The prodomain of BDNF plays an important role in the secretion and anterograde transport of proBDNF (36). For example, methionine substitution at codon 66 reduces BDNF transport and activity-dependent secretion (3). This polymorphism also causes reduction of hippocampal volume, impairment of episodic learning (45, 46), and a number of neurological disorders (47–49). However, the mechanism underlying the defect in BDNF trafficking remains unknown. Poly(Q) huntingtin (Htt) reduces BDNF mRNA expression and axonal transport of BDNF (50–52) by disrupting the interaction with HAP1 and p150glued (23), and the phosphorylation of Ser421 in poly(Q) Htt rescues the defect in BDNF transport (53). In a previous study, we have demonstrated that HAP1 may regulate axonal transport of proBDNF by interacting with the prodomain of BDNF (54). In the present study, we have further characterized the role of HAP1 and sortilin in sorting and trafficking of proBDNF and found that HAP1 and sortilin form a complex with proBDNF, preventing proBDNF degradation and modulating its targeting to endosomes, microtubules, and neurites.
EXPERIMENTAL PROCEDURES
Animals
All of the procedures involving animals were approved by the Animal Welfare Committee of Flinders University and undertaken according to the guidelines of the National Health and Medical Research Council of Australia. The use of genetically modified animals was approved by the Biosafety Committee of Flinders University. HAP1 knock-out mice were generated as described previously (55). All of the animals were kept under standardized barrier breeding conditions (12-h light/12-h dark cycle) with free access to water and food. PCR genotyping of HAP1 knock-out mice was carried out using primers 5′-TTTTGGAGGTCTGGTCTCGCTCTG-3′/5′-CGTCTTCCATCTTAGTGCGTTCAC-3′ for wild type and TTTTGGAGGTCTGGTCTCGCTCTG-3′/5′-CTTCATGTGGATGCTAGGGATCC-3′ for knock-out animals.
Plasmid Constructs
The constructs of rat mature BDNF, proBDNF, and prodomain CFP or YFP were made by PCR with primers (mature primer set, 5′-GCGAATTCATGCACTCCGACCCT-3′/5′-ATGGCGACCGGTGGATCCCT-3′; proBDNF primer set, 5′-GCGAATTCCACCAGGTG-3′/5′-ATGGCGACCGGTGGATCCCT-3′; and the prodomain primer set, 5′-GCGAATTCCACCAGGTG-3′/5′-ATGGATCCCGCCGAACCCT-3′), amplifying EcoRI and BamHI fragment from proBDNF-EGFP (a gift from Dr. Masami Kojima, Research Institute for Cell Engineering, National Institute of Advanced Industrial Science and Technology, Ikeda, Osaka, Japan) and cloning into pECFP-N1 or pEYFP-N1 (Clontech). The pcDNA3.1 constructs of prodomain, proBDNF, and mature BDNF were generated by PCR, amplifying the respective region with primers (prodomain primer set, 5′-TGCAAGCTTGCCACCATGACCATCCTTTTCCTTACT-3′/5′-TGCCTCGAGCGTGGCGCCGAACCCTCATAG-3′; proBDNF primer set, 5′-TGCAAGCTTGCCACCATGACCATCCTTTTCCTTACT-3′/5′-TGCCTCGAGCTCTTCCCCTTTTAATGGTC-3′; and BDNF primer set, 5′-TGCAAGCTTGCCACCATGCACTCCGACCCYGCCCGC-3′/5′-TGCCTCGAGCTCTTCCCCTTTTAATGGTC-3′) from AAproBDNF-EGFP (a furin-resistant construct with amino acids 129 and 130, RR to AA mutations from Dr. Masami Kojima). HAP1-CFP-N1 and HAP1-YFP-N1 were derived by cloning a PCR product generated from PRK-HAP1 (from Dr. Li) using appropriate primers (5′-TAGCTAGCATGCGCCCGAAGGAC-3′ and 5′-GAGGTACCAGGGTTGATGATCGGTAGC-3′) in the NheI and KpnI sites of pECFP-N1 and pEYFP-N1 (Clontech). The integrity of all cloned constructs was determined by DNA sequence analysis in both directions. p75CFP-YFP was a gift from Dr. E. Coulson (The University of Queensland, Chancellors Place, QLD 4067, Australia).
Cell Culture
HEK293 cells and rat pheochromocytoma cells (PC12) were obtained from the American Type Culture Collection (Manassas, VA) and maintained in DMEM (Invitrogen) supplemented with 10% FBS and 2 mm glutamine. Mouse cortical neurons were prepared from the brains of wild type or HAP1−/− neonates as described (56) with minor modifications. Briefly, cortical neurons were dissociated with 10 units/ml of papain (Worthington LS 03126) for 30 min at 37 °C, triturated, and plated at 1.5 × 105 cells/cm2 on coverslips or 35-mm FluoroDishes (World Precision Instruments, Inc.) coated with poly-d-lysine (Sigma). Neuronal cultures were maintained in neurobasal medium (Invitrogen) supplemented with 2% B27, 1% N2, 1% FBS, 100 units of penicillin/ml, and 100 μg of streptomycin/ml.
Transfection of Cell Lines and Neurons
PC12 or HEK293 cells (1 × 105) were seeded on poly-d-lysine-treated coverslips in 24-well plates and cultured in antibiotic-free DMEM supplemented with 10% FBS and 2 mm glutamine for 12 h. Mouse cortical neurons were cultured in 35-mm FluoroDishes for 3 days. Plasmid DNA constructs were transfected into cells using Lipofectamine 2000TM (Invitrogen) according to the manufacturer's recommendation. After transfection for 36 h, the cells were ready for FRAP- and FRET-related experiments. For co-immunoprecipitation (Co-IP) and Western blot, HEK293 cells were seeded into 10-cm culture dishes or six-well plates with 80% confluence and cultured in antibiotic-free DMEM supplemented with 10% FBS and 2 mm glutamine for 12 h. Plasmid constructs were transfected into cells using Lipofectamine 2000TM (Invitrogen) according to the manufacturer's recommendation. Transfection with YFP or CFP constructs served to monitor transfection efficiency. The percentage of fluorescent cells was calculated using IX71 microscopy and subjected to normalization.
Quantitative Real Time PCR
Total RNA (30 mg) was extracted from the cortex and dorsal root ganglia of neonatal wild type and HAP1−/− mice using a RNAeasy mini kit (Qiagen). The RNA concentration and purity were determined using a NanoDrop® ND-1000 UV-visible spectrophotometer (Thermo Fisher Scientific). The first strand cDNA was synthesized from 0.3 μg of total RNA using Qiagen QuantiTect reverse transcription kit. The SYBR Green real time PCR was performed using a Rotorgene 3000 (Corbett Research, Mortlake, NSW 2137, Australia) in triplicate using Qiagen PCR kit with specific primers for mouse proBDNF (forward primer, 5′-ATGACCATCCTTTTCCTTACT-3′; and reverse primer, 5′-GCGCCGAACCCTCATAGAC-3′) and normalized against the housekeeping gene GAPDH (forward primer, 5′-AACATCATCCCTGCATCCAC-3′; and reverse primer, 5′-TTGAAGTCRCAGGAGACAAC-3′). Real time quantitative PCR analysis was then performed using Q-Gene software (57) with the amplification efficiency applied to the relative concentration analyses of both the genes of interest and the housekeeping gene (GAPDH). Gene of interest expression data were normalized by dividing the corresponding levels of GAPDH for each sample.
Co-IP and GST Pulldown/Competition Assay
HEK293 cells co-transfected with HAP1A-CFP, sortilin-pcDNA3.1/Myc (a gift from Dr. C. Morales), and/or proBDNF-pcDNA3.1/Myc (RRH furin cleavage site mutant) were prepared in radio immunoprecipitation assay buffer containing 2 mm phenylmethanesulfonyl fluoride and protease inhibitors (Roche Applied Science) for cell lysates. The protein concentration of the lysates was determined using BCA protein assay kit (Thermo Scientific, Rockford, IL). All of the lysates were stored at −80 °C until use. To reduce the nonspecific binding to protein G beads, the cell lysate was precleared with protein G beads according to the manufacturer's instructions (Sigma). The precleared lysates (250 μg) of HEK293 cells were then incubated at 4 °C for 1 h with protein G bead-immobilized anti-sortilin (rabbit, ab16640; Abcam, Cambridge, MA), anti-HAP1 (rabbit, a gift from Dr. M. DiFiglia, Harvard Medical School, Boston, MA), anti-proBDNF (sheep), or anti-GFP (rabbit, ab290; Abcam). The beads were washed five times and boiled in loading buffer for Western blot using anti-HAP1, anti-sortilin, or anti-proBDNF. For negative control, rabbit or sheep IgG was used in the same way for validation.
GST-HAP1 constructs (HAP1 1–350, HAP1 280–445, HAP1 371–599, HAP1 328–599, HAP1 240–599, HAP1 215–599, and HAP1 153–599) were gifts from Dr. Josef Kittler (Department of Neuroscience, Physiology, and Pharmacology, University College London, London, UK). GST-HAP1 fusion proteins were produced in Escherichia coli BL21 (Invitrogen) and purified with glutathione-agarose beads (Sigma). The proBDNF lysates were incubated with GST-HAP1 fusion protein (2 μg) coupled to 40 μl of glutathione agarose beads at 4 °C for 2 h. After washing with radioimmune precipitation assay buffer five times, the proteins bound to the beads were subjected to Western blot analysis with rabbit anti-GFP (Abcam) or mouse anti-Myc antibodies (Invitrogen).
For the competition assay, the proBDNF lysates were incubated with proBDNF peptides (proBDNF, 44ESVNGPKAGSRGLTSLA60; proBDNF, 55GLTSLADTFEHVIEELLDED74; proBDNF, 65HVIEELLDEDQKVRPN80; proBDNF, 75KVRPNEENNKDADLY90; and proBDNF, 85KDADLYTSRVMLSSQV100) (Peptides International) and one nonspecific prostate-specific membrane antigen (PSMA) peptide (NH-PQSGAAVVHEIVRSFG-OH, accession number NP001014986) (Auspep, Victoria, Australia), respectively, at 4 °C for 1 h prior to the addition of GST-HAP1 fusion proteins. Then GST-HAP1 fusion protein (2 μg) coupled to 40 μl of glutathione-agarose beads was supplemented for a further 2 h of incubation. The beads were washed five times with radioimmune precipitation assay buffer and subjected to Western blot with rabbit anti-GFP antibody (Abcam).
Western Blot
Lysates of transfected HEK293 cells were prepared using radioimmune precipitation assay buffer supplemented with 2 mm phenylmethanesulfonyl fluoride and protease inhibitors (Roche Applied Science). The protein concentration of the lysates was determined using BCA protein assay kit (Thermo Scientific). Lysate proteins (50 μg) were analyzed by 10% SDS-PAGE and transferred to nitrocellulose membrane (Hybond ECL; GE Healthcare). Corresponding primary antibodies (1:1000) were incubated with blots at 4 °C overnight. HRP-conjugated secondary antibodies (1:2000) were used for detection. β-Actin was used as a loading control. Imaging was performed using ECL (GE Healthcare). Image J (National Institutes of Health) was used for quantitative analysis.
Immunocytochemistry
Antibodies to proBDNF were generated by immunization of sheep with synthetic peptide, corresponding to the 14 amino acids of the preregion sequence of proBDNF, which were conjugated to keyhole limpet hemocyanin (58, 59). proBDNF monoclonal antibody (PB17-2A) was prepared by immunization of BALB/c mice with the same peptide. The antibody was thoroughly characterized for specificity and binding capacity by Western blot and immunohistochemistry in parallel with sheep proBDNF antibody. This antibody only recognizes proBDNF but does not stain for mature BDNF. CD71 (endosome marker, goat, sc-7087), secretogranin II (rabbit), HAP1 polyclonal (rabbit, sc-30126), and monoclonal (mouse, sc166245) antibodies were purchased from Santa Cruz (Santa Cruz, CA). Tau (ab80579), MAP2 (Neuronal marker, ab32454), and GM130 (cis-Golgi marker, ab1299) antibodies were purchased from Abcam. Monoclonal p150Glued antibody (mouse 612709) was purchased from BD Biosciences. Sortilin antibody (rabbit, OSC-203) was purchased from Osenses (Adelaide, Australia). All of the fluorescent conjugated secondary antibodies were purchased from Sigma. For single and multiple labeling of mouse cortical neurons or transfected cells, appropriate combinations of primary and secondary antibodies were used. The images were acquired and analyzed by BX50 fluorescence microscope (Olympus) and Leica SP5 confocal microscope.
FRAP in Live Cells
Mouse cortical neurons were co-transfected with proBDNF-YFP alone or proBDNF-YFP·HAP1A or proBDNF-YFP·HAP1A·sortilin for 36 h. The culture medium was then replaced with CO2-independent medium. The cells in FluoroDishes were imaged using a Leica SP5 spectral confocal microscope equipped with a 37 °C heating device, according to the manufacturer's recommendation. Briefly, the fluorescence of YFP was assessed before and after photobleaching in a time series. Two scans were set before bleaching. To bleach YFP, the laser was set to 100% for both 496 and 514 nm, and six bleaches were taken. A total of 30 scans were performed at 3-s intervals after bleaching. For background correction and fluorescence loss, regions of interest were also selected from unbleached cell regions and outside of the cells. For each wild type or HAP1−/− genotype, six neurons were examined, and the fluorescent data were averaged at each time point.
FRET Acceptor Bleaching
PC12 cells were transfected with proBDNF-CFP and HAP1A-YFP plasmids or control plasmids. FRET was performed using a Leica SP5 spectral confocal microscope according to the manufacturer's recommendation (Leica). Briefly, acceptor bleaching was used to compare the donor fluorescence intensity in the same sample before and after photo bleaching the acceptor. If there is FRET, the intensity of the donor fluorescence increases after photo-bleaching the acceptor. The efficiency of FRET can be determined as: FRETeff = (Dpost − Dpre)/Dpost × 100%, where Dpost is the fluorescence intensity of the Donor after acceptor photo bleaching, and Dpre is the fluorescence intensity of the donor before acceptor photo bleaching. The FRETeff is considered positive when FRETeff is higher than background and negative controls. Based on the FRET principle, the higher the FRETeff, the closer the two molecules are. The p75CFP-YFP was used as a positive control. A pair of proteins such as proBDNF-CFP and pEYFP that show no interaction was used as a negative control. In addition, the signal of proBDNF-CFP from the surrounding cells in an adjacent region that expressed proBDNF-CFP alone was used as background. Each value of FRETeff from the tested samples including positive, negative, and background controls were determined according to a mean of six individual tests from different viewing fields. The results were summarized from three different experiments.
Statistical Analysis
All of the statistical analyses were performed with SPSS software (version 18.0). The data were expressed as the means ± S.E., and p < 0.05 was considered significant. Variables between groups were determined by independent or paired t test.
RESULTS
Prodomain of BDNF Interacts with HAP1
To strengthen the finding that HAP1 directly associates with proBDNF, we performed Co-IP (Fig. 1A) and FRET experiments (Fig. 1B). We co-transfected HEK293 cells with HAP1A-YFP and proBDNF-CFP or prodomain-CFP. It was shown that the HAP1A-YFP fusion protein was detected in either proBDNF or the prodomain immunoprecipitated complexes (Fig. 1A, lanes 1 and 2) but not found in negative control (Fig. 1A, lane 3), compared with positive control (Fig. 1A, lane 4).
FIGURE 1.
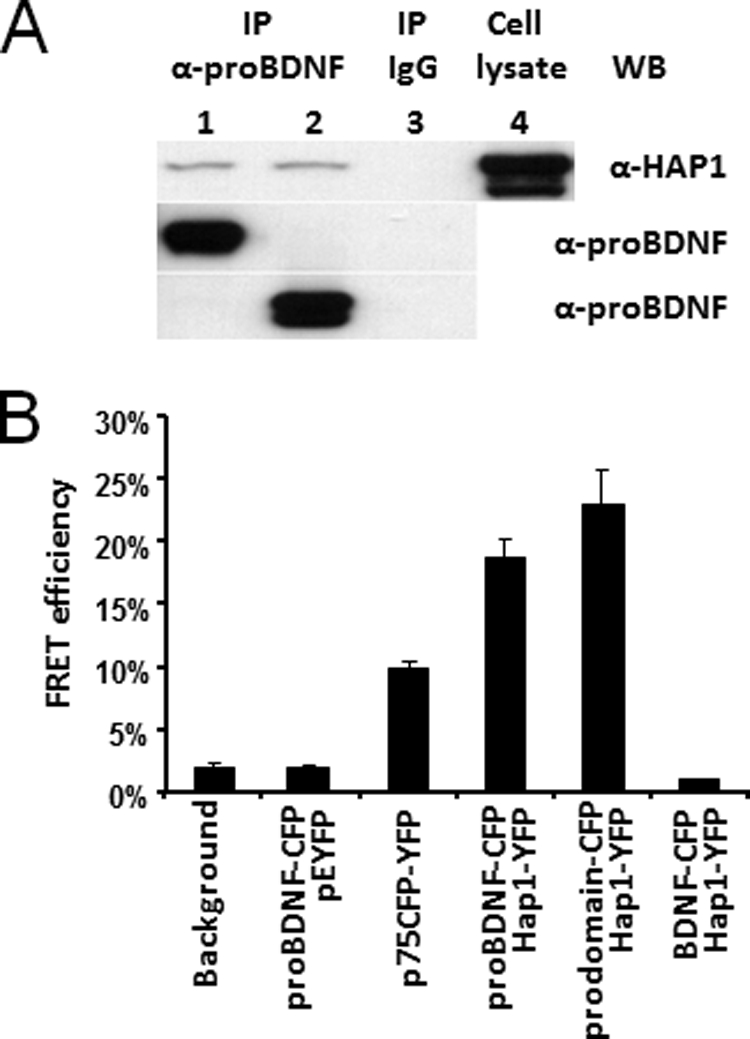
The prodomain of BDNF interacts with HAP1. A, proBDNF and the prodomain interact with HAP1 in co-transfected HEK293 cells. HEK293 co-transfected with HAP1A-YFP·proBDNF-CFP (lane 1) or HAP1A-YFP·prodomain-CFP (lane 2), respectively, were used for immunoprecipitation. Lane 1 and 2, Co-IP with sheep anti-prodomain antibodies (α-proBDNF) and Western blot (WB) with mouse anti-HAP1 (α-HAP1); HAP1A-YFP was detected (top panel). proBDNF-CFP and prodomain-CFP were blotted from the same samples used for Co-IP (middle and bottom panel), respectively. Lane 3, Co-IP with sheep IgG for negative control; no targeted protein was detected from the lysates used in lanes 1 and 2. Lane 4, co-transfected cell lysate input for probing HAP1A-YFP. B, FRET analysis of proBDNF interaction with HAP1A in co-transfected PC12 cells. The proBDNF-CFP·pEYFP were performed for the negative control. The p75CFP-YFP plasmid was used as a positive control. FRETeff was determined from a photobleached and targeted vesicle. The bars represent the means ± S.E. (n = 6).
Given that the background can cause false FRET signals, controls were used for quantifying the FRET efficiency (FRETeff) in transfected PC12 cells. The transfection using proBDNF-CFP·pEYFP plasmids for negative control resulted in a 2% FRETeff, which was similar to the background. The positive control, p75CFP-YFP fusion protein, showed an elevated FRETeff up to 10% and a 5-fold increase compared with background and negative controls. Therefore, the 2% of FRETeff was used as a cut-off in this study. The FRETeff from proBDNF-CFP·HAP1A-YFP was 18% when the measuring area was focused on the vesicle, suggesting a close proximity between proBDNF and HAP1A. Furthermore, a higher FRETeff was seen from prodomain-CFP·HAP1A-YFP, evident by a 23% of FRETeff directly from the targeted vesicle, suggesting that the prodomain is closer to HAP1A. In contrast, the FRETeff between the mature BDNF-CFP and HAP1A-YFP was less than 2%, indicating no direct interaction between this pair. Taken together, these data confirm our previous findings and indicate that HAP1A is a component of the proBDNF complex.
HAP1A Is Specifically Co-localized with proBDNF-containing Vesicles and Lack of HAP1 Impairs Neuritic proBDNF Distribution
To investigate whether the interaction between HAP1 and proBDNF has any physiological significance, we examined the distribution of HAP1 and proBDNF in live PC12 cells co-transfected with HAP1A-YFP and proBDNF-CFP or prodomain-CFP using the Nikon Biostation. As shown in merged panels (yellow), HAP1A (green) was highly co-localized with proBDNF (red) (90%) and the prodomain of BDNF (red) (87%) (Fig. 2A). To rule out the possibility of nonspecific binding of protein caused by overexpression, we stained endogenous proBDNF and HAP1 in cultured and fixed mouse cortical neurons with specific antibodies to proBDNF. It was shown that proBDNF was highly co-localized with HAP1 in both soma and neurites of wild type neurons (91%), whereas proBDNF was only present in cell soma or proximal neurites of HAP1−/− neurons compared with MAP2 staining (Fig. 2A). However, the stigmoid-like bodies found in transfected PC12 cells were not seen in cortical neurons. It is possible that the overexpression of HAP1 recruited proBDNF in large aggregates. To quantitatively analyze the transport of neuritic proBDNF, we measured the length of neurites fluorescently stained by anti-proBDNF and anti-MAP2 (the total length of neurites). The ratio of proBDNF and MAP2 was plotted in Fig. 2A (bottom left). The ratio is significantly lower in HAP1−/− neurons than in WT neurons. However, we found no significant change in the neuritic distribution of secretogranin II-containing vesicles between HAP1−/− and WT neurons (Fig. 2B). To see whether the lower level of proBDNF in neurites was due to down-regulation of proBDNF mRNA transcription by HAP1 knock-out, we examined whether proBDNF mRNA is normally expressed in HAP1−/− neurons. The proBDNF mRNA levels in the cortex and dorsal root ganglia of WT and HAP1−/− mice were analyzed by a quantitative real time RT-PCR. The data showed no significant difference in BDNF mRNA expression between WT and HAP1−/− mice (Fig. 2C, top panel). Interestingly, proBDNF protein level of the cortex was significantly lower in HAP1−/− mice than in WT mice (Fig. 2C, bottom panel). However, the decreased level of proBDNF alone does not fully explain the observation by immunohistochemistry that proBDNF is lacking in the neurites. We postulate that a mechanism other than protein expression may result in deficiencies in proBDNF transport in the HAP1−/− neurons. Taken together with the findings that mRNA levels were not changed, the data suggest that proBDNF protein may be less stable in HAP−/− cortex.
FIGURE 2.

HAP1 is physiologically associated with proBDNF. A, proBDNF co-localization with HAP1 in co-transfected PC12 cells and mouse cortical neurons. PC12 cells were co-transfected with proBDNF-CFP (red) and HAP1A-YFP (green) (top row). PC12 cells were co-transfected with prodomain of BDNF-CFP, designed as prodomain-CFP (red) and HAP1A-YFP (green) (second row). Mouse WT cortical neurons (third row) were immunostained for proBDNF (red) and HAP1 (green). Mouse WT cortical neurons (fourth row) were immunostained for proBDNF (red) and MAP2 (green). Mouse HAP1 −/− cortical neurons (bottom row) were immunostained for proBDNF (red) and MAP2 (green). HAP1 co-localization with proBDNF or the prodomain was performed using a SP5 confocal microscope and indicated in merged panels (yellow). Snapshots of co-localization are shown in the right panels. The transport of proBDNF in neurites was quantitatively determined by measuring the distance from the soma of proBDNF fluorescence as a ratio of total neurite length (MAP2) (bottom panel). B, HAP1-mediated proBDNF transport is unrelated to secretogranin II vesicle. Mouse WT (upper panel) and HAP1 −/− (bottom panel) cortical neurons immunostained for proBDNF (red) and secretogranin II (SgII, green). C, real time quantitative RT-PCR of proBDNF mRNA level (top). The mRNA expression level was determined in Hap1−/− cortex or dorsal root ganglia (n = 3, p > 0.05). Western blot analysis of proBDNF protein level in cortex of WT and HAP1−/− (bottom). The protein level of proBDNF was significantly lower in HAP1−/− samples (n = 3, p < 0.05).
Mapping proBDNF and HAP1 Binding Sites
To identify the proBDNF-binding sites in HAP1, we performed a GST-HAP1 pull-down assay. Seven GST-HAP1 constructs (HAP1 1–350, HAP1 280–445, HAP1 371–599, HAP1 328–599, HAP1 240–599, HAP1 215–599, and HAP1 153–599) (31) were incubated with proBDNF-containing cell lysates. Except HAP1 1–350, all other constructs pulled down proBDNF (Fig. 3A). Based on overlapping, the region of HAP1 between amino acids 371 and 445 contained proBDNF-binding sites. The flanking region structure may be involved in the binding as well because a lack of these regions decreased the interaction (Fig. 3A). To determine the binding region of proBDNF to HAP1, we added proBDNF peptides as competitors in the pulldown assay. Interestingly, all five proBDNF peptides except the negative control of PSMA showed inhibitory effects, but the greatest inhibition (>80%) resulted from two peptides, proBDNF 65–80 and proBDNF 75–90 (Fig. 3B). These results suggest that the region between amino acids 65 and 90 of the prodomain has a key structure for HAP1 binding.
FIGURE 3.

Mapping proBDNF and HAP1 interacting region. A, GST-HAP1 pulldown assay for mapping proBDNF binding region. The columns indicate seven GST-HAP1 constructs (HAP1 1–350, HAP1 280–445, HAP1 371–599, HAP1 328–599, HAP1 240–599, HAP1 215–599, and HAP1 153–599). proBDNF-YFP lysate was used for incubation with GST-HAP1 1–350 fusion protein and proBDNF-Myc lysate for incubation with all other GST-HAP1 fusion proteins in GST-pulldown assays. proBDNF-YFP used for GST-HAP1 1–350 pulldown assay is indicated by an arrow (top) and blotted with rabbit anti-GFP antibody (a-GFP). proBDNF-Myc used for all other GST-HAP1 pulldown assays is indicated by an arrow from pulled down and input samples (bottom), which was blotted with mouse anti-Myc antibody (a-Myc). The proBDNF binding region is indicated between dashed lines. B, the proBDNF peptides competition assay. Five specific proBDNF peptides (proBDNF 44–60, proBDNF 55–74, proBDNF 65–80, proBDNF 75–90, and proBDNF 85–100) and one nonspecific PSMA peptide used for competing proBDNF-YFP binding to GST-HAP1 153–599 in GST pulldown assay are listed at the top. The positive band of proBDNF-YFP is shown in the Input lane. The arrow indicates proBDNF-YFP either pulled-down by GST-HAP1 153–599 in the competition assay or input samples. On the right, the average inhibition of the proBDNF peptides on the interaction between proBDNF-YFP and GST-HAP1 153–599 from three individual experiments is indicated as a percentage over the nonspecific PSMA peptide, which was calculated as 0% inhibition. WB, Western blot.
Deficient proBDNF Trafficking in HAP1−/− Neurons
To confirm our immunostaining results on co-localization and transport of proBDNF in neurons, we examined proBDNF trafficking in WT and HAP1−/− neurons using FRAP. We transiently transfected cultured WT and HAP1−/− cortical neurons with proBDNF-YFP and measured the movement of proBDNF-YFP in the neurites of mouse cortical neurons. After bleaching proBDNF-YFP, the maximum recovery of its fluorescence was 87% in WT neurons and 78% in HAP1−/− neurons (Fig. 4A). When measuring the recovery velocity (time for 50% of fluorescent recovery), HAP1−/− neurons took longer (24 s) than WT neurons (15 s), showing that the difference was significant (p < 0.05) (Fig. 4B). To see whether the reintroduction of HAP1 to HAP−/− neurons could rescue the phenotype, we co-transfected HAP−/− neurons with both HAP1A-CFP and proBDNF-YFP plasmids. We found that the time for 50% of fluorescent recovery was 5 s shorter in dual co-transfected HAP−/− neurons (Fig. 4B). Interestingly, the recovery was further improved by co-transfection in conjunction with sortilin cDNA plasmids, showing that the time for 50% of fluorescent recovery was similar to that in WT (p > 0.05) (Fig. 4B). Therefore, it was evident that the exogenous expression of HAP1A in conjunction with sortilin was able to rescue the defect in proBDNF trafficking in HAP1−/− neurons.
FIGURE 4.

Lack of HAP1 impacts on the trafficking of proBDNF-YFP in mouse cortical neurons. A, FRAP of proBDNF-YFP trafficking in living neurons. Mouse cortical neurons were transfected with proBDNF-YFP. The trafficking of proBDNF-YFP in neurites was examined using FRAP under a Leica SP5 confocal microscope. The initial fluorescence is indicated as 100%. B, time for 50% fluorescence recovery was determined by FRAP in transfected mouse cortical neurons. The four bars on the left are in HAP1−/− neurons, and the bars on the right are in WT neurons. The data are represented as the means ± S.E. from six transfected neurons. The labels under each bar indicate different plasmids that are used for transfection. Less time is needed for WT neurons to get a recovery of the fluorescence of proBDNF-YFP compared with HAP1−/− neurons. Co-transfection of HAP1-CFP in HAP1−/− neurons, but not in WT neurons, reduced the time needed for recovery, whereas co-transfection of both HAP1-CFP and sortilin completely rescued the defect in fluorescence recovery.
The proBDNF·HAP1 Complex Is Present in Different Organelles
To investigate how proBDNF is intracellularly transported, we analyzed the distribution and association of proBDNF·HAP1 complex with Golgi network, microtubules, endosomes, and p150Glued structures in cortical neurons. We found that both HAP1 and proBDNF were distributed in Golgi apparatus and that a number of yellow spots from overlap between proBDNF and HAP1 were observed on one side of the Golgi apparatus in WT neurons (Fig. 5A, top panel), suggesting that HAP1 may be involved in sorting proBDNF to the TGN and secretory pathway. In contrast, most proBDNF gathered around cis-Golgi apparatus in HAP1−/− neurons (Fig. 5A, bottom panel). More than 90% of HAP1 or proBDNF was also co-localized with endosomal marker CD71 in WT neurons (Fig. 5B, top panel), indicating that endosomes may participate in sorting of the HAP1·proBDNF complex via TGN or may be involved in the transport of proBDNF·HAP1 complex. However, a decreased (65%) co-localization of proBDNF with endosomal marker CD71 was found in HAP1−/− neurons (Fig. 5B, bottom panel), compared with WT neurons. Our results showed that the proBDNF·HAP1 complex was localized with microtubule marker Tau in both axons and soma (Fig. 5C). In addition, 96% of proBDNF was co-localized with molecular motor protein p150Glued in WT neurons, whereas only 71% was observed in HAP1−/− neurons (Fig. 5D), suggesting that HAP1 contributes to targeting proBDNF to molecular motor proteins.
FIGURE 5.

Differential distribution of organelle markers in HAP1−/− cortical neurons. A, top panel, proBDNF and HAP1 are present in cis-Golgi apparatus of WT neurons. Primary cultured WT mouse cortical neurons were immunostained for proBDNF (red), HAP1 (green), and cis-Golgi (GM130, blue). A 4× enlargement of the white box shown in the merged panels is presented in the lower left corner. Bottom panel, primary cultured HAP1−/− mouse cortical neurons were immunostained for proBDNF (red) and cis-Golgi (blue). A 3× enlargement of the white box showed in merged panels was present on the right. B, proBDNF and HAP1 are co-localized with CD71, a marker for endosome. Top panel, mouse cortical neurons were immunostained for proBDNF (red), endosome marker CD71 (green), HAP1 (blue). The merged image is shown in white. A snapshot of co-localization between CD71 and proBDNF or HAP1 is shown on the right. Bottom panel, HAP1−/− mouse cortical neurons were immunostained for proBDNF (red) and endosome marker, CD71 (green), with the merged image in yellow. A snapshot of co-localization between CD71 and proBDNF is present on the right. The quantitative co-localizations are represented as the means ± S.E. (n = 5 neurons). p < 0.05. C, proBDNF and HAP1 are co-localized to microtubule. Primary cultured mouse cortical neurons were immunostained for proBDNF (red), HAP1 (green), and Tau (blue). A 6× enlargement of the white box showed in the merged panels is present in the lower left corner. D, proBDNF is co-localized with p150Glued in cortical neuron. Top panel, primary cultured mouse cortical neurons were immunostained for proBDNF (red), p150Glued (green), and DAPI (blue), and the merged image is in yellow. A snapshot of co-localization is present on the right. Bottom panel, primary cultured HAP1−/− mouse cortical neurons were immunostained for proBDNF (red), p150Glued (green), and DAPI (blue) with the merged image in yellow. A snapshot of co-localization is present on the right. The quantitative co-localizations are represented as the means ± S.E. (n = 5 neurons). p < 0.05.
proBDNF, HAP1, and Sortilin Form a Complex
Our results revealed that proBDNF was highly co-localized with HAP1 at the TGN. proBDNF is known to interact with HAP1 and sortilin (36). Therefore, we postulated that proBDNF, HAP1, and sortilin might form a complex that could be involved in sorting of proBDNF at TGN. To test this hypothesis, we firstly investigated whether HAP1 associated with sortilin, because 90% sortilin is found in trans-Golgi network (39, 42). Sortilin is also reported to co-localize with HAP1 in stigmoid bodies, a type of protein aggregation structure of unknown function found in neurons of healthy animals (35). We found that ∼91% of endogenous HAP1 and sortilin were co-localized in cortical neurons (Fig. 6A, middle row). This high co-localization was similar to that (93%) of proBDNF with HAP1 (Fig. 6A, top row) and 85% of proBDNF with sortilin (Fig. 6A, bottom row). The result suggested that proBDNF, HAP1, and sortilin could form a complex in cortical neurons. To further confirm this idea, we performed Co-IP using co-transfected HEK293 cell lysate. We found that proBDNF and sortilin were present in the sample immunoprecipitated with HAP1 antibody (Fig. 6B, rows d and e). Alternatively, we also found that HAP1 and sortilin were present in the samples immunoprecipitated with proBDNF antibody (Fig. 6B, rows b and f). Additionally, we found that HAP1 and proBDNF were present in the samples immunoprecipitated with sortilin antibody (Fig. 6B, rows a and c). No specific band was found when nonspecific immunoglobulin was used for Co-IP. Thus, we conclude that proBDNF, sortilin, and HAP1 form a complex in neurons.
FIGURE 6.

Association of sortilin, HAP1, and proBDNF. A, co-localization of proBDNF, HAP1, and sortilin in cultured mouse cortical neurons (WT). Top panel, cortical neurons were immunostained for proBDNF (red), HAP1 (green), and DAPI (blue), with the merged image in yellow. Middle panel, cortical neurons were immunostained for HAP1 (red), sortilin (green), and DAPI (blue), with the merged image in yellow. Bottom panel, cortical neurons were immunostained for proBDNF (red), sortilin (green), and DAPI (blue) with the merged image in yellow. A snapshot of co-localization image for each panel is present on the right. The scale bar (10 μm) was shown on the bottom right corner of the images. B, interaction of proBDNF, HAP1, and sortilin in the co-transfected HEK293. Co-IP was used for identification of the interaction among proBDNF, HAP1, and sortilin. Cell lysates were prepared from HEK293 cells co-transfected with proBDNF-Myc, HAP1A-YFP, and sortilin-Myc plasmids. Antibodies and control IgG used for Co-IP and positive controls from input are indicated on the top of each blot. Antibodies used for probing each sample by Western blot (WB) are marked on the right side of the panel. On the left, molecule names and arrows are listed to mark each blot. Row a, Co-IP of HAP1A-YFP by sortilin with rabbit sortilin antibodies (α-sort). Rabbit IgG was used as a negative control. Mouse anti-HAP1 (α-HAP1) was used for Western blot. HAP1A-YFP is indicated by the arrow. Row b, co-IP of HAP1A-YFP by proBDNF with sheep proBDNF antibodies (α-proB). Sheep IgG was used as a negative control. Mouse anti-HAP1 (α-HAP1) was used for Western blot. HAP1A-YFP is indicated by the arrow. Row c, Co-IP of proBDNF-Myc by sortilin with rabbit sortilin antibodies (α-sort). Rabbit IgG was used as a negative control. Sheep anti-proBDNF (α-proB) was used for Western blot. proBDNF-Myc is indicated by the arrow. Row d, Co-IP of proBDNF-Myc by HAP1 with mouse HAP1 antibodies (α-HAP1). Mouse IgG was used as a negative control. Sheep anti-proBDNF (α-proB) was used for Western blot. proBDNF-Myc is indicated by the arrow. Row e, Co-IP of sortilin-Myc by HAP1A with mouse HAP1 antibodies (α-HAP1). Mouse IgG was used as a negative control. Rabbit anti-sortilin (α-sort) was used for Western blot. Sortilin-Myc is indicated by the arrow. Row f, Co-IP of sortilin-Myc by proBDNF with sheep proBDNF antibodies (α-proB). Sheep IgG was used as a negative control. Rabbit anti-sortilin (α-sort) was used for Western blot. Sortilin-Myc is indicated by the arrow.
proBDNF Degradation Was Prevented by Co-expression of Sortilin in Transfected HEK293 Cells
Sortilin was found to improve proBDNF trafficking in HAP−/− neurons (Fig. 4B) and also formed a complex with proBDNF and HAP1 (Fig. 6B), suggesting that the intracellular sortilin may have an important function in proBDNF trafficking and stabilization. To demonstrate the role of sortilin in the formation of proBDNF·HAP1 complex, we analyzed the efficiency of proBDNF·HAP1 immunoprecipitation in the presence or absence of sortilin by Co-IP. We found that proBDNF was less immunoprecipitated by HAP1 antibody in the absence of sortilin, showing a significant difference (p < 0.05) (Fig. 7A). These data suggest that sortilin may be involved in proBDNF stabilization. To further test this hypothesis, we co-transfected HEK 293 cells with proBDNF and HAP1 with or without sortilin in the presence or absence of cycloheximide (CHX), a chemical that blocks protein synthesis. Anti-GFP, anti-proBDNF, and anti-BDNF antibodies were used for specific validation. We found that the protein level of proBDNF was positively correlated with the increased amount of sortilin plasmids. (Fig. 7B, top panel). All β-actin and HAP1 for internal controls or loading variations were consistent with each other, ruling out the possibility of a loading variation. The results generated by blotting with anti-GFP were similar to that probed with anti-proBDNF and anti-mature BDNF, suggesting that the detection of proBDNF and BDNF was specific.
FIGURE 7.

The degradation of proBDNF protein is prevented by co-expression of sortilin in transfected HEK293. A, Co-IP of proBDNF-Myc using co-transfected HEK 293 cells with or without sortilin-Myc plasmids. The addition of plasmids for transfection is listed on the top (left panel). Rabbit anti-GFP (a-GFP) was used for immunoprecipitation of HAP1A-YFP, and mouse anti-Myc (a-Myc) was used for Western blot (WB) of proBDNF-Myc fusion protein. The Co-IP lysate (10 μg) was blotted with the antibody to β-actin (a-βactin), which was used for normalization (bottom). Quantitation data of immunoprecipitated proBDNF-Myc are presented as the means ± S.E. of three independent experiments (right panel). P/H/S, proBDNF-Myc·HAP1A-YFP·sortilin-Myc and P/H, proBDNF-Myc·HAP1A-YFP. B, Western blot of proBDNF in a dose response of sortilin expression. The addition of plasmids for transfection is indicated on the top, and untransfected HEK293 as negative control on the right. Rabbit anti-GFP (α-GFP), sheep anti-proBDNF (α-proBDNF), and sheep anti-mature BDNF (α-BDNF) were used for Western blot. The bottom panel was probed with anti-β-actin, anti-GFP, and anti-Myc. HEK293 was treated with CHX (100 μg/ml) for 4 h before harvesting. C, time course of proBDNF degradation. Western blot of proBDNF-YFP was performed in the presence or absence of 100 ng of sortilin-Myc plasmid in the transfection (top panel) and β-actin for control (bottom panel). The quantitative data are shown as the means ± S.E. of three independent experiments (bottom panel).
To further examine whether proBDNF was more stable in the presence of HAP1 and sortilin, we performed a time course for Western blot to measure the degradation of synthesized proBDNF in HEK293 cells co-transfected with all three plasmids and treated with CHX. We found that the level of proBDNF protein was much less in the absence of sortilin plasmid after CHX treatment for 2 and 4 h, showing a significant difference (p < 0.05) between the cells with and without sortilin (Fig. 7C). The decrease of proBDNF was decelerated at 12 h but continued for 24 h after CHX treatment (Fig. 7C). The consistent β-actin level ruled out the loading variation but suggested that the proBDNF complex lacking sortilin might be easily exposed to proteasome and degraded.
The proBDNF·HAP1·Sortilin Complex Favors Furin Cleavage and Production of Mature BDNF
We showed that the level of mature BDNF increased in the cell lysate co-transfected with proBDNF, HAP1A, and sortilin, compared with the cell lysate without sortilin (Fig. 7B). To investigate whether the complex takes advantage of furin cleavage to release mature BDNF, we examined furin processing of proBDNF in the presence of HAP1 and sortilin or proBDNF alone in vitro (Fig. 8). We performed Western blotting with sheep anti-proBDNF and sheep anti-BDNF antibodies to detect the ratio of BDNF/proBDNF. We found that the ratio was significantly higher in the digestion mixture containing proBDNF-YFP, HAP1A-CFP, and sortilin-Myc (lane 2), compared with the mixture containing proBDNF alone (lane 1), suggesting that the complex of proBDNF·HAP1·sortilin might facilitate furin cleavage to release mature BDNF in vivo.
FIGURE 8.

proBDNF·HAP1·sortilin complex stabilized BDNF release. proBDNF·HAP1·sortilin complex favors BDNF yield by furin digestion in vitro. HEK293 cells were transfected with proBDNF-YFP, HAP1A-CFP, and sortilin-Myc expression plasmid separately. After 36 h transfection, the cells were harvested in cold PBS and sonicated on ice. Lysate (100 μg) containing proBDNF-YFP mixed with HAP1A-CFP and sortilin-Myc lysates or BSA alone was incubated with 4 units of furin at 30 °C for 6 h. The addition in the presence or absence of furin is listed on the top. Western blot (WB) was performed with sheep anti-proBDNF (α-proB) and sheep anti-BDNF (α-BDNF). Lane 1, proBDNF-YFP + BSA. Lanes 2 and 3, proBDNF-YFP·HAP1A-CFP·sortilin-Myc mix. β-Actin was used to confirm equal loading and for normalization. The quantitative data are presented as the means ± S.E. of the ratio of BDNF-YFP·proBDNF-YFP from three independent experiments.
DISCUSSION
BDNF is synthesized as a precursor (proBDNF), which is anterogradely transported via axon to presynaptic nerve terminals for release (60, 61). However, the mechanism of how proBDNF is anterogradely transported remains unclear because of a highly regulated and complex process. Our previous study revealed that HAP1 interacts with the prodomain of BDNF, which was possibly involved in regulating proBDNF axonal trafficking and secretion (54). In this study, we sought to extend our understanding of proBDNF trafficking and its molecular insights. Our data demonstrated that 1) proBDNF associated with both HAP1 and sortilin and formed a complex with these two molecules during its trafficking; 2) HAP1 and sortilin increased proBDNF targeting to dendrites and axonal organelles; and 3) sortilin stabilized intracellular proBDNF protein and may facilitate furin cleavage to release mature BDNF.
HAP1 and Sortilin Form a Complex with proBDNF, Which Is Important for proBDNF Intracellular Trafficking
The prodomain of BDNF is involved in intracellular transport of proBDNF toward both axonal nerve terminals and dendritic trees for the regulatory release of BDNF. The sorting of proBDNF to secretory pathway requires the interaction between the prodomain of BDNF and sortilin at the trans-Golgi network (36). The importance of the prodomain in proBDNF trafficking is demonstrated by a polymorphism that replaces a valine by methionine at codon 66 in the prodomain (62), which dramatically decreases the secretion of BDNF. In addition to these reports, we found that the prodomain could directly interact with HAP1. HAP1 knock-out in neurons caused a defect in proBDNF transport in sciatic nerve in vivo and in dendrites in vitro (50). We further mapped the binding sites between proBDNF and HAP1, showing that HAP1 amino acids 371–445 contain a key binding site for proBDNF. The flanking sequences may also play a role in the binding affinity because more proBDNF was pulled down in the presence of the flanking region (Fig. 3A). It is possible that the broad region is required for high conformational changes in protein-protein interaction. The binding site of proBDNF to HAP1 was determined between amino acids 65 and 90 (Fig. 3B). The V66M mutation in this region dramatically decreases dendritic targeting and the secretion of BDNF (3, 62). The V66M mutation also causes a significant reduction in the binding affinity of the prodomain to sortilin (49). It is likely that HAP1 acts as a bridge or adaptor linking proBDNF to motor protein such as p150glued subunit of dynactin, which facilitates anterograde axonal transport of proBDNF along the microtubule (63). The V66M mutation might impair the connection leading to insufficient trafficking of proBDNF to dendrites and nerve terminals. An intense FRET signal from the prodomain·HAP1 and proBDNF·HAP1 but not from mature BDNF·HAP1 (Fig. 1B) suggests that the molecular mechanism of proBDNF trafficking is different from mature BDNF.
The interaction between proBDNF and sortilin has been reported (36). Interestingly, we found that sortilin interacts not only with proBDNF but also with HAP1 (Fig. 6B), suggesting that proBDNF forms a complex with both HAP1 and sortilin intracellularly. HAP1 is a cytoplasmic protein containing a number of binding protein partners. Our findings also show its interaction with sortilin. The co-localization between proBDNF, HAP1, and sortilin was seen not only in co-transfected PC12 cells but also in mouse cortical neurons (Figs. 2A and 6A). Our immunoprecipitation and co-localization data showed that proBDNF, HAP1, and sortilin are always in the same Co-IP samples or subcellular vesicles, suggesting that the complex formation may be important for proBDNF functions. More interestingly, the lack of HAP1 did not impact on secretogranin vesicle transport (Fig. 2B), indicating that HAP1 specifically regulated proBDNF vesicle transport. In addition, the loss of HAP1 does not down-regulate the proBDNF mRNA expression (Fig. 2C), whereas Htt−/− does (50, 51, 64), suggesting that HAP1 has a different mechanism in regulation of proBDNF trafficking. Based on the current studies, we suggest that proBDNF trafficking is likely governed by its binding proteins in neurons. HAP1 may participate with sortilin and other co-factors in regulating proBDNF axonal trafficking (30, 33). The proBDNF·HAP1·sortilin complex may be required for proBDNF trafficking from Golgi to the nerve terminal. This hypothesis is confirmed by FRAP of HAP1 rescue studies, showing that reintroduction of HAP1 and sortilin cDNA into HAP1−/− neurons improves the defective trafficking of proBDNF-YFP (Fig. 4, A and B). Given that the regulatory mechanism of proBDNF transport is a complex process, other co-factors in addition to HAP1 and sortilin may be recruited for efficient transport of proBDNF. It is reported that HAP1 interacts with the p150Glued subunit of dynactin (63, 65) and the kinesin light chain (66). p150Glued and kinesin light chain are components of dynein motor (24, 27, 28) and kinesin motor (24, 25), respectively. HAP1 has been reported to interact with p150Glued (63), which mediates Golgi transport vesicles to microtubules (29). Indeed, our results have shown that proBDNF, HAP1, and microtubules are highly co-localized (Fig. 5C), and 96% of proBDNF and p150Glued are co-localized in WT neurons (Fig. 5D, top panel). In contrast, this co-localization was significantly decreased in HAP1−/− neurons (Fig. 5D, bottom panel), reflecting a defect of proBDNF trafficking without HAP1 in the complex. The V66M mutation may destabilize proBDNF·HAP1·sortilin complex, resulting in dissociation with other trafficking factors such as p150Glued (23), 14-3-3 protein (67), HIP-14 (68), HIP1R (61), carboxypeptidase E (69), and Htt (34, 45). Hence, impairment of the proBDNF·HAP1·sortilin complex formation may lead to inefficient transport of proBDNF-containing vesicles and reduction of proBDNF secretion.
Sortilin is engaged in Golgi-endosome transport (42). Our data show that HAP1 interacted with sortilin (Fig. 6B) and co-localized with proBDNF in Golgi area (Fig. 5A), suggesting that Golgi may be the first place where HAP1 and sortilin are recruited into the complex with proBDNF for its anterograde trafficking. Our results also demonstrate that HAP1 and proBDNF are co-localized with other intracellular organelles such as CD71 (endosome marker), Tau (microtubule marker), and neurites in WT cortical neurons (Fig. 5, B and C). In contrast, lack of HAP1 leads to the retention of proBDNF in Golgi and a decreased co-localization of proBDNF with endosome marker CD71. Therefore, the Golgi region may provide sites for proBDNF, HAP1, and sortilin to form a functional motility complex. The ∼95% co-localization among HAP1, proBDNF, and endosomal marker CD71 suggests that Golgi to/from endosomal transport is a part of proBDNF trafficking process. Endosomes can sort and transport proteins from vesicles to trans-Golgi network or directly release the luminal vesicle from the cell via exosomes (70) before they reach lysosomes for degradation (71). It is known that p150Glued is associated with sorting nexins 5 and 6, which are components of retromer subcomplexes mediating endosomes to Golgi transport (72). In addition, sortilin through sorting nexin 1 or GGA sorting protein mediates endosome to/from Golgi transport (42, 73). It is likely that the complex of proBDNF·HAP1·sortilin may be sorted back to trans-Golgi network via interacting with retromer proteins, preventing them from targeting lysosomes or proteasomes for degradation.
proBDNF·HAP1·Sortilin Complex Prevents proBDNF Degradation and Facilitates Mature BDNF Release by Furin Cleavage
We demonstrated that proBDNF forms a complex with HAP1 and sortilin, which is required not only for proBDNF axonal trafficking and sorting to the secretory pathway but for preventing proBDNF degradation. Our data showed that the exogenous expression of proBDNF was degraded in co-transfected HEK293 cells without the expression of sortilin (Fig. 7). Indeed, this result was in agreement with the study that secreted sortilin forms a complex extracellularly with the secreted proBDNF in vitro, which reduces its degradation in the presence of plasmin (74). In fact, we have found a sortilin dose-dependent preservation of proBDNF in the presence of protein synthesis inhibitor, and mature BDNF production is likely increased at higher doses of sortilin. We also found that sortilin prevented degradation of proBDNF and facilitated cleavage by furin (Fig. 8), suggesting that the complex may be required for the intracellular stabilization of proBDNF and for its proper processing to produce mature BDNF. Indeed, it is evident that the protein level of proBDNF is significantly lower in HAP1−/− than in WT (Fig. 2C, bottom panel). Because proBDNF and mature BDNF have differential functions, the regulation of proBDNF and mature BDNF production caused by the complex stabilization may have biological significance during development and in neurological disorders. However, how the proBDNF·HAP1·sortilin complex modulates proBDNF processing in vivo needs further elucidation.
In conclusion, our findings provide new insights into dendritic and axonal targeting of proBDNF in cortical neurons. The interaction between proBDNF, HAP1, and sortilin may be the key to form the functional motility complex. Our results, for the first time, suggest that Golgi and endosomes may be involved in proBDNF trafficking. The impairment of formation of proBDNF·HAP1·sortilin complex in vivo leading to proBDNF degradation may underlie the reduction in BDNF secretion in human neurological disorders.
Acknowledgments
We thank Dr. M. Kojima (National Institute of Advanced Industrial Science and Technology, Ikeda, Japan) for providing wild type and V66M BDNF plasmids, Dr. E. Coulson from University of Queensland for p75-CFP-YFP plasmids, Dr. J. Kittler from University College London for GST-HAP1 constructs, and Dr. C. Morales from McGill University for sortilin plasmid. We also thank Dr. Jennifer Clarke and Dr. Michael Jackson from Flinders University for critical reading of the manuscript.
The project was supported by National Health and Medical Research Council Grants 375110 and 488022.
- BDNF
- brain-derived neurotrophic factor
- proBDNF
- precursor of BDNF
- HAP1
- Huntingtin-associated protein-1
- FRAP
- fluorescence recovery after photobleaching
- TGN
- trans-Golgi network
- Co-IP
- co-immunoprecipitation
- PSMA
- prostate-specific membrane antigen
- CHX
- cycloheximide
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- FRET
- fluorescence resonance energy transfer.
REFERENCES
- 1. Bibel M., Barde Y. A. (2000) Genes Dev. 14, 2919–2937 [DOI] [PubMed] [Google Scholar]
- 2. Chao M. V. (1992) Neuron 9, 583–593 [DOI] [PubMed] [Google Scholar]
- 3. Egan M. F., Kojima M., Callicott J. H., Goldberg T. E., Kolachana B. S., Bertolino A., Zaitsev E., Gold B., Goldman D., Dean M., Lu B., Weinberger D. R. (2003) Cell 112, 257–269 [DOI] [PubMed] [Google Scholar]
- 4. Hefti F. (1994) J. Neurobiol. 25, 1418–1435 [DOI] [PubMed] [Google Scholar]
- 5. Huang E. J., Reichardt L. F. (2001) Annu. Rev. Neurosci. 24, 677–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poo M. M. (2001) Nat. Rev. Neurosci. 2, 24–32 [DOI] [PubMed] [Google Scholar]
- 7. Seidah N. G., Benjannet S., Pareek S., Savaria D., Hamelin J., Goulet B., Laliberte J., Lazure C., Chrétien M., Murphy R. A. (1996) Biochem. J. 314, 951–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. MacQueen G. M., Ramakrishnan K., Croll S. D., Siuciak J. A., Yu G., Young L. T., Fahnestock M. (2001) Behav. Neurosci. 115, 1145–1153 [DOI] [PubMed] [Google Scholar]
- 9. Goodman L. J., Valverde J., Lim F., Geschwind M. D., Federoff H. J., Geller A. I., Hefti F. (1996) Mol. Cell Neurosci. 7, 222–238 [DOI] [PubMed] [Google Scholar]
- 10. Mowla S. J., Farhadi H. F., Pareek S., Atwal J. K., Morris S. J., Seidah N. G., Murphy R. A. (2001) J. Biol. Chem. 276, 12660–12666 [DOI] [PubMed] [Google Scholar]
- 11. Matsumoto T., Rauskolb S., Polack M., Klose J., Kolbeck R., Korte M., Barde Y. A. (2008) Nat. Neurosci. 11, 131–133 [DOI] [PubMed] [Google Scholar]
- 12. Haubensak W., Narz F., Heumann R., Lessmann V. (1998) J. Cell Sci. 111, 1483–1493 [DOI] [PubMed] [Google Scholar]
- 13. Mowla S. J., Pareek S., Farhadi H. F., Petrecca K., Fawcett J. P., Seidah N. G., Morris S. J., Sossin W. S., Murphy R. A. (1999) J. Neurosci. 19, 2069–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. von Bartheld C. S., Wang X., Butowt R. (2001) Mol. Neurobiol. 24, 1–28 [DOI] [PubMed] [Google Scholar]
- 15. Yang J., Siao C. J., Nagappan G., Marinic T., Jing D., McGrath K., Chen Z. Y., Mark W., Tessarollo L., Lee F. S., Lu B., Hempstead B. L. (2009) Nat. Neurosci. 12, 113–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou X. F., Rush R. A. (1996) Neuroscience 74, 945–953 [DOI] [PubMed] [Google Scholar]
- 17. Kafitz K. W., Rose C. R., Thoenen H., Konnerth A. (1999) Nature 401, 918–921 [DOI] [PubMed] [Google Scholar]
- 18. Altar C. A., Cai N., Bliven T., Juhasz M., Conner J. M., Acheson A. L., Lindsay R. M., Wiegand S. J. (1997) Nature 389, 856–860 [DOI] [PubMed] [Google Scholar]
- 19. Butowt R., von Bartheld C. S. (2001) J. Neurosci. 21, 8915–8930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ginty D. D., Segal R. A. (2002) Curr. Opin. Neurobiol. 12, 268–274 [DOI] [PubMed] [Google Scholar]
- 21. Hibbert A. P., Kramer B. M., Miller F. D., Kaplan D. R. (2006) Mol. Cell Neurosci. 32, 387–402 [DOI] [PubMed] [Google Scholar]
- 22. Miller F. D., Kaplan D. R. (2001) Neuron 32, 767–770 [DOI] [PubMed] [Google Scholar]
- 23. Gauthier L. R., Charrin B. C., Borrell-Pagès M., Dompierre J. P., Rangone H., Cordelières F. P., De Mey J., MacDonald M. E., Lessmann V., Humbert S., Saudou F. (2004) Cell 118, 127–138 [DOI] [PubMed] [Google Scholar]
- 24. Gunawardena S., Goldstein L. S. (2004) J. Neurobiol. 58, 258–271 [DOI] [PubMed] [Google Scholar]
- 25. Hirokawa N., Takemura R. (2004) Curr. Opin. Neurobiol. 14, 564–573 [DOI] [PubMed] [Google Scholar]
- 26. Goldstein L. S., Yang Z. (2000) Annu. Rev. Neurosci. 23, 39–71 [DOI] [PubMed] [Google Scholar]
- 27. Schroer T. A. (2004) Annu. Rev. Cell Dev. Biol. 20, 759–779 [DOI] [PubMed] [Google Scholar]
- 28. Vallee R. B., Williams J. C., Varma D., Barnhart L. E. (2004) J. Neurobiol. 58, 189–200 [DOI] [PubMed] [Google Scholar]
- 29. Vaughan K. T. (2005) Biochim. Biophys. Acta 1744, 316–324 [DOI] [PubMed] [Google Scholar]
- 30. Li X. J., Li S. H. (2005) Trends Pharmacol. Sci. 26, 1–3 [DOI] [PubMed] [Google Scholar]
- 31. Twelvetrees A. E., Yuen E. Y., Arancibia-Carcamo I. L., MacAskill A. F., Rostaing P., Lumb M. J., Humbert S., Triller A., Saudou F., Yan Z., Kittler J. T. (2010) Neuron 65, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kittler J. T., Thomas P., Tretter V., Bogdanov Y. D., Haucke V., Smart T. G., Moss S. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 12736–12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rong J., McGuire J. R., Fang Z. H., Sheng G., Shin J. Y., Li S. H., Li X. J. (2006) J. Neurosci. 26, 6019–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu L. L., Zhou X. F. (2009) Cell Adh. Migr. 3, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gutekunst C. A., Torre E. R., Sheng Z., Yi H., Coleman S. H., Riedel I. B., Bujo H. (2003) J. Histochem. Cytochem. 51, 841–852 [DOI] [PubMed] [Google Scholar]
- 36. Chen Z. Y., Ieraci A., Teng H., Dall H., Meng C. X., Herrera D. G., Nykjaer A., Hempstead B. L., Lee F. S. (2005) J. Neurosci. 25, 6156–6166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lou H., Kim S. K., Zaitsev E., Snell C. R., Lu B., Loh Y. P. (2005) Neuron 45, 245–255 [DOI] [PubMed] [Google Scholar]
- 38. Sarret P., Krzywkowski P., Segal L., Nielsen M. S., Petersen C. M., Mazella J., Stroh T., Beaudet A. (2003) J. Comp. Neurol. 461, 483–505 [DOI] [PubMed] [Google Scholar]
- 39. Petersen C. M., Nielsen M. S., Nykjaer A., Jacobsen L., Tommerup N., Rasmussen H. H., Roigaard H., Gliemann J., Madsen P., Moestrup S. K. (1997) J. Biol. Chem. 272, 3599–3605 [DOI] [PubMed] [Google Scholar]
- 40. Morris N. J., Ross S. A., Lane W. S., Moestrup S. K., Petersen C. M., Keller S. R., Lienhard G. E. (1998) J. Biol. Chem. 273, 3582–3587 [DOI] [PubMed] [Google Scholar]
- 41. Mazella J. (2001) Cell Signal 13, 1–6 [DOI] [PubMed] [Google Scholar]
- 42. Nielsen M. S., Madsen P., Christensen E. I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., Petersen C. M. (2001) EMBO J. 20, 2180–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Feng D., Kim T., Ozkan E., Light M., Torkin R., Teng K. K., Hempstead B. L., Garcia K. C. (2010) J. Mol. Biol. 396, 967–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Willnow T. E., Petersen C. M., Nykjaer A. (2008) Nat. Rev. Neurosci. 9, 899–909 [DOI] [PubMed] [Google Scholar]
- 45. Hariri A. R., Goldberg T. E., Mattay V. S., Kolachana B. S., Callicott J. H., Egan M. F., Weinberger D. R. (2003) J. Neurosci. 23, 6690–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pezawas L., Verchinski B. A., Mattay V. S., Callicott J. H., Kolachana B. S., Straub R. E., Egan M. F., Meyer-Lindenberg A., Weinberger D. R. (2004) J. Neurosci. 24, 10099–10102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ventriglia M., Bocchio Chiavetto L., Benussi L., Binetti G., Zanetti O., Riva M. A., Gennarelli M. (2002) Mol. Psychiatry 7, 136–137 [DOI] [PubMed] [Google Scholar]
- 48. Momose Y., Murata M., Kobayashi K., Tachikawa M., Nakabayashi Y., Kanazawa I., Toda T. (2002) Ann. Neurol. 51, 133–136 [DOI] [PubMed] [Google Scholar]
- 49. Chen Z. Y., Jing D., Bath K. G., Ieraci A., Khan T., Siao C. J., Herrera D. G., Toth M., Yang C., McEwen B. S., Hempstead B. L., Lee F. S. (2006) Science 314, 140–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zuccato C., Ciammola A., Rigamonti D., Leavitt B. R., Goffredo D., Conti L., MacDonald M. E., Friedlander R. M., Silani V., Hayden M. R., Timmusk T., Sipione S., Cattaneo E. (2001) Science 293, 493–498 [DOI] [PubMed] [Google Scholar]
- 51. Zuccato C., Cattaneo E. (2007) Prog. Neurobiol. 81, 294–330 [DOI] [PubMed] [Google Scholar]
- 52. del Toro D., Canals J. M., Ginés S., Kojima M., Egea G., Alberch J. (2006) J. Neurosci. 26, 12748–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zala D., Colin E., Rangone H., Liot G., Humbert S., Saudou F. (2008) Hum. Mol. Genet. 17, 3837–3846 [DOI] [PubMed] [Google Scholar]
- 54. Wu L. L., Fan Y., Li S., Li X. J., Zhou X. F. (2010) J. Biol. Chem. 285, 5614–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li S. H., Yu Z. X., Li C. L., Nguyen H. P., Zhou Y. X., Deng C., Li X. J. (2003) J. Neurosci. 23, 6956–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hilgenberg L. G., Smith M. A. (2007) J. Vis. Exp. 10, 562, 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Simon P. (2003) Bioinformatics 19, 1439–1440 [DOI] [PubMed] [Google Scholar]
- 58. Zhou X. F., Song X. Y., Zhong J. H., Barati S., Zhou F. H., Johnson S. M. (2004) J. Neurochem. 91, 704–715 [DOI] [PubMed] [Google Scholar]
- 59. Wang H., Wu L. L., Song X. Y., Luo X. G., Zhong J. H., Rush R. A., Zhou X. F. (2006) Eur. J. Neurosci. 24, 2444–2452 [DOI] [PubMed] [Google Scholar]
- 60. Katz L. C., Shatz C. J. (1996) Science 274, 1133–1138 [DOI] [PubMed] [Google Scholar]
- 61. Carreno S., Engqvist-Goldstein A. E., Zhang C. X., McDonald K. L., Drubin D. G. (2004) J. Cell Biol. 165, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen Z. Y., Patel P. D., Sant G., Meng C. X., Teng K. K., Hempstead B. L., Lee F. S. (2004) J. Neurosci. 24, 4401–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Engelender S., Sharp A. H., Colomer V., Tokito M. K., Lanahan A., Worley P., Holzbaur E. L., Ross C. A. (1997) Hum. Mol. Genet. 6, 2205–2212 [DOI] [PubMed] [Google Scholar]
- 64. Zuccato C., Tartari M., Crotti A., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B. R., Hayden M. R., Timmusk T., Rigamonti D., Cattaneo E. (2003) Nat. Genet. 35, 76–83 [DOI] [PubMed] [Google Scholar]
- 65. Li S. H., Hosseini S. H., Gutekunst C. A., Hersch S. M., Ferrante R. J., Li X. J. (1998) J. Biol. Chem. 273, 19220–19227 [DOI] [PubMed] [Google Scholar]
- 66. McGuire J. R., Rong J., Li S. H., Li X. J. (2006) J. Biol. Chem. 281, 3552–3559 [DOI] [PubMed] [Google Scholar]
- 67. Rong J., Li S., Sheng G., Wu M., Coblitz B., Li M., Fu H., Li X. J. (2007) J. Biol. Chem. 282, 4748–4756 [DOI] [PubMed] [Google Scholar]
- 68. Singaraja R. R., Hadano S., Metzler M., Givan S., Wellington C. L., Warby S., Yanai A., Gutekunst C. A., Leavitt B. R., Yi H., Fichter K., Gan L., McCutcheon K., Chopra V., Michel J., Hersch S. M., Ikeda J. E., Hayden M. R. (2002) Hum. Mol. Genet. 11, 2815–2828 [DOI] [PubMed] [Google Scholar]
- 69. Park J. J., Cawley N. X., Loh Y. P. (2008) Mol. Cell Neurosci. 39, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Keller S., Sanderson M. P., Stoeck A., Altevogt P. (2006) Immunol. Lett. 107, 102–108 [DOI] [PubMed] [Google Scholar]
- 71. Mellman I. (1996) Annu. Rev. Cell Dev. Biol. 12, 575–625 [DOI] [PubMed] [Google Scholar]
- 72. Wassmer T., Attar N., Harterink M., van Weering J. R., Traer C. J., Oakley J., Goud B., Stephens D. J., Verkade P., Korswagen H. C., Cullen P. J. (2009) Dev. Cell 17, 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mari M., Bujny M. V., Zeuschner D., Geerts W. J., Griffith J., Petersen C. M., Cullen P. J., Klumperman J., Geuze H. J. (2008) Traffic 9, 380–393 [DOI] [PubMed] [Google Scholar]
- 74. Teng H. K., Teng K. K., Lee R., Wright S., Tevar S., Almeida R. D., Kermani P., Torkin R., Chen Z. Y., Lee F. S., Kraemer R. T., Nykjaer A., Hempstead B. L. (2005) J. Neurosci. 25, 5455–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]