

Abstract
Studies of post-mortem tissue have shown that the location of fibrillar tau deposits, called neurofibrillary tangles (NFT), matches closely with regions of massive neuronal death1,2, severe cytological abnormalities3, and markers of caspase activation and apoptosis4–6, leading to the idea that tangles cause neurodegeneration in Alzheimer’s disease and tau-related frontotemporal dementia. However, using in vivo multiphoton imaging to observe tangles and activation of executioner caspases in living tau transgenic mice (Tg4510 strain), we find the opposite: caspase activation occurs first, and precedes tangle formation by hours to days. New tangles form within a day. After a new tangle forms, the neuron remains alive and caspase activity seems to be suppressed. Similarly, introduction of wild-type 4-repeat tau (Tau-4R) into wild-type animals triggered caspase activation, tau truncation and tau aggregation. Adeno-associated virus-mediated expression of a construct mimicking caspase-cleaved tau into wild-type mice led to the appearance of intracellular aggregates, tangle-related conformational- and phospho-epitopes, and the recruitment of full-length endogenous tau to the aggregates. On the basis of these data, we propose a new model in which caspase activation cleaves tau to initiate tangle formation, then truncated tau recruits normal tau to misfold and form tangles. Because tangle-bearing neurons are long-lived, we suggest that tangles are ‘off pathway’ to acute neuronal death. Soluble tau species, rather than fibrillar tau, may be the critical toxic moiety underlying neurodegeneration.
To test the hypothesis that neurofibrillary tangles are a marker of cell injury or death, we used in vivo multiphoton imaging through a craniotomy, with topical application of thioflavin S to detect tangles and a fluorescent dye that binds to activated caspases, in Tg4510 tau transgenic mice that develop both tangles and substantial neuronal loss by 7 months of age7–10. We expected relatively few cells to have active caspases at any moment, and indeed relatively few neurons (under 1% of all neurons) were observed to be caspase-positive at any imaging session. Even though tangles occupied only 15% of neurons in the superficial neocortical region imaged at this age (Supplementary Fig. 1a), caspase-positive neurons were overwhelmingly likely (87.6%) to be in a tangle-bearing neuron (Supplementary Fig. 1c), confirming a close relationship between tangles and apoptosis-related markers. On the other hand, caspase activation is rare: only a few percent of tangles are caspase-positive and well over 90% of tangle-containing neurons did not contain observable caspase activation (Supplementary Fig. 1b). We were interested in the fate of neurons that contain tangles, and especially in the fate of both the caspase-positive, tangle-positive neurons and the extremely rare caspase-positive, tangle-negative neurons, reasoning that these might represent a subclass of neurons undergoing neurodegeneration.
We identified over 500 tangle-bearing neurons in four (7–9 months old) Tg4510 mice, and carried out repeated imaging of these mice 2–5 days later, re-identifying the neurons in the initial imaging session by their location and proximity to fiduciary markers such as blood vessels. All of the tangle-bearing neurons followed were still present 2–5 days later. In the same animals, we identified 20 neurons that were caspase-positive and tangle-positive at the first imaging sessions. Caspase-positive neurons with tangles were also universally present 2–5 days later (Fig. 1) (although we were unable to continue to monitor caspase activation in these cells because the caspase dye fades quickly). Postmortem histological studies confirmed that, in the cortex, all tangles were present in neurons with an intact nucleus. Thus, the presence of a tangle, even a tangle in a neuron containing caspase activation, was not associated with acute toxicity.
Figure 1. Tangle-bearing neurons can be imaged for several days in the living brain, and caspase activation is not associated with acute neuronal death.
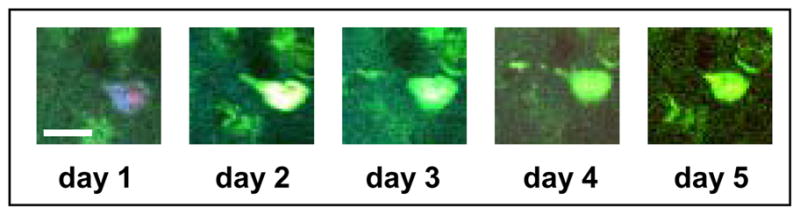
Neurons were followed for 5 days of consecutive imaging. The presence of tangles (labelled with thioflavin S, green) and activated caspases (stained with poly-caspase indicator, red) in the same neuron does not lead to neuronal death within days. Scale bar, 10μm.
We next focused on the infrequent neurons that were caspase-positive and tangle-negative (as defined by thioflavin S) during the initial imaging session. Because these neurons were quite rare, rather than imaging randomly selected fields we sought out caspase-positive, tangle-negative cells and noted their location. In three animals, in over 24 imaging sites we found 22 such neurons. Unexpectedly, repeat imaging 1 day later showed that a new tangle had formed in 20 (91%) of these 22 cells sometime in the 24 h period after the first imaging session (Fig. 2a–c). We repeated this experiment, extensively imaging four additional mice, who received topically, along with the other markers, a solution of Hoechst to estimate the number of neurons per volume. We found 19 caspase-positive, tangle-negative neurons, and the next day 16 of these neurons had a new tangle (85%). Taken together (Fig. 2d), in seven animals and 56 imaging sites, we observed 41 caspase-positive, tangle-negative neurons; 36 of these developed a tangle within 24 h (88%). We confirmed these observations with an alternative dye to image tangles, X-34 (refs 11, 12), a fluorescent derivative of Congo Red that can be administered systemically (Supplementary Fig. 2) and found similarly that caspase-positive, X-34-negative cells on day 1 develop tangles within a day. By contrast to the observation that 88% of caspase-positive tangle-negative cells develop a tangle in the next 24 h, only 1.7% of Hoechst-positive neurons that did not have caspase activation in the first imaging session were found to have a new tangle on re-imaging 1 day later. We interpret these few cells as ones in which we missed the caspase activation period, indicating that it may last less than 24 h. This finding strongly indicates that new tangles form in less than a day, and that caspase activation precedes tangle formation.
Figure 2. New tangles form very rapidly in cells with activated caspases.

Thioflavin S (green) and caspase indicator (red) were applied on the surface of the brain on day 1. Images (in z-stacks) of the same cells were collected on day 1 and day 2. a, b, Some rare tangle-free cells (arrow in a, thioflavin S) showed caspase activation (arrow in b, merged thioflavin S and caspase indicator) on the initial day of surgery. c, d, The vast majority formed a tangle within the next day (arrow in c, thioflavin S), and quantification (d) shows that 88% of caspase-positive tangle-free cells at day 1 (red) formed a tangle by day 2 (green). Scale bar, 10μm.
Because essentially all tangles are ‘born’ in a caspase-positive neuron, but (at any moment) >90% of tangle-containing neurons are not caspase-positive, we suggest that tangle-bearing cells survive the initial caspase attack and become caspase-negative. We attempted to image caspase activation repeatedly over time, but could not successfully remove the imaging coverslip to re-apply the caspase indicator reagent on subsequent imaging sessions. Unlike thioflavin S, which has a long half-life in the neuropil and remains available for imaging 24 h after application, the caspase indicator is not present at later imaging sessions.
We never observed a caspase-positive neuron in 48 imaging sites of six wild type mice or two APP (Amyloid Precursor Protein)- overexpressing mice. We therefore considered the possibility that the soluble P301L tau expression in Tg4510 mice led to caspase activation independently of the presence of a tangle. To test this hypothesis we examined caspase activation in tangle-bearing neurons, in the presence or absence of soluble tau expression. The Tg4510 mice are designed with a regulatable promoter, in which transgene expression can be suppressed with doxycycline. We treated 7–8-month-old animals with doxycycline and examined the presence of tangles and activation of caspases at baseline (n = 6 transgenic and n = 2 wild type animals), or after a brief (2 weeks, n = 2 transgenic and n = 2 wild type animals) or long (6 weeks, n = 4 transgenic and n = 2 wild type animals) doxycycline treatment, by analysing random sites throughout the imaging volume.
Transgene suppression reduced the incidence of caspase activation in tangle-bearing cells in a time dependent fashion from 5–6% at baseline to 0.3% after 6 weeks, a reduction of nearly 20 fold (Supplementary Fig. 3). As observed previously7, innumerable tangles remain after 6 weeks of transgene suppression, indicating that they are stable and long-lived aggregates. No caspase-positive, tangle-negative cells were observed after transgene suppression. These data indicate that soluble tau molecules, rather than tangles, are upstream of caspase activation.
Caspases cleave tau preferentially at aspartate 421 (D421), creating a truncated molecule that can be recognized by neoepitope-specific antibodies13,14. Caspase-cleaved tau colocalizes with tangles and correlates with the progression in Alzheimer’s disease15,16 and in animal models of tauopathy14,17. Both caspase 3 and caspase 6 have been implicated in this cleavage5,18,19. We repeated our in vivo imaging using caspase probes selective for caspase 6, caspase 3/7, and a pan-caspase indicator. Both caspase 6 and caspase 3/7 probes, as well as the pan caspase probe, co-localized with tangles in vivo. Immediately after in vivo imaging with the robust in vivo pan-caspase indicator, we sacrificed the animals and examined the brains using an antibody directed against the tau-D421 neoepitope. The bright fluorescent pan-caspase indicator always correlated with the presence of immunohistochemically defined tau truncated at D421 (Fig. 3a–c). Thus, the caspase activation we observe in vivo is associated with generation of a truncated form of tau.
Figure 3. Caspase activation in Tg4510 correlates with the presence of truncated forms of tau, which appears in multiple models of tauopathy.

a, Pan-caspase fluorescent indicator applied in vivo can be detected in post-mortem tissue (red). b, c, Tau truncated at Asp 421 (labelled with Alexa Fluor 488, b) was detected using immunochemistry in the same cells that were positive for caspase activation (merged in c). All caspase-positive neurons were also positive for tau-D421. d, In wild-type (WT) animals, injection of AAV encoding tau-4R resulted in caspase activation detected in post-mortem sections by the persistent fluorescent indicator. e, f, Presence of truncated tau (labelled with Alexa Fluor 488, e) was detected in the same neuronal cell bodies using immunochemistry (merged in f). i, Tau-D421-positive neurons were observed in the hippocampus of 14-month-old hTau animals. h–g, The immunodetection of tau-D421 in neurons of the tau-4R virus-infected (h) and hTau (i) brains seems very comparable to signal detected in the neurons infected by the AAV encoding tau-D421 (g). Scale bars, 10 μm.
Intracerebral injections of adeno-associated virus (AAV)-vector encoding for wild-type tau-4R were performed in two wild-type mice (13 months old) and analysed 6 months later. We chose to use wild-type tau rather than the P301L mutant tau overexpressed in the Tg4510 mice because wild-type tau represents the situation in sporadic Alzheimer disease, and most frontotemporal dementia. By in vivo imaging, 8 to 10 caspase-positive cells per brain were detected around the injection sites (Supplementary Fig. 4), consistent with a recent study describing caspase 3 activation after a comparable tau-4R virus infection20. On post-mortem tissue, the active caspase -positive cells correlated with the presence of caspase-cleaved tau (Fig. 3d–f). Truncated tau was occasionally associated with Alz50-reactive neurons (Supplementary Fig. 5a–c). Caspase-truncated tau was similarly detected in 14-month-old hTau mice21 that carry the non-mutated human tau gene (Fig. 3h, i). Taken together, these data confirm in three separate models (Tg4510 mice, wild-type mice overexpressing tau-4R, and hTau mice) the hypothesis that soluble tau molecules trigger caspase activation that leads to tau truncation at D421.
Tau truncated products are prone to aggregation, and have been previously shown to augment nucleation efficiency directly and accelerate fibrillization of tau in vitro22–24. We propose that this cleavage-induced acceleration of nucleation and aggregation also can occur in vivo, leading to tangle formation in Tg4510 mice. We injected a virus encoding a construct of wild-type tau truncated at position 421 into wild-type mice to test the hypothesis that this form of truncated tau would seed aggregation in vivo. We found that about 35% of neurons expressing truncated tau developed accumulations of Alz50-positive immunoreactivity, representing conformational changes in tau in the cell body (Supplementary Fig. 5d–f). PHF1 and AT8-positive phosphorylation changes were also detected, indicating that the presence of truncated tau was sufficient to induce Alzheimer-like conformational and phosphorylation changes. Endogenous tau, immunolocalized with a carboxy-terminal antibody, was found to mislocalize to the cell body and colocalize with truncated tau in Alz50-positive structures (Fig. 4). These data are reminiscent of the results observed by transgenic expression of a truncated tau fragment centred on the repeat domains, that developed aggregates containing endogenous mouse tau25. This is consistent with the idea that cleavage of tau (by caspase or non-caspase proteolysis26,27) precedes, and is sufficient to cause, misfolding of tau into a conformation that can nucleate and recruit additional tau molecules to the neuronal cell body.
Figure 4. Caspase-cleaved tau can nucleate pathological conformation of tau, and can recruit additional tau molecules.

a–d, Immunochemistry showed that truncated tau (labelled with Cy3 (Cyanine dye), a) colocalized with endogenous mouse tau (labelled with Alexa Fluor 488, b) and Alz50 (labelled with AMCA (aminomethylcoumarin acetate)-conjugated secondary antibodies, c; merged in d), indicating accumulation of endogenous mouse tau in cell bodies in response to the presence of truncated tau. Scale bars, 10μm.
Taken together, these data indicate that, rather than being the cause of neurodegenerative cascades, tau fibrillar deposits are the consequence of a cellular degenerative process marked by caspase activation (Supplementary Fig. 6). Tangles form quickly, in a time frame of hours—but persist apparently indefinitely. Tangles seem to always start in neurons that contain active caspases, but at a later time, the vast majority of tangle-positive neurons are not caspase-positive. Thus, direct visualization of caspase activation and tangle formation show three results unanticipated from cross-sectional histopathological studies: (1) caspase activation precedes and leads to tangle formation over less than 24 h, (2) neurons can have activated caspases but not die acutely, and (3) on the basis of our in vivo imaging and the results of the long term transgene suppression, tangles are long-lived species. Tangle-bearing neurons may represent survivors of an attack of enzymes usually associated with acute apoptotic death and, therefore, represent a marker, rather than a direct cause, of neurodegenerative processes. How neurons survive caspase activation, and whether tangles are a protective factor, a simple bystander, or are associated with slow or delayed toxic effects will be important questions to address as therapeutics aimed at tangle formation enter clinical trials.
METHODS SUMMARY
Animals
We used 7–9-months-old transgenic Tg4510 mice overexpressing human four-repeat tau gene containing the frontotemporal dementia-associated P301L mutation that can be suppressed with doxycycline7. By 7 months of age, Tg4510 mice have developed a large number of tangles in the cortex.
Surgery and imaging
Tg4510 mice and littermate controls were anaesthetized with isoflurane, a craniotomy was performed, and thioflavin S and caspase indicators (FLICA – fluorescent inhibitor of caspase reagent, Invitrogen) applied topically before sealing the craniotomy with a coverslip. In vivo multiphoton microscopy was used to image living neurons, existing tangles, activated caspases and formation of new tangles9,10.
Viral injection
Intracranial injection of an adeno-associated virus (AAV2) encoding human tau-4R was performed on two non-transgenic animals at 13 months of age, and left for 6 months. Similarly, an AAV2 encoding human tau truncated at aspartate 421 was performed on three non-transgenic animals at 13 months of age, and left for 10 weeks. Post-mortem studies were carried out by immunohistochemistry on fixed frozen sections.
All animal experiments were carried out under national (United States National Institutes of Health) and institutional guidelines (Massachusetts General Hospital subcommittee for research animal care).
METHODS
Animals
To express tau in rTg4510 mice7, the human tau gene (also known as MAPT) with the P301L mutation, placed downstream of a tetracycline-operon responsive element is co-expressed with an activator transgene consisting of the tet-off open reading frame28, which is downstream of Ca2+-calmodulin kinase II promoter elements, resulting in P301L expression restricted to forebrain structures29. Tg4510 littermate mice, lacking the activator transgene, served as controls.
Surgery
Cranial window surgery was done under isoflurane anaesthesia (0.5–2%, Baxter) in balanced oxygen. A 6 mm diameter-wide craniotomy was performed on the top of the skull and dura was resected from the exposed cortical surface. A solution containing fluorescent markers (described for individual experiments later) was applied for 20 min. A glass window was then sealed on top of the craniotomy, as described previously30.
The animal’s temperature was monitored through a flexible rectal temperature-sensing probe with Homeothermic Blanket Control Unit (Harvard Apparatus). The animal’s temperature is continuously displayed on a front panel LCD display. Temperature was maintained as close as possible to 37 °C with heating stereotaxic frame and blanket.
Imaging NFT -positive and NFT-negative neurons
A sterile PBS solution containing thioflavin S (0.025%, Sigma) and Hoechst 33342 (1 mM, Invitrogen) was applied on the surface of the brain. On the day following surgery, images from randomly selected fields throughout the window area were taken at 800 nm excitation to detect thioflavin S aggregates. Images of the same sites were taken at 750 nm excitation to detect Hoechst-positive nuclei. As an alternative to thioflavin S, 300μl of a PBS solution containing X-34 (0.03%, gift from W. E. Klunk) was injected in the tail vein, 2 h before imaging. X-34 aggregates were detected at 800 nm.
Imaging caspase activation in NFT-positive neurons
A sterile PBS solution containing thioflavin S (0.025%, Sigma) and a red fluorescent indicator of caspase activation (FLICA indicators, 5× concentration, pan-caspase, caspase 6 or caspase 3/7 indicator, Invitrogen) was applied on the surface of the brain. The exposed brain area was randomly imaged on the day of the surgery at 800 nm excitation where both fluorophores were detected.
Imaging formation of new tangles
The same solution described above with thioflavin S and red fluorescent caspase indicator was applied, a removable window was temporarily sealed for the first imaging session where caspase-positive and NFT-negative neurons were recorded throughout the entire imaging volume. After the imaging session, the coverslip was removed, a second dose of thioflavin S (0.025%) was applied to the brain, and a new coverslip was permanently sealed. Observations of the same neurons on the following day were possible using xy coordinates recorded for each high resolution (5×) image of cells of interest; lower resolution (1×) images of the surface of the brain were captured and used as reference to identify the same site based on blood vessels map; 2× images of the same focal plans provided an overview of the area with neighbouring cells.
Transgene suppression
Doxycycline administration in the chow (Harlan Tekland) was used to suppress the tau transgene for 2–6 weeks. Control mice were also treated with doxycycline.
Multiphoton imaging
In vivo imaging was performed using a Bio-Rad 1024ES multiphoton microscope (Bio-Rad Laboratories) (Ti:Sapphire laser: Maitai, Spectra-Physics) as described previously. For quantification of cell death markers, images were taken from randomly selected fields throughout the imaging volume. z-stacks of images were collected in 0.5–2μm steps with resolutions ranging from 0.1μm per pixel to 0.4μm per pixel. Images presented are two-dimensional projections of z-stacks.
Image post-processing
Images were taken as z-stacks. All images were processed with ImageJ, and presented as z-projections. Intensity, contrast and colour balances were used to pseudo-colour the images and enhance quality for some images.
Viral injection
Two non-transgenic animals were injected at 13 months of age with an AAV2 encoding human wild-type tau-4R, and three non-transgenic animals were injected at 13 months of age with an AAV2 encoding human tau truncated at aspartate 421, mimicking caspase cleavage at this site. Vectors were constructed in-house and packaged by the Harvard Gene Therapy Initiative.
Post-mortem studies
After completion of the last imaging experiment, or 10 weeks after tau-D421 virus infection (at 15 months old), or 6 months after tau-4R virus infection (18 months old), animals were euthanized with CO2, brains dissected into 4% PFA (paraformaldehyde) + 15% glycerol, and post-fixed for 48 h. Sections (40μm) were cut on a freezing microtome (Leica Microsystems) and standard immunofluorescence techniques were used to label tau-D421 (cleaved at Asp421; mouse monoclonal antibody TauC3, Invitrogen), C-terminal tau (beyond the 421 truncation site; rabbit polyclonal antibody Ab-3, Thermo Scientific), the conformation-specific Alz50 epitope (mouse monoclonal IgM antibody Alz50, courtesy P. Davies), the hyperphosphorylated tau-specific AT8 epitope (mouse monoclonal antibody human PHF-tau, clone AT8, Thermo Scientific) and PHF1 epitope (mouse monoclonal antibody, courtesy P. Davies).
To determine the level of overexpression associated with pathological changes in AAV injected animals, sections from one AAV-injected animal were co-stained with pan-tau antibody Tg-5 (which labels both human and mouse tau; mouse monoclonal antibody, courtesy Peter Davies) and Alz50. Image stacks were acquired on a Zeiss LSM 510 META confocal system with collection parameters identical for all images. The intensity of Tg-5 immunostaining was measured in z-projections in ImageJ (NIH) for cells with Alz-50 staining (n = 19) and cells far from the injection site that had no tau-D421 expression (n = 20). The mean intensity of Alz50 positive neurons divided by the mean intensity of non-infected neurons was calculated to estimate the fold overexpression of truncated tau in neurons with pathological changes; this analysis indicates that virally mediated transduction led to a 2.7-fold overexpression of tau in Alz50-positive neurons.
Images for figures were collected on a Zeiss LSM 510 META confocal/2-photon system mounted on an inverted Zeiss Axiovert 200 microscope (Zeiss) for fluorescent staining (Fig. 4; Supplementary Fig. 5d–f), and on an upright Olympus BX51 microscope (Olympus America) for fluorescent staining (Fig. 3; Supplementary Fig. 5a–c).
Supplementary Material
Acknowledgments
This work was supported by AG08487, AG 026249, K99 AG033670-01A1, Alzheimer’s disease Drug Discovery Foundation, Harvard Medical School Shore Award. We thank W. E. Klunk for his generous gift of compound X-34. A.d.C. is a student in the B2M programme (Université Pierre and Marie Curie, Paris, France) and the results in this manuscript will be presented in her thesis.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Author Contributions A.d.C. and L.M.F. carried out the experiments described and helped design the experiments; A.d.C. wrote the manuscript. T.L.S.-J. developed the caspase reagents and contributed to experimental design and analysis and manuscript preparation. R.P. and G.A.C. provided the Tg4510 animals and contributed to characterization of this line. B.J.B. contributed to experimental design and development of multiphoton imaging protocols to allow multicolour, multiday imaging. B.T.H. assisted with conception and design of the experiments, assisted with data analysis, manuscript preparation, and provided funding for the study.
References
- 1.Ramsden M, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gómez-Isla T, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 3.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 4.Ramalho RM, et al. Apoptosis in transgenic mice expressing the P301L mutated form of human tau. Mol Med. 2008;14:309–317. doi: 10.2119/2007-00133.Ramalho. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo H, et al. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohn TT, et al. Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer’s disease. Am J Pathol. 2001;158:189–198. doi: 10.1016/S0002-9440(10)63957-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santacruz K, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spires TL, et al. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spires-Jones TL, et al. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28:862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Calignon A, Spires-Jones TL, Pitstick R, Carlson GA, Hyman BT. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68:757–761. doi: 10.1097/NEN.0b013e3181a9fc66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem. 2000;48:1223–1232. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 12.Velasco A, et al. Detection of filamentous tau inclusions by the fluorescent Congo red derivative FSB [(trans, trans)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene] FEBS Lett. 2008;582:901–906. doi: 10.1016/j.febslet.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rissman RA, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Zhang X, Sun A. Truncated tau at D421 is associated with neurodegeneration and tangle formation in the brain of Alzheimer transgenic models. Acta Neuropathol. 2009;117:687–697. doi: 10.1007/s00401-009-0491-6. [DOI] [PubMed] [Google Scholar]
- 15.Basurto-Islas G, et al. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470–483. doi: 10.1097/NEN.0b013e31817275c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillozet-Bongaarts AL, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging. 2005;26:1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Delobel P, et al. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol. 2008;172:123–131. doi: 10.2353/ajpath.2008.070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horowitz PM, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillozet-Bongaarts AL, et al. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem. 2006;97:1005–1014. doi: 10.1111/j.1471-4159.2006.03784.x. [DOI] [PubMed] [Google Scholar]
- 20.Jaworski T, et al. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS One. 2009;4:e7280. doi: 10.1371/journal.pone.0007280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andorfer C, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 22.Gamblin TC, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin H, Kuret J. C-terminal truncation modulates both nucleation and extension phases of tau fibrillization. FEBS Lett. 2006;580:211–215. doi: 10.1016/j.febslet.2005.11.077. [DOI] [PubMed] [Google Scholar]
- 24.Ding H, Matthews TA, Johnson GV. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem. 2006;281:19107–19114. doi: 10.1074/jbc.M511697200. [DOI] [PubMed] [Google Scholar]
- 25.Mocanu MM, et al. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park SY, Tournell C, Sinjoanu RC, Ferreira A. Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience. 2007;144:119–127. doi: 10.1016/j.neuroscience.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayford M, et al. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 30.Skoch J, Hickey GA, Kajdasz ST, Hyman BT, Bacskai BJ. In: Amyloid proteins: methods and protocols. Sigurdsson EM, editor. Humana Press; 2004. pp. 349–364. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.