

Summary
A child with homozygous protein C deficiency was treated at age 20 months by orthotopic hepatic transplantation. Postoperatively there was complete reconstitution of protein C activity and resolution of the thrombotic condition.
Introduction
Protein C is a vitamin-K-dependent anticoagulant glycoprotein which, once activated by thrombin, inhibits activated factors V and VIII and stimulates fibrinolysis.1 Current evidence suggests that protein C deficiency is inherited as an autosomal codominant trait.2–4 Patients heterozygous for protein C deficiency usually have about 50% of normal protein C activity. An increased incidence of serious thrombosis has been reported amongst the heterozygous relatives of several kindreds.2,5–8 Patients with homozygous protein C deficiency typically present shortly after birth with purpura fulminans, retinal haemorrhage, and evidence of central nervous system or renal thrombosis.9–17 Protein C activity levels have been undetectable in all reported cases, and the disease is invariably fatal if left untreated.
Although fresh frozen plasma (FFP), coumarin anticoagulants, and factor IX concentrates have been used successfully in the treatment of homozygous protein C deficiency, all existing treatments have substantial drawbacks. The biological half-life of injected protein C (<8 h) necessitates unacceptably frequent administration of plasma products, and poor access to veins is often another limiting factor. The thrombotic tendency can be controlled with coumarin anticoagulants, but only in doses that severely restrict normal childhood activities and carry a risk of fatal haemorrhage.
Several lines of evidence suggest that the liver is the major site of protein C synthesis. First, other vitamin-K-dependent factors (II, VII, IX, and X) are synthesised in the liver; second, protein C levels are reduced in liver disease;18 third, synthesis of soluble protein C by a human hepatoma line has been demonstrated in vitro;19 and, fourth, cDNA inserts in a gt11 library prepared from human liver mRNA have been shown to code for protein C.20 These observations led us to attempt orthotopic hepatic transplantation as primary therapy for a patient with protein C deficiency.
Case-report and Methods
The patient, born at full term, was the child of first cousins. Details of his clinical course and of protein C levels in the pedigree have been presented elsewhere.17 Retinal and vitreous haemorrhages (ultimately resulting in complete blindness7) were noted shortly after birth. Purpuric skin lesions were first observed at 24 h of age. Protein C antigen was present at 17% of normal levels, but protein C activity was undetectable. Therapy with FFP was instituted at 48 h of age, and was continued until months of age. Two attempts at withdrawal of FFP were associated with renal failure, microangiopathic haemolytic anaemia, and fibrinolysis. At age months, warfarin (‘Coumadin’) therapy was successfully instituted during phased withdrawal of FFP. At age 15 months an intracranial haematoma developed after a fall from bed. Warfarin was discontinued and treatment with FFP was reinstituted for six weeks. Warfarin was then resumed, and the haematoma resolved without neurological sequelae.
At age 20 months, the patient was transferred to the Children’s Hospital of Pittsburgh for orthotopic hepatic transplantation. Before surgery, he had been receiving warfarin 0·5 mg/kg per day—3·0 mg in the morning and 2·5 mg at night. The last dose (2·5 mg) was administered 6 h before induction of anesthesia. At the time of induction the prothrombin and partial thromboplastin times were 25·6 s (control 11·8) and 57·2 s (control 25), respectively. 5 mg vitamin K1 was given intramuscularly, followed by an intravenous infusion of 10 ml/kg FFP. The protein C antigen and activity levels immediately after the infusion were 29% and 24%, respectively. Maximum reduction of the prothrombin time was achieved h after induction, at which time the prothrombin time was 14·8 s with a partial thromboplastin time of 31·6 s. Revascularisation of the graft was complicated by hepatic artery and portal vein thrombosis. Heparin, 3 mg/kg as a single intravenous bolus, was administered immediately before thrombectomy of both vessels; however, thrombectomy of the portal vein failed, necessitating placement of a vein graft from the confluence of the splenic and superior mesenteric arteries to the hilum of the liver. A Roux-en-Y choledochojejunostomy was performed.
Postoperatively, the patient was maintained on aspirin 40 mg/kg once daily, dipyridamole 1 mg/kg orally twice daily, tapering doses of prednisone, and cyclosporin adjusted to maintain serum levels of approximately 500 ng/ml as measured by radioimmunoassay. Protein C levels improved steadily after reaching a nadir 24 h postoperatively (figure). In the second week after transplantation an episode of presumed rejection characterised by lymphocytic infiltration of the hepatic parenchyma and portal areas and mild increases in serum transaminases was treated with OKT3 monoclonal antibody. The liver enzymes fell, but four weeks after transplantation the patient became feverish and irritable, with atypical lymphocytosis and recurrent modest rises in transaminases. A cerebrospinal fluid mononuclear cell pleocytosis reached a maximum of 247/μl six weeks after transplantation; this remained unexplained and the patient improved spontaneously. He has been treated subsequently for two episodes of pneumococcal septicaemia without meningitis, the second within three weeks of administration of pneumococcal vaccine (‘Pnu-immune’). He remains symptom-free with regard to his initial coagulation disorder. Protein C levels 7 months after hepatic transplantation are normal.
Figure. Preoperative and postoperative protein C levels in relation to factor X.
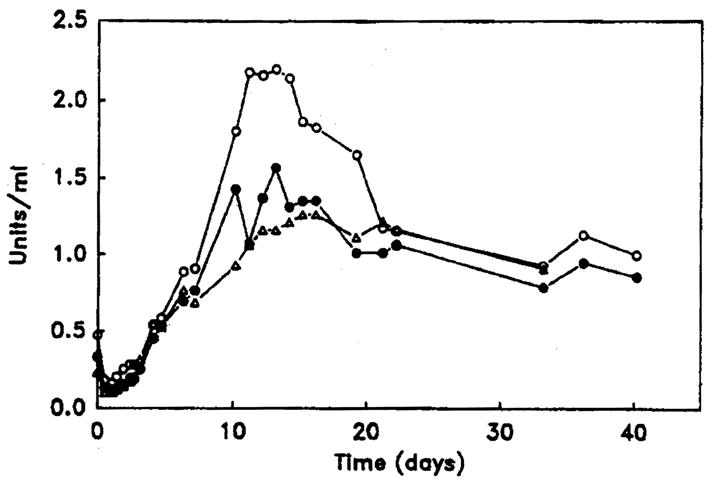
Levels of protein C activity (○) and antigen (●) are shown in relation to factor X levels (△) before (time 0) and after hepatic transplantation.
Laboratory Methods
Coagulation factors were assayed by published methods.21–23 Protein C functional assay was done by the method of Bertina4 and of Comp et al24 (reagents from American Diagnostica Inc, Greenwich, CT). A venom derivative, ‘Protac C’, converts human protein C to the active protease (aPC) which is then measured on a chromogenic substrate, ‘Spectrozyme-aPC’. Protein C immunological assay was done by the rocket technique in prepared plates from Helena Laboratories (Beaumont, TX). For both kinetic and immunological assays, the normal ranges were 65–145% of laboratory normal standard. Protein S immunological assay was likewise done with the rocket technique, antibody being purchased from American Diagnostica. The normal range was 50–150% of laboratory normal standard. The C4 bound protein was precipitated with polyethylene glycol at 3·75%. Crossed immunoelectrophoresis25 was used if the result with rocket assay was low.
Results
The accompanying figure shows post-transplantation levels of factor X and functional and antigenic protein C, and the table gives results of additional coagulation assays. The time-course and pattern of protein C recovery did not differ noticeably from that of other vitamin-K-dependent procoagulant factors. There were no thrombotic complications in the postoperative period.
Table.
LEVELS OF ADDITIONAL COAGULATION FACTORS PREOPERATIVELY AND POSTOPERATIVELY★
| Time | Factor
|
Protein S | |||||||
|---|---|---|---|---|---|---|---|---|---|
| II | V | VII | VIII | IX | X | XI | XII | ||
| Day I (preop) | 0·29 | 0·76 | 0·33 | 2·70 | 0·39 | 0·23 | 0·69 | 0·96 | |
| Day 1 (postop) | 0·09 | 0·13 | 0·07 | 0·65 | 0·02 | 0·10 | 0·03 | 0·36 | |
| Day 2 | 0·13 | 0·27 | 0·08 | 1·20 | 0·31 | 0·12 | 0·07 | 0·15 | <0·12 |
| Day 3 | 0·31 | 0·36 | 0·29 | 1·30 | 0·61 | 0·20 | 0·18 | 0·13 | 0·64 |
| Day 4 | 0·37 | 0·46 | 0·32 | 1·60 | 0·78 | 0·31 | 0·27 | 0·13 | |
| Day 5 | 0·56 | 0·74 | 0·42 | 2·25 | 1·10 | 0·54 | 0·42 | 0·32 | 1·37 |
| Day 6 | 0·54 | 1·00 | 0·58 | 2·55 | 1·10 | 0·64 | 0·55 | 0·47 | 1·66 |
| Day 8 | 0·54 | 0·80 | 0·64 | 2·75 | 1·05 | 0·68 | 0·72 | 0·69 | 1·37 |
| Day 10 | 1·60 | 1·50 | 0·60 | 3·70 | 1·50 | 0·90 | 0·96 | 1·05 | |
All assays are functional, with normal values of 50–150%, expressed as units/ml.
Discussion
The successful reconstitution of protein C activity in this patient establishes hepatic transplantation as a useful treatment for an otherwise catastrophic illness. The results also unequivocally confirm that the liver is a major site of protein C synthesis. Preoperatively, there was cause for concern not only about the ability of the transplanted liver to synthesise protein C but also about the pattern of reconstitution. If protein C had been synthesised more slowly than the procoagulant proteins, for example, then an imbalance between procoagulant and anticoagulant activities might have led to postoperative thromboses. A similar imbalance may account for the episodes of skin necrosis that occur in heterozygotes for protein C deficiency who arc receiving warfarin.26,27 The explanation, presumably, is that protein C concentrations fall quickly after initiation of warfarin, because of the short half-life of the protein in vivo, whereas most of the procoagulant proteins have much longer half-lives. Fortunately, it seems that protein C is elaborated on a time-course similar to that of other vitamin-K-dependent factors.
The occurrence of intraoperative thrombosis in this patient suggests that perioperative management of his protein C abnormality could have been improved upon, although thrombosis of the hepatic arteries and portal veins has also been seen in liver-transplant patients without protein C deficiency. Perhaps continued anticoagulation during the operative procedure will turn out to be the best course despite the added risks. Alternatively, higher doses of FFP before and during the operation might provide enough protein C to prevent thromboses.
Severe homozygous protein C deficiency joins the growing list of metabolic disorders including alpha-1-antitrypsin deficiency,28 Wilson’s disease,29 hereditary tyrosinaemia,30 type 1 and 4 glycogen storage31,32 disease, Crigler-Najjar syndrome,33 porphyria,34 familial hypercholesterolaemia,35 factor VIII36,37 deficiency, and primary hyperoxaluria38 that are correctable by hepatic transplantation. Yet, protein C deficiency is distinguished from all but the last three in that hepatic failure is not a primary feature of the disease. In fact, to our knowledge, protein C is the first coagulation defect in which liver transplantation has been done to correct the specific disorder and not end-stage liver disease related to previous blood product therapy. Removal of the need for continuous warfarin therapy or frequent infusions of plasma products has resulted in a sustained improvement in the quality of life for this child, despite the need for continuing immunosuppression.
Further observation of this and other patients will be necessary before liver transplantation can be declared the treatment of choice for protein C deficiency. Future difficulties with this approach may be encountered. For example, protein C, like many other proteins, may exhibit polymorphisms.4,39,40 Therefore, protein C synthesised by donor livers may differ from that formerly synthesised by the recipient’s liver. As a consequence, the patient’s immune system might respond by forming antibodies against the foreign protein C. In the future, other treatment methods will need to be pursued—in view of the inherent risks of liver transplantation and the fact that early morbidity such as blindness cannot conceivably be prevented by transplantation. Ultimately, a genetic approach is likely to be most successful, either through carrier detection in high-risk populations or through gene therapy.
Acknowledgments
We thank Dr Richard A. Marlar and Dr Robert R. Montgomery (Blood Center of Southeastern Wisconsin) for performing the initial protein C determinations. They and Dr William H. Zinkham and Dr George J. Dover (Johns Hopkins School of Medicine) and Dr William Hathaway (University of Colorado School of Medicine) provided helpful comments. This work was supported in part by NIH NHLBI Clinical Investigator Award HL 01341 to J. F. C. and AM 29961 to T. E. S.
References
- 1.Clouse LH, Comp PC. The regulation of hemostasis: The protein C system. N Engl J Med. 1986;314:1298–1304. doi: 10.1056/NEJM198605153142006. [DOI] [PubMed] [Google Scholar]
- 2.Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–73. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broekmans AW, Veltkamp JJ, Bertina RM. Congenital protein C deficiency and venous thromboembolism. N Engl J Med. 1983;309:340–44. doi: 10.1056/NEJM198308113090604. [DOI] [PubMed] [Google Scholar]
- 4.Bertina RM, Broekmans AW, Krommenhoek-van Es C, Van Wijngaarden A. The use of a functional and immunologic assay for plasma protein C in the study of the heterogeneity of congenital protein C deficiency. Thromb Haemostas. 1984;51:1–5. [PubMed] [Google Scholar]
- 5.Horellou MH, Conard J, Bertina RM, Samama M. Congenital protein C deficiency and thrombotic disease in nine French families. Br Med J. 1984;289:1285–87. doi: 10.1136/bmj.289.6454.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pabinger-Fasching I, Bertina RM, Lechner K, Noessner H, Korninger C. Protein C deficiency in two Austrian families. Thromb Haemostas (Stuttgart) 1983;50:810–13. [PubMed] [Google Scholar]
- 7.Broekmans AW. Hereditary protein C deficiency. Haemostasis. 1985;15:233–40. doi: 10.1159/000215154. [DOI] [PubMed] [Google Scholar]
- 8.Marlar RA. Protein C in thromboembolic disease. Semin Thromb Hemo. 1985;11:387–93. doi: 10.1055/s-2007-1004399. [DOI] [PubMed] [Google Scholar]
- 9.Branson HE, Marble R, Katz J, Griffin JH. Inherited protein C deficiency and courmarin-responsive chronic relapsing purpura fulminans in a newborn infant. Lancet. 1983;ii:1165–68. doi: 10.1016/s0140-6736(83)91216-3. [DOI] [PubMed] [Google Scholar]
- 10.Seligsohn U, Berger A, Abend M, Rubin L, Attias D, Zivelin A, Rapaport SI. Homozygous protein C deficiency manifested by massive venous thrombosis in the newborn. N Engl J Med. 1984;310:559–62. doi: 10.1056/NEJM198403013100904. [DOI] [PubMed] [Google Scholar]
- 11.Estellés A, Garcia-Plaza I, Dasi A, Azner J, Durant M, Sanz G, Perez-Requejo JL, Espana F, Jimenez C, Abeledo G. Severe inherited “homozygous” protein C deficiency in a newborn infant. Thromb Haemostas. 1984;52:53–56. [PubMed] [Google Scholar]
- 12.Sills RH, Marlar RA, Montgomery RR, Deshpande GN, Humbert JR. Severe homozygous protein C deficiency. J Pediatr. 1984;105:409–13. doi: 10.1016/s0022-3476(84)80013-x. [DOI] [PubMed] [Google Scholar]
- 13.Marciniak E, Wilson HD, Marlar RA. Neonatal purpura fulminans: a genetic disorder related to the absence of protein C in blood. Blood. 1985;65:15–20. [PubMed] [Google Scholar]
- 14.Yuen P, Cheung A, Lin HJ, et al. Purpura fulminans in a Chinese boy with congenital protein C deficiency. Pediatrics. 1986;77:670–76. [PubMed] [Google Scholar]
- 15.Wehinger H, Geiger E, Freudenberg V, Schurmann J, Alexandrakis E, Witt I. Schwerer hereditärer protein-C-mangel bei einem neugeborenen mit purpura fulminans—erfolgreiche behandlung mit phenprocoumon. Klin Pädiat. 1985;197:116–20. doi: 10.1055/s-2008-1033940. [DOI] [PubMed] [Google Scholar]
- 16.Rappaport ES, Speights VO, Helbert B, Trowbridge A, Koops B, Montgomery RR, Marlar RA. Protein C deficiency. South Med J. 1987;80:240–42. doi: 10.1097/00007611-198702000-00025. [DOI] [PubMed] [Google Scholar]
- 17.Peters C, Casella JF, Marlar RA, Montgomery RR, Zinkham WH. Homozygous protein C deficiency: observations on the nature of the molecular abnormality and the effectiveness of warfarin therapy. Pediatrics. 1988;81:272–76. [PubMed] [Google Scholar]
- 18.Mannucci PM, Vigano S. Deficiencies of protein C, an inhibitor of blood coagulation. Lancet. 1982;ii:463–66. doi: 10.1016/s0140-6736(82)90494-9. [DOI] [PubMed] [Google Scholar]
- 19.Fair DS, Marlar RA. Biosynthesis and secretion of factor VII, protein C, protein S, and the protein C inhibitor from a human hepatoma cell line. Blood. 1986;67:64–70. [PubMed] [Google Scholar]
- 20.Foster D, Davie EW. Characterization of a cDNA coding for human protein C. Proc Natl Acad USA. 1984;81:4766–70. doi: 10.1073/pnas.81.15.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis JH. Coagulation defects. JAMA. 1961;178:1014–20. doi: 10.1001/jama.1961.73040490010008. [DOI] [PubMed] [Google Scholar]
- 22.Lewis JH. Hemostasis and hemorrhage. Sci Clin. 1971;1:1–66. [Google Scholar]
- 23.Lewis JH, Spero JA, Hasiba U. Bleeding disorders: discussions in patient management. Garden City, NY: Medical Exam Publishing; 1978. Diagnostic methods: laboratory tests; pp. 22–34. [Google Scholar]
- 24.Comp PC, Nixon RR, Cooper MR, Esmon CT. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest. 1984;74:2082–88. doi: 10.1172/JCI111632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Comp PC, Doray D, Patton D, Esmon CT. An abnormal plasma distribution of protein S occurs in functional protein S deficiency. Blood. 1986;67:504–08. [PubMed] [Google Scholar]
- 26.Broekmans AW, Bertina RM, Loeliger EA, Hofmann V, Klingemann HG. Protein C and the development of skin necrosis during anticoagulant therapy. Thromb Haemostas. 1983;49:251. [PubMed] [Google Scholar]
- 27.McGehee WG, Klotz TA, Epstein DJ, Rapaport SI. Coumarin necrosis associated with hereditary protein C deficiency. Ann Intern Med. 1984;101:59–60. doi: 10.7326/0003-4819-101-1-59. [DOI] [PubMed] [Google Scholar]
- 28.Hood JM, Koep LJ, Peters RL, et al. Liver transplantation for advanced liver disease with alpha-1-antitrypsin deficiency. N Engl J Med. 1980;302:272–75. doi: 10.1056/NEJM198001313020505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groth CG, Dubois RS, Corman J, et al. Metabolic effects of hepatic replacement in Wilson’s disease. Transplant Proc. 1973;5:829–33. [PMC free article] [PubMed] [Google Scholar]
- 30.Van Thiel DH, Gartner LM, Thorp FK, et al. Resolution of clinical features of tyrosinemia following orthotopic liver transplantation for hepatoma. J Hepatol. 1986;3:42–48. doi: 10.1016/s0168-8278(86)80144-1. [DOI] [PubMed] [Google Scholar]
- 31.Malatack JJ, Finegold DN, Iwatsuki S, et al. Liver transplantation for type I glycogen storage disease. Lancet. 1983;i:1073–74. doi: 10.1016/s0140-6736(83)91910-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon RD, Shaw BW, Jr, Iwatsuki S, Esquivel CO, Starzl TE. Indications for liver transplantation in the cyclosporine era. Surg Clin N Am. 1986;66:541–56. doi: 10.1016/s0039-6109(16)43939-3. [DOI] [PubMed] [Google Scholar]
- 33.Kaufman SS, Wood RP, Shaw BW, Jr, et al. Orthotopic liver transplantation for type I Crigler-Najjar syndrome. Hepatology. 1986;6:1259–62. doi: 10.1002/hep.1840060606. [DOI] [PubMed] [Google Scholar]
- 34.Wells MM, Golitz LE, Bender BJ. Erythropoietic protoporphyria with hepatic cirrhosis. Arch Dermatol. 1980;116:429–32. [PubMed] [Google Scholar]
- 35.Starzl TE, Bilheimer DW, Bahnson HT, et al. Heart-liver transplantation in a patient with familial hypercholesterolaemia. Lancet. 1984;i:1382–83. doi: 10.1016/s0140-6736(84)91876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis JH, Bontempo FA, Spero JA, Rajni MV, Starzl TE. Liver transplantation in hemophiliac. N Engl J Med. 1985;312:1189–90. doi: 10.1056/NEJM198505023121812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bontempo FA, Lewis JH, Gorenc TJ, et al. Liver transplantation in hemophilia A. Blood. 1987;69:1721–24. [PMC free article] [PubMed] [Google Scholar]
- 38.Watts RWE, Calne RY, Kolles K, et al. Successful treatment of primary hyperoxaluria type I by combined hepatic and renal transplantation. Lancet. 1987;ii:474–75. doi: 10.1016/s0140-6736(87)91791-0. [DOI] [PubMed] [Google Scholar]
- 39.Comp PC, Nixon RR, Esmon CT. Determination of functional levels of protein C, an antithrombotic protein, using thrombin-thrombomodulin complex. Blood. 1984;63:15–21. [PubMed] [Google Scholar]
- 40.Barbui T, Finazzi G, Mussoni L, et al. Hereditary dysfunctional protein C (protein C Bergamo) and thrombosis. Lancet. 1984;ii:819. doi: 10.1016/s0140-6736(84)90750-5. [DOI] [PubMed] [Google Scholar]