

Abstract
Slowly, but surely, Alzheimer’s disease (AD) patients lose their memory, their cognitive abilities and even their personalities may change dramatically. These changes are due to the progressive dysfunction and death of nerve cells that are responsible for the storage and processing of information. While drugs can temporarily improve memory, at present there are no treatments that can stop or reverse the inexorable neurodegenerative process. But rapid progress towards understanding the cellular and molecular alterations that are responsible for the neuron’s demise is increasing optimism that effective preventative and therapeutic strategies are in the offing.
Alzheimer’s disease (AD) is a neurodegenerative disorder that currently affects nearly 2% of the population in industrialized countries; the risk of AD dramatically increases in individuals beyond the age of 70 and it is predicted that the incidence of AD will triple within the next 50 years (www.alz.org). There can be other causes of memory loss and definitive diagnosis of AD therefore requires postmortem examination of the brain, which must contain sufficient numbers of “plaques” and “tangles” to qualify as AD1, 2. Plaques are extracellular deposits of fibrils and amorphous aggregates of amyloid β-peptide (Aβ); diffuse deposits of Aβ are also present in high amounts. Neurofibrillary tangles are intracellular fibrillar aggregates of the microtubule-associated protein tau which exhibit hyperphosphorylation and oxidative modifications. Plaques and tangles are present mainly in brain regions involved in learning and memory and emotional behaviors such as the entorhinal cortex, hippocampus, basal forebrain and amygdala. Brain regions with plaques typically exhibit reduced numbers of synapses, and neurites associated with the plaques are often damaged, suggesting that Aβ damages synapses and neurites. Neurons that employ glutamate or acetylcholine as neurotransmitters appear to be particularly affected, but neurons that produce serotonin and norepinephrine are also damaged. This review focuses on the molecular and cellular abnormalities that occur in the brain in AD, how they result in synaptic dysfunction and cell death, and how they can be counteracted. Central to the disease is altered proteolytic processing of the amyloid precursor protein (APP) resulting in the production and aggregation of neurotoxic forms of Aβ. Neurons that degenerate in AD exhibit increased oxidative damage, impaired energy metabolism and perturbed cellular calcium homeostasis; Aβ appears to be an important instigator of these abnormalities. Genetic and environmental factors can determine one’s risk for and an understanding of these risk factors and how they modify the amyloid cascade is leading to the development of interventions for the prevention and treatment of AD that range from changes in diet and lifestyle, to vaccines and drugs.
Pathways Towards Alzheimer’s Disease
You cannot choose your parents
The genes passed on to you from your parents may or may not have a major role in determining whether you develop AD. The discoveries of genetic aberrancies that either cause or increase the risk of AD during the past 15 years heralded a rapid increase in knowledge of the molecular and cellular alterations responsible for neuronal degeneration and cognitive dysfunction in AD3. Once the amino acid sequence of Aβ was determined, the gene encoding APP was cloned and localized to chromosome 21. DNA samples from pedigrees in which dominantly inherited AD occurred were then screened for mutations in APP, and several causative mutations were found4. Subsequent linkage analysis identified a region of chromosome 14 as the locus of a mutation(s) responsible for inherited AD in several different pedigrees, and then the presenilin-1 (PS1) gene was identified as the affected gene5. Mutations in a gene on chromosome 1 with high homology to PS1, now called presenilin-2 (PS2), were then shown to cause a few cases of inherited AD6.
The vast majority of cases of AD are sporadic – they do not run in families. Nevertheless, molecular genetic analyses suggest that there are likely to be many genes that influence one’s susceptibility to AD. The first such susceptibility gene identified was apolipoprotein E for which there are 3 alleles that encode 3 different isoforms of apolipoprotein E (E2, E3 and E4). Individuals that produce the E4 isoform are at increased risk of AD7. The mechanism whereby E4 promotes AD is not established, but there is evidence that E4 enhances Aβ aggregation and reduces Aβ clearance. In addition, data suggest that E4 might increase the risk of AD by enhancing amyloidogenic processing of APP, promoting cerebrovascular pathology, increasing oxidative stress and impairing neuronal plasticity. A second susceptibility locus for late onset AD has been localized to chromosome 10, but the gene responsible has not yet been established8.
Risk factors
As with other age-related diseases (cardiovascular disease, diabetes, cancers, etc), there are likely to be behavioral, dietary and other environmental factors that may affect the risk of AD. However, this area of research has not yet matured to a point where clear recommendations can be made. Epidemiological findings suggest that a low education level, history of head trauma, consumption of high calorie – high fat diets and a sedentary lifestyle may each increase the risk of AD9,10. When rodents are maintained in cognitively stimulating environment or on a dietary restriction regimen neurons in their hippocampus are more resistant to death and neurogenesis (the production of new neurons from stem cells) is increased10–12. Similarly, regular physical exercise enhances hippocampal synaptic plasticity and neurogenesis, and is neuroprotective22.
Specific dietary components may affect the risk of AD. Individuals with low dietary folate intakes are at increased risk of AD, as an apparent consequence of increased levels of homocysteine; studies of mouse models of AD have demonstrated adverse effects of low dietary folate levels and high homocysteine levels on the disease process10. Other dietary factors implicated as risk factors for include lipids and metals such as copper and iron9,13. However, despite accumulating data suggesting that dietary factors may influence disease risk, no definite links between caloric intake, or any specific dietary component, and AD been established.
Cleaving APP and unleashing Aβ
A fundamental abnormality that plays a pivotal role in the dysfunction and death of neurons in AD is altered proteolytic processing of APP resulting in increased production and accumulation in the brain of neurotoxic forms of Aβ. The evidence supporting the “amyloid hypothesis” of AD is extensive and has recently been reviewed4. In every case of autosomal dominant early-onset familial AD where the genetic abnormality has been identified (mutations in the APP, PS1 and PS2 genes), the defective gene causes an increase in the production of the long 42 amino acid form of Aβ (Aβ42) in patients, in cultured cells and in transgenic mice14,15. Three different enzyme activities have been identified that determine whether and which form of Aβ is produced (Box 1). The production of Aβ requires sequential cleavages of APP by β-secretase (BACE) at the N-terminus of Aβ and by γ-secretase at the C-terminus of Aβ. Alternatively, an enzyme called α-secretase cleaves APP within the Aβ sequence thereby precluding production of Aβ. Mutations in APP that cause familial AD result in one or two amino acid changes within or immediately adjacent to Aβ that enhance its cleavage by BACE and γ-secretase, while presenilin mutations alter γ-secretase activity4. The causes of altered APP metabolism and Aβ deposition in sporadic cases of AD are not understood, but may include age-related increases in oxidative stress, impaired energy metabolism and perturbed cellular ion homeostasis.
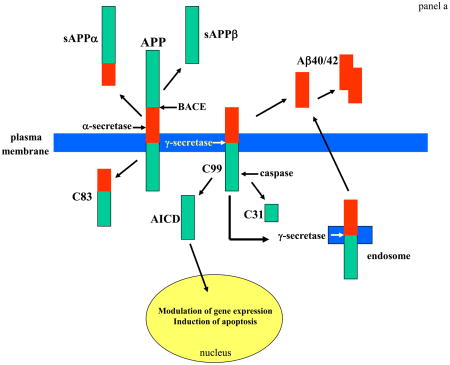
Box 1. APP cut down to size.
APP is widely expressed in cells throughout the body where the amount produced is influenced by the developmental and physiological state of the cells. APP is an integral membrane protein with a single membrane-spanning domain, a large extracellular glycosylated N-terminus and a shorter cytoplasmic C-terminus – Aβ is located at the cell surface (or on the lumenal side of ER and Golgi membranes) with part of the peptide embedded in the membrane (panel a). APP is produced in several different isoforms ranging in size from 695 to 770 amino acids. The most abundant form in brain (APP695), is produced mainly by neurons, and differs from longer forms of APP in that it lacks a kunitz-type protease inhibitor sequence in its ectodomain4,16. Enzyme activities involved in cleavage of APP at the α-, β- and γ-secretase sites are being identified (ref. 3 for review). The identity of α-secretase remains unclear although TACE (an enzyme responsible for cleavage of members of the TNF receptor family at the cell surface) and ADAM9 and ADAM10 are candidates64,65. Cleavage of APP by α-secretase releases sAPPα from the cell surface and leaves an 83 amino acid C-terminal APP fragment (C83). Production of sAPPα increases in response to electrical activity and activation of muscarinic acetylcholine receptors, suggesting that neuronal activity increases α-secretase cleavage of APP16. Amyloidogenic processing of APP involves sequential cleavages by BACE and γ-secretase at the N- and C-termini of Aβ, respectively. The 99 amino acid C-terminal fragment of APP generated by BACE cleavage can be internalized and further processed by γ-secretase to produce Aβ40/42 in endocytic compartments. Cleavage of C99 by γ-secretase liberates an APP intracellular domain (AICD) that can translocate to the nucleus where it may regulate gene expression including the induction of apoptotic genes66. Cleavage of APP/C99 by caspases produces a neurotoxic peptide (C31)67.
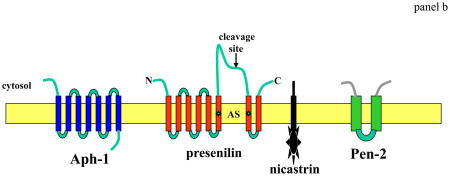
γ-secretase, which cleaves APP within a transmembrane region, involves four different proteins, presenilin, nicastrin, Aph-1 and Pen-2 (panel b) The active site (AS) of γ-secretase requires the aspartyl protease activity of PS1 conferred by aspartate residues in adjacent transmembrane domains of the C- and N-terminal cleavage fragments of PS1 (*). Nicastrin, Pen-2 and Aph-1 are each critical components of γ-secretase and each may modify enzyme activity in specific ways and in response to physiological stimuli68–70. APP is only one of several proteins that is cleaved by γ-secretase, with Notch-1 being another well-studied γ-secretase substrate4. Notch-1 is a cell surface receptor that, when activated by ligands such as Jagged and Delta, is cleaved at the membrane resulting in the release of an intracellular domain of Notch (NICD). NICD then translocates to the nucleus where it regulates the transcription of various genes. γ-secretase-mediated Notch-1 signaling plays an essential role in the regulation of cell fate during development of many organ systems including the brain as indicated by embryonic lethality and defective neurogenesis that is identical in Notch-1- and PS1-deficient mice71.
The normal functions of APP are not fully understood, but increasing evidence suggests it plays important roles in regulating neuronal survival, neurite outgrowth, synaptic plasticity and cell adhesion16. APP is transported along axons to presynaptic terminals where it accumulates at relatively high levels which can result in Aβ deposition at synpases72. One possible function of full-length APP is as a cell surface receptor that transduces signals within the cell in response to an extracellular ligand73. However, neither a ligand nor downstream signaling cascades for APP have been clearly established. Physiological roles for sAPPα are supported by data showing that sAPPα is released from presynaptic terminals in response to electrical activity, and that sAPPα regulates neuronal excitability and enhances synaptic plasticity and learning and memory, possibly by activating a cell surface receptor that modulates the activity of potassium channels and also activates the transcription factor NF-κB16.
The initial experiments showing that synthetic fragments of Aβ can kill cultured neurons74, led to a series of studies that have revealed the chemical and cell biological bases for the synaptic dysfunction and death of neurons in AD. Aβ may be most toxic when it is in the form of soluble oligomers in the earliest stages of aggregation75,76. Synapses may be particularly susceptible to the adverse effects of aggregating forms of Aβ as suggested by the ability of Aβ to impair synaptic ion and glucose transporters, and by electrophysiological studies showing that Aβ impairs synaptic plasticity16,77. Aβ may damage neurons by inducing oxidative stress and disrupting cellular calcium homeostasis16. Coincident with the increased production of Aβ in AD is a decrease in the amount of sAPPα produced, which may contribute to the demise of neurons because sAPPα is known to increase the resistance of neurons to oxidative and metabolic insults16. In APP mutant mice memory deficits become apparent relatively early in the process of Aβ deposition, consistent with the neurotoxic effects of Aβ occurring during the formation of oligomers of the peptide78. Adding to the evidence that Aβ deposition is a pivotal event in AD is the remarkable finding that immunization of APP mutant mice with human Aβ42 results in the removal of Aβ deposits from the brain44 which may result in reversal of cognitive deficits17.
In addition to the histopathological and genetic evidence supporting a central role of APP mismetabolism in AD pathogenesis, experimental findings in cell culture and animal models have identified adverse consequences of altered APP metabolism consistent with its involvement in synaptic dysfunction and nerve cell death in AD (Box 1). The increased Aβ deposition that occurs in AD most likely contributes to the demise of neurons because Aβ can be directly toxic to neurons and also greatly increases their vulnerability to oxidative and metabolic stress, and excitotoxicity16. The ability of immunization-based therapies that remove Aβ from the brains of APP mutant mice to improve synaptic function provides further evidence that Aβ plays a pivotal role in AD and also suggests that the process might be reversible (ref. 4 and 17; and see “Pathways away from AD” below).
Cellular stress: renegade radicals and power cuts
Increased oxidative stress (uncontrolled production of highly reactive oxygen radicals) and impaired cellular energy metabolism are features of many prominent age-related diseases, and AD is no exception. Cells in the brains of AD patients exhibit abnormally high amounts of oxidatively modified proteins, lipids and DNA; .such free radical-mediated molecular damage is particularly prominent in the environment of plaques and in neurofibrillary tangle-bearing neurons suggesting roles for oxidative stress in amyloid-mediated neuronal damage and neurofibrillary pathologys18. Several sources of oxidative stress in AD have been proposed, with Aβ and redox-active metals such as Fe2+ and Cu+ being two such sources13,18,19. During the process of aggregation Aβ generates hydrogen peroxide, a process which requires oxygen and that is greatly potentiated by Fe2+ and Cu+ (ref. 13, 18). Lipid peroxidation induced by Aβ impairs the function of ion-motive ATPases, glucose and glutamate transporters, and GTP-binding proteins as the result of covalent modification of the proteins by the aldehyde 4-hydroxynonenal16. By disturbing cellular ion homeostasis and energy metabolism, relatively low levels of membrane-associated oxidative stress can render neurons vulnerable to exitotoxicity and apoptosis. The dysfunction and degeneration of synapses in AD may involve Aβ-induced oxidative stress because exposure of synapses to Aβ impairs the function of membrane ion and glutamate transporters and compromises mitochondrial function by an oxidative stress-mediated mechanism (Fig. 2).
Figure 2. The neurotoxic action of Aβ involves generation of reactive oxygen species and disruption of cellular calcium homeostasis.

Interactions of Aβ oligomers and Fe2+ or Cu+ generates H2O2. When Aβ aggregation occurs at the cell membrane, membrane-associated oxidative stress (MAOS) results in lipid peroxidation and the consequent generation of 4-hydroxynonenal (4HNE) a neurotoxic aldehyde that covalently modifies proteins on cysteine, lysine and histidine residues. Some of the proteins oxidatively modified by this Aβ-induced oxidative stress include membrane transporters (left), receptors (R), GTP-binding proteins (g) and ion channels. Oxidative modifications of tau by 4HNE and other reactive oxygen species (ROS) can promote its aggregation and may thereby induce the formation of neurofibrillary tangles. Aβ can also cause mitochondrial oxidative stress and dysregulation of Ca2+ homeostasis resulting in impairment of the electron transport chain (ETC), increased production of superoxide anion radical. and decreased production of ATP. Superoxide is converted to H2O2 by the activity of superoxide dismutases (SOD) and superoxide can also interact with nitric oxide (NO) to produce peroxynitrite (ONOO*). Interaction of H2O2 with Fe2+ or Cu+ generates hydroxyl radical (OH*) a highly reactive oxyradical and potent inducer of MAOS which contributes to the dysfunction of the endoplasmic reticulum (ER).
Brain imaging studies have demonstrated deficits in glucose utilization in living AD patients, an abnormality that may occur before the onset of clinical symptoms20. The activities of cytochrome c oxidase, pyruvate dehydrogenase complex and α-ketoglutarate dehydrogenase complex, critical enzymes in energy metabolism, are decreased in brain cells of AD patients20. Altered proteolytic processing of APP may contribute to impaired energy metabolism, because brain glucose metabolism is decreased in cognition-related brain regions of APP mutant mice in association with increased amounts of Aβ in their brains21,22. Moroever, hypoxic tolerance is significantly decreased in presymptomatic APP mutant mice suggesting an early role for perturbed energy metabolism in the pathogenic action of altered APP processing23. There is increasing evidence that a systemic abnormality in glucose regulation, and insulin resistance in particular, is a risk factor for AD24, although the possible link between diabetes and AD requires further investigation. The ability of dietary restriction, which enhances insulin sensitivity, to protect neurons in experimental models relevant to AD supports a role for perturbed glucose metabolism in AD pathogenesis10, although this remains to be established in humans. Because oxidative stress and impaired energy metabolism can induce amyloidogenic processing of APP resulting in accumulation of potentially neurotoxic forms of Aβ, it has been suggested that such cellular stresses may promote amyloidogenesis25. The latter mechanism could contribute to increased amyloid production in late onset forms of AD, since oxidative stress and metabolic impairment increase with advancing age.
Uncontrollable calcium
The calcium ion (Ca2+) plays fundamental roles in learning and memory and is also involved in neuron survival and death. The inability of neurons to regulate calcium homeostasis is an aspect of AD pathogenesis that appears to be intimately involved in the dysfunction and death of neurons25. Studies of AD patients and of the pathogenic actions of APP and PS1 mutations support a role for perturbed calcium regulation in AD. For example, neurofibrillary tangle-bearing neurons exhibit high amounts of Ca2+ and evidence of hyperactivation of Ca2+-dependent proteases and Ca2+-activated kinases27. AD-causing mutations in PS1 and APP perturb cellular calcium homeostasis in cultured neurons and transgenic mice. Perturbed processing of APP may destabilize calcium homeostasis in neurons by increasing production of Aβ42 and by decreasing levels of sAPPα25. Aβ may perturb calcium regulation by inducing oxidative stress which impairs membrane calcium pumps and enhances calcium influx through voltate-dependent channels and ionotropic glutamate receptors (Fig. 2). Other findings suggest that Aβ can promote Ca2+ influx by forming channels in membranes or by activating cell surface receptors coupled to calcium influx26,28.
Presenilin mutations and amyloidogenic processing of APP can destablize Ca2+ homeostasis in astrocytes, oligodendrocytes and microglia which might contribute to white matter damage and local inflammatory processes26. AD-causing PS1 mutations have been shown to alter Ca2+ regulation by increasing the amount of Ca2+ that can be released from the ER in neurons stimulated by glutamate, membrane depolarization or agonists that activate receptors coupled to IP3 production29. Moreover, a defect in capacitative calcium entry has been documented in cells expressing mutant PS1. However, presenilin mutations are a very rare cause of AD, and there is no evidence that a Ca2+-regulating action of PS1 is involved in sporadic forms of AD. Disturbances of Ca2+ regulation in AD may not be limited to neurons. Nevertheless, recent findings from studies of neurons lacking PS1 suggest that wild-type PS1 may normally serve a Ca2+-regulating function in the ER30. Interestingly, studies of lymphocytes from patients with familial or sporadic AD, and from APP and PS1 mutant mice, have demonstrated abnormalities in calcium signaling reminiscent of those in neurons31. Thus, perturbed cellular Ca2+ homeostasis appears to be a widespread abnormality in both familial and sporadic forms of AD that may contribute to the disease process.
Feeding in the fats
Lipids are structural components of cell membranes, serve as intra- and inter-cellular signaling molecules and can modify the functions of many different proteins. Individuals that consume diets high in cholesterol and those with elevated cholesterol levels may be at increased risk of AD, whereas those who take cholesterol-lowering drugs (statins) may be at reduced risk32,33. The well-known adverse effects of high cholesterol diets on the vasculature could contribute an increased risk of AD. However, accumulating data suggest that cholesterol may feed more directly into the amyloid cascade by promoting amyloidogenic processing of APP. Statin treatment decreases Aβ levels and plaque formation in APP mutant transgenic mice34, and data showing that high levels of cholesterol and shifts in subcellular cholesterol metabolism can increase the production of Aβ in cultured cells and APP mutant transgenic mice32. Alterations in cholesterol metabolism might also promote neuronal degeneration by perturbing membrane fluidity and signal transduction. Increasing evidence suggests that diets high in saturated fats may increase the risk of AD, whereas diets rich in mono- and poly-unsaturated fatty acids and n-3 fatty acids may decrease disease risk, and several studies suggest that diets rich in n-3 fatty acids, such as those found in fish, can reduce the risk of AD35. While the emerging data linking cholesterol and fatty acids to AD are encouraging, the potential of dietary modifications of fat intake to affect disease risk remains to be established.
Microdomains of plasma membranes called lipid rafts, in which the outer leaflet of the lipid bilayer is enriched in cholesterol and sphingomyelin may be sites at which several molecular events implicated in AD pathogenesis occur including synaptic signal transduction, APP processing and the initiation of apoptosis36. Analyses of raft lipids in brain tissue samples from AD and control subjects have demonstrated increased amounts of cholesterol and ceramides in AD which are associated with increased levels of membrane-associated oxidative stress37. Exposure of neurons to Aβ or other oxidative insults results in increased accumulation of cholesterol and ceramides in the neurons and these alterations can be prevented by treating the neurons with vitamin E and inhibitors of ceramide37 suggesting that increased membrane oxidative stress may induce the production of cytotoxic ceramides in AD. Further studies in animal models and AD patients are required to clarify the roles of perturbed lipid metabolism in AD.
Synaptic and axonal alterations short circuit cognition
Learning and memory requires information exchange between neurons in circuits that pair at least two different environmental inputs in space and time. For example, when we meet someone new visual and auditory pathways are activated and this information is routed to the hippocampus, a brain region in which the convergent activity leads to enduring changes in synaptic strength so that the next time we see the person or hear their voice we remember their name. For reasons that remain elusive, neuronal circuits critical for learning and memory are particularly vulnerable to dysfunction and degeneration in AD. Emerging evidence implicates subtle changes in the function and structure of synapses and axons in these brain regions as being early and pivotal events in the pathogenesis of the neurodegenerative process in AD (Box 2). Consistent with this possibility, drugs that enhance activation of those synapses, such as acetylcholinesterase inhibitors, can improve cognition during the early stages of clinical disease38.
Box 2. The weakest link?
Elaborate neuronal circuits with intricate morphologies and multiple synaptic communication sites allow the brain to rapidly process and store large amounts of information. Axons and dendrites that extend long distances from the cell body are exposed to many different environmental conditions and signaling molecules. In order to survive and function properly, synapses and axons possess local homeostatic mechanisms which protect them against adverse conditions including those associated with aging and disease. Several factors may render synapses and axons vulnerable in AD including their high content of the disease-related proteins APP and presenilins and tau (axons), and their high metabolic and oxidative loads (panel a). Synaptic activity regulates APP processing and Aβ released from synaptic terminals may, in turn, modify synaptic plasticity79. Because APP is axonally transported and processed in presynaptic terminals, synapses are sites where oligomers of Aβ may accumulate in high amounts80. Soluble oligomers of Aβ42 inhibit long-term potentiation (LTP; a memory-related form of synaptic plasticity) in the hippocampus of rodents, and increasing evidence suggests that such oligomers are responsible for memory impairment in AD patients81. In addition to Aβ, increased production of C-terminal fragments of APP may promote synaptic dysfunction and degeneration in AD82. In transgenic mouse models of AD, synaptic dysfunction and memory impairment can occur in the absence of any overt evidence of amyloid deposition or neuronal degeneration81, 83, 84. Synapses are likely to be the sites at which neuronal death is initiated in AD because they contain most of the biochemical machinery for the initiation and execution of apoptosis, and Aβ can induce apoptotic cascades in synapses (ref. 85 and see Box 3).
Many of the neurons affected in AD are relatively large and have long axons. Analyses of AD patients suggest that damage to axons of such neurons may occur relatively early in the disease process86. Alterations in axonal transport have been suggested to play a role in AD. APP is axonally transported to synaptic terminals in brain regions affected in AD suggesting a role for axonal transport in plaque biogenesis87. Other findings suggest that abnormalities in tau may contribute to axonal transport deficits in AD88, consistent with tau’s function as an axonal protein that regulates microtubule stability. Perhaps the most compelling evidence that tau and axonal microtubule abnormalities play a fundamental role in the neurodegenerative process in AD comes from the causal role for tau mutations in familial frontotemporal lobe dementia with Parkinsonism linked to chromosome 17 and elucidation of the pathogenic actions of these tau mutations in cell culture and animal models89. Finally, alterations in the amounts and localization of synaptic proteins in vulnerable brain regions in AD patients have been documented90, and may be involved in the early dysfunction of synapses. In particular, evidence is emerging that the process of synaptic vesicle recycling may be abnormal in AD91. Several proteins that regulate clathrin-mediated vesicle recycling are reduced in AD including synaptotagmin, dynamin, AP2 and AP180. Impairment of vesicle recycling might cause the enlargement of axon terminals documented in ultrastructural studies of AD and might also result in impaired neurotrophic factor signaling which involves clathrin-mediated endocytosis of activated receptors.
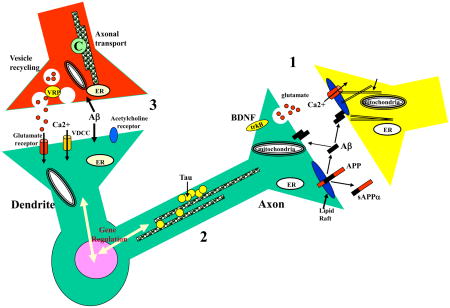
Panel. The molecules that regulate synaptic function and axonal transport and how their alterations may contribute to cognitive impairment and neuronal degeneration in AD. Perturbed processing of APP resulting in increased production of Aβ at synapses may be an early event in AD. 1. APP is axonally transported and Aβ therefore likely accumulates at synapses in high amounts in AD. Aβ can have multiple adverse effects on the functions and integrity of both pre-and post-synaptic terminals including inducing oxidative stress, impairing calcium homeostasis and perturbing the functions of mitochondria and the ER. 2. Abnormalities in axons may result from adverse effects of Aβ on tau and microtubules resulting in neurofibrillary tangle formation and cell death. 3. Presynaptic disturbances in synaptic vesicle trafficking and axonal transport may also contribute to the dysfunction and death of neurons in AD. APP, amyloid precursor protein; C, cargo; ER, endoplasmic reticulum; VDCC, voltage-dependent calcium channel; VRP, vesicle recycling proteins.
A neuron’s demise
Loss of neurons in the entorhinal cortex, hippocampus, frontal, parietal and temporal cortices of AD patients has been documented39. Neurons in layer II of the entorhinal cortex and hippocampal CA1 neurons are particularly vulnerable. The reason(s) for this selective vulnerability remains uncertain, but might be related to the expression of genes that either promote or prevent neuronal death including glutamate receptors, calcium-binding proteins and neurotrophic factors. The pattern of neuronal loss in AD overlaps with, but is not identical to, that of normal aging suggesting that AD pathogenesis is not simply an acceleration of normal brain aging. While large numbers of neurofibrillary tangle-bearing neurons occur in AD, neuronal loss exceeds numbers of neurofibrillary tangles suggesting that tangle-bearing neurons are removed and/or that some neurons die without forming tangles40.
The death of populations of neurons in the brain regions affected in AD apparently occurs over a prolonged time period of many years, which suggests that a relatively small number of neurons are dying at any one time. Such a spatio-temporal pattern of cell death is characteristic of a form of programmed cell death called apoptosis and contrasts with necrosis in which cells die en mass. Increasing evidence suggests that many neurons may die by apoptosis in AD (Box 3; and ref. 41), although it may not be the only form of cell death..
Box 3. The death of neurons.
Apoptosis is a form of programmed cell death that involves changes in the cytoplasm, ER, mitochondria and nucleus. It typically includes the production and/or activation of proteins such as Bax and Bad that increase the permeability of mitochondrial and ER membranes resulting in the release into the cytoplasm of cytochrome C from the mitochondria and calcium from the ER92. The latter events then activate enzymes called caspases that cleave various protein substrates to sculpt morphological and biochemical aspects of the cell death process. For example, cleavage of cytoskeletal proteins and ion channel proteins causes cell shrinkage and may prevent necrosis, while cleavage of DNA degrades chromosomes. Evidence that many neurons undergo apoptosis in AD includes the presence of high levels of activated apoptotic proteins such as caspase-3 and Bax in neurons that exhibit neurofibrillary tangle pathology93,94. In addition, DNA damage and upregulation of the pro-apoptotic proteins p53 and Bax occur in vulnerable neuronal populations at a relatively early stage in the disease process. FAD mutations in presenilins render neurons vulnerable to apoptosis induced by Aβ, trophic factor deprivation and other stimuli93, consistent with an apoptotic mode of neuronal death in patients with these PS mutations. APP mutations are also sufficient to trigger apoptosis in cultured cells95.
The triggers of cell death in AD may include Aβ, activation of glutamate receptors, increased oxidative stress, DNA damage and elevation of intracellular calcium levels. Once triggered apoptosis proceeds with the production and/or activation of proteins such as p53, Bax, Bad and Par-4 that induce mitochondrial membrane permeability changes. Release of cytochrome c and AIF (apoptosis-inducing factor) from mitochondria and release of calcium from the ER occurs, followed by activation of the apoptosome, a protein complex that includes cytochrome C, Apaf-1 and caspases 9 and 3. Finally, cleavage of various substrate proteins by caspases and endonucleases occurs. Neurotoxic forms of Aβ may be a trigger of apoptosis in AD because pro-apoptotic proteins are associated with Aβ deposits in the brains of AD patients and Aβ can induce apoptosis of cultured neurons93,94. Exposure of neurons to Aβ induces an apoptotic cascade that involves upregulation of p53, Bax and Par-4; mitochondrial membrane permeability transition and release of cytochrome C; activation of the apoptosome resulting in caspase-3 activation; and nuclear chromatin fragmentation. Neurons treated with inhibitors of p53, agents that stabilize mitochondrial and ER membranes or caspase inhibitors are resistant to being killed by Aβ93,94. In addition to Aβ, C-terminal proteolytic products of APP have been implicated in neuronal apoptosis67.
In addition to mitochondrial alterations, abnormalities in the ER and nucleus have been documented in studies of AD patient tissue and cell culture and animal models. FAD PS1 mutations have been shown to render neurons vulnerable to apoptosis, apparently by altering ER stress responses and ER calcium regulation29,84. ER-associated caspases may play an important role in AD because neurons from mice lacking caspase-12 are resistant to Aβ-induced apoptosis96. Apoptosis in AD may involve DNA damage response pathways and inappropriate activation of cell cycle pathways involving the cyclin-dependent kinase 5 and its neuron-specific activator p35. As evidence, proteolytic cleavage of p35 produces p25 which accumulates in the brains of AD patients and in cultured neurons exposed to Aβ1-42 (ref. 97), and Aβ-induced neuronal death is mediated by the ATM-dependent DNA damage response pathway98. Increasing evidence suggests that ubiquitin-proteasomal degradation of proteins is impaired in AD resulting in the abnormal accumulation of damaged proteins in neurons (UBB+1 mutations)99. In this regard it was recently reported that a ubiquitin-conjugating enzyme called E2-25K/Hip-2 mediates Aβ-induced inihibition of proteasome activity which is required for Aβ-induced apoptosis100. There are several mechanisms that can protect neurons against apoptosis, including activation of cell surface receptors coupled to anti-apoptotic pathways, such as those activated by neurotrophic factors and sAPPα16, and agonists that activate the cyclic AMP second messenger pathway101.
Of glia and microglia
Glial cells (astrocytes, oligodendrocyes and microglia) greatly outnumber neurons in the brain; they express many of the genes linked to AD and are subjected to many of the same environmental conditions as are neurons. Alterations in each of the three types of glial cells have been documented in studies of AD patients and animal and cell culture models of AD, and available data suggest that Aβ plays a role in inducing many of the alterations in glial cells42. Abnormalities in astrocytes that may contribute to synaptic dysfunction and neuronal death include impaired glutamate transport, perturbed calcium regulation and production of pro-inflammatory cytokines. White matter consists of axons and oligodendrocytes, the cells that wrap around and thereby insulate the axons and thereby facilitate rapid conduction of action potentials. Degeneration of cells in white matter is prominent in the brains of AD patients, and oligodendrocytes are vulnerable to being damaged and killed by Aβ43.
Microglia are macrophage-like cells that play important roles in responses of the brain to injury and infection. In AD, activated microglia congregate around amyloid plaques and degenerating neurons, and may produce toxins and inflammatory cytokines that contribute to the neurodegenerative process42. Both innate and cell-mediated immune mechanisms are involved in the pathogenesis of AD. In addition to activation of microglia, the innate immune response includes engagement of the classical complement cascade and induction of chemokines and pentraxins42. Evidence for participation of cell- and antibody-mediated responses in AD pathogenesis comes from recent studies demonstrating the ability of the immune system to generate antibodies against Aβ which may promote removal of Aβ from the brain44,45.
While many of the changes in glial cells in AD may promote neuronal degeneration, some of the changes may represent adaptive responses aimed at promoting neuronal plasticity and survival. For example, microglia may also remove Aβ, a potentially beneficial action of these immune cells46. In addition, although synapses degenerate in vulnerable neuronal circuits, the remaining synapses may increase in size to compensate and astrocytes may play a role in this process47,48. Moreover, the production of neurotrophic factors such as basic fibroblast growth may increase in astrocytes associated with Aβ deposits49 and such neurotrophic factors as well as certain cytokines50 may counteract the neurodegenerative process in AD.
Pathways Away From Alzheimer’s Disease
Diet and Lifestyle
Potentially effective means of decreasing disease risk, and of inhibiting or even reversing the AD disease process in symptomatic patients, are emerging from recent studies (Fig. 3). Although not yet fully established, available data suggest that cognitively stimulating environments, physical exercise and diets low in calories and “bad” fats (cholesterol and saturated fats) may reduce the risk of AD9,10,51, The latter epidemiological findings are supported by animal studies showing that cognitively stimulating environments, physical exercise and dietary restriction regimens increase the resistance of neurons in the brain to degeneration, enhance neurogenesis and improve learning and memory10–12. Exercise, cognitive stimulation and dietary restriction factors may exert their beneficial effects through a similar mechanism involving increased production of brain-derived neurotrophic factor (BDNF)10–12, although the importance of BDNF in AD remains to be determined. Vitamin supplementation may reduce disease risk as suggested by a recent prospective study demonstrating reduced prevalence and incidence of AD in individuals taking vitamins C and E52, and the association with high folic acid levels and decreased homocysteine levels with reduced disease risk10,53. Although the possibility that one’s risk for AD can be reduced by modifications of diet and lifestyle is of considerable interest, if, and to what extent, this is true remains to be determined.
Figure 3. Strategies and targets for the prevention and treatment of AD.

Approaches that are being tested in clinical and/or primary prevention trials include Aβ immunization, Cu+/Fe2+ chelation, cholesterol-lowering drugs (statins), anti-inflammatory agents, antioxidants and folic acid. Epidemiological and animal studies suggest the potential benefits of cognitive stimulation, regular physical exercise and dietary restriction, but these remain to be critically examined in controlled prospective studies or clinical trials.
Targeted therapeutics
Drugs are in development that target specific sites in the neurodegenerative cascade. Because the production of neurotoxic forms of Aβ from APP appears to be a pivotal event in AD pathogenesis, there is intense interest in developing drugs that block the β- or γ-secretase enzymes. Specific γ-secretase inhibitors that decrease Aβ production have been produced54, but their use in humans may be compromised by side effects resulting from blockade of γ-secretase cleavage of Notch and other protein substrates. BACE inhibitors55 may prove beneficial in reducing Aβ production without major side effects, because BACE-deficient mice have reduced Aβ production do not exhibit any overt abnormal phenotypes56. Another approach to reducing amyloid accumulation in the brain is agents that chelate copper and iron13,57; such chelators would also be expected to reduce oxidative stress in neurons. The emerging link between cholesterol levels and AD has led to trials of cholesterol-lowering statins in AD patients (www.alzforum.org/dis/tre/drc).
One of the most promising approaches for preventing and treating AD is based upon stimulating the immune system to remove Aβ from the brain. The initial reports that immunization with human Aβ42 (ref. 58) or passive immunization with Aβ antibodies59 result in the clearance of Aβ plaques from the brains of APP mutant transgenic mice were followed by reports that such immunization approaches can also ameliorate memory deficits in the mice17, 60. Results of an initial clinical trial suggest that Aβ immunization may be an effective treatment for AD, although adverse reactions occurred in some patients and refinement of the immunization methods will be required61. In addition, the rate of cognitive decline in the AD group that received the vaccine but did not respond by plaque clearance, was much greater than is typically seen in AD patients. Therefore, although the vaccine responders were cognitively better that the non-responders, their rate of cognitive was similar to that typical for patients with sporadic AD. It should also be noted that there have been many failures of trials in human patients treatments that worked well in animal models of other neurodegenerative disorders including Parkinson’s disease and stroke. Nevertheless, an effective vaccine would have a major impact on the disease, and is currently one of the most exciting areas of AD research.
Other therapeutic approaches being tested include anti-inflammatory agents such as COX-2 inhibitors and aspirin62, and steroids that decline during normal aging such as estrogen and testosterone63. Drugs that target specific sites in neurodegenerative cascades have, to date, only been employed in cell culture and animal models of AD, and their potential in the clinic remains unclear. Finally, the ability to identify individuals at risk for AD, based upon genetic (Apo E4 genotype, for example) or environmental (overweight sedentary lifestyle) factors, will allow the application of more aggressive interventions in those individuals.
Figure 1. The pathology of Alzheimer’s disease .
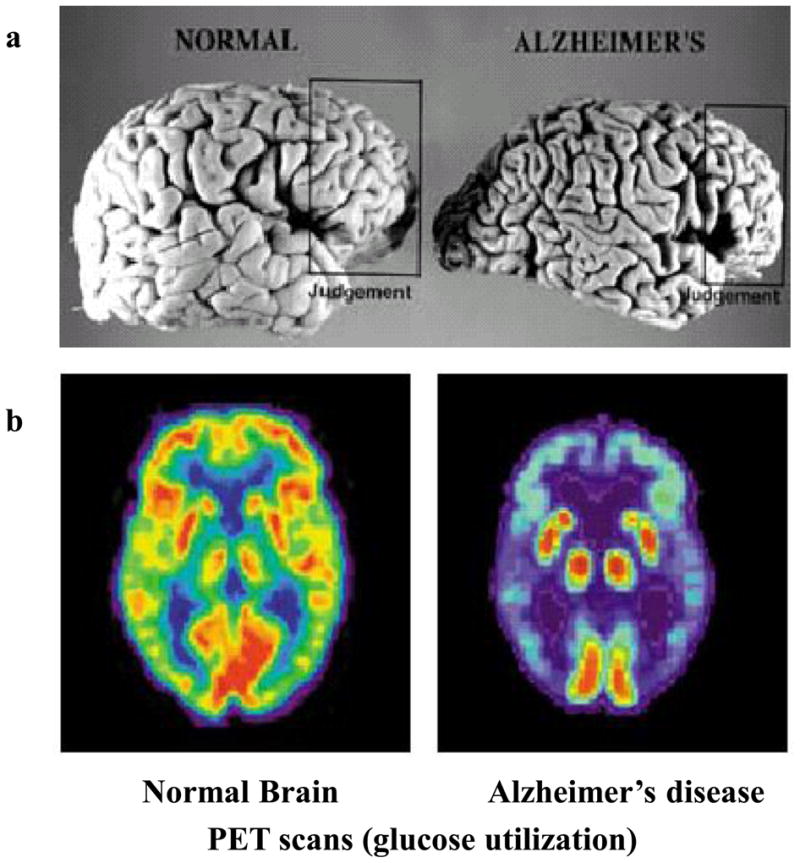
amyloid plaques, neurofibrillary tangles and degeneration of multiple neurotransmitter systems. a. A tissue section from region CA1 of the hippocampus of an Alzheimer’s patient exhibits many neurofibrillary tangle-bearing pyramidal neurons and neuritic plaques, examples of which are demarcated. In addition, diffuse amyloid deposits (yellow) are present. b. Hippocampal pyramidal neurons receive synaptic inputs from several different types of neurotransmitters including excitatory glutamatergic and inhibitory GABAergic inputs from intrinsic hippocampal neurons, and modulatory inputs from cholinergic neurons in the basal forebrain, serotonergic neurons in the raphe nucleus and noradrenergic neurons in the locus ceruleus..
References
- 1.Dickson DW. Neuropathological diagnosis of Alzheimer’s disease: a perspective from longitudinal clinicopathological studies. Neurobiol Aging. 1997;18:S21–26. doi: 10.1016/s0197-4580(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer’s disease. J Neural Transm Suppl. 1998;53:127–140. doi: 10.1007/978-3-7091-6467-9_11. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 5.Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 6.Levy-Lahad E, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 7.Roses AD. A model for susceptibility polymorphisms for complex diseases: apolipoprotein E and Alzheimer disease. Neurogenetics. 1997;1:3–11. doi: 10.1007/s100480050001. [DOI] [PubMed] [Google Scholar]
- 8.Tanzi RE, Bertram L. New frontiers in Alzheimer’s disease genetics. Neuron. 2001;32:181–184. doi: 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- 9.Mayeux R. Epidemiology of neurodegeneration. Ann Rev Neurosci. 26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- 10.Mattson MP. Gene-diet interactions in brain aging and neurodegenerative disorders. Ann Intern Med. 139:441–444. doi: 10.7326/0003-4819-139-5_part_2-200309021-00012. [DOI] [PubMed] [Google Scholar]
- 11.Young D, et al. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]
- 12.Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- 13.Bush AI, Masters CL, Tanzi RE. Copper, beta-amyloid, and Alzheimer’s disease: tapping a sensitive connection. Proc Natl Acad Sci USA. 2003;100:11193–11194. doi: 10.1073/pnas.2135061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haass C, et al. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- 15.Scheuner D, et al. Secreted amyloid beta-protein similar to that in senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 16.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 17.Morgan D, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield DA, et al. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 19.Smith MA, et al. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blass JP. Brain metabolism and brain disease: is metabolic deficiency the proximate cause of Alzheimer dementia? J Neurosci Res. 2001;66:851–856. doi: 10.1002/jnr.10087. [DOI] [PubMed] [Google Scholar]
- 21.Dodart JC, et al. Early regional cerebral glucose hypometabolism in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Neurosci Lett. 1999;277:49–52. doi: 10.1016/s0304-3940(99)00847-2. [DOI] [PubMed] [Google Scholar]
- 22.Reiman EM, et al. Tracking Alzheimer’s disease in transgenic mice using fluorodeoxyglucose autoradiography. Neuroreport. 2000;11:987–991. doi: 10.1097/00001756-200004070-00018. [DOI] [PubMed] [Google Scholar]
- 23.Buchner M, Huber R, Sturchler-Pierrat C, Staufenbiel M, Riepe MW. Impaired hypoxic tolerance and altered protein binding of NADH in presymptomatic APP23 transgenic mice. Neuroscience. 114:285–289. doi: 10.1016/s0306-4522(02)00280-4. [DOI] [PubMed] [Google Scholar]
- 24.Watson GS, Craft S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs. 2003;17:27–45. doi: 10.2165/00023210-200317010-00003. [DOI] [PubMed] [Google Scholar]
- 25.Gabuzda D, Busciglio J, Chen LB, Matsudaira P, Yankner BA. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem. 269:13623–13628. [PubMed] [Google Scholar]
- 26.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 27.Saito K, et al. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Y, et al. Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci. 2001;21:RC123. doi: 10.1523/JNEUROSCI.21-02-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaFerla FM. Calcium dyshomeostasis and intracellular signaling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Cook DG. Presenilin-1 deficiency impairs glutamate-evoked intracellular calcium responses in neurons. Neuroscience. 124:501–506. doi: 10.1016/j.neuroscience.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 31.Eckert A, et al. Alzheimer’s disease-like alterations in peripheral cells from presenilin-1 transgenic mice. Neurobiol Dis. 2001;8:331–342. doi: 10.1006/nbdi.2000.0378. [DOI] [PubMed] [Google Scholar]
- 32.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 33.Wolozin B, et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 34.Fassbender K, et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalmijn S. Fatty acid intake and the risk of dementia and cognitive decline: a review of clinical and epidemiological studies. J Nutr Health Aging. 2000;4:202–207. [PubMed] [Google Scholar]
- 36.Ehehalt R, et al. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cutler RG, et al. Involvment of perturbed ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farlow M. A clinical overview of cholinesterase inhibitors in Alzheimer’s disease. Int Psychogeriatr. 2002;14:93–126. doi: 10.1017/s1041610203008688. [DOI] [PubMed] [Google Scholar]
- 39.West MJ, et al. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 41.Gomez-Isla T, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 42.McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol. 2002;8:529–538. doi: 10.1080/13550280290100969. [DOI] [PubMed] [Google Scholar]
- 43.Xu J, et al. Amyloid-beta peptides are cytotoxic to oligodendrocytes. J Neurosci. 2001;21:RC118. doi: 10.1523/JNEUROSCI.21-01-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 45.Bard F, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 46.Jantzen PT, et al. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 48.Murai KK, et al. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci. 2003;6:153–160. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- 49.Cummings BJ, et al. Neuritic involvement within bFGF immunopositive plaques of Alzheimer’s disease. Exp Neurol. 1993;124:315–325. doi: 10.1006/exnr.1993.1202. [DOI] [PubMed] [Google Scholar]
- 50.Barger SW, et al. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pope SK, Shue VM, Beck C. Will a healthy lifestyle help prevent Alzheimer’s disease? Annu Rev Public Health. 24:111–132. doi: 10.1146/annurev.publhealth.24.100901.141015. [DOI] [PubMed] [Google Scholar]
- 52.Zandi PP, et al. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61:82–88. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 53.Wang HX, et al. Vitamin B(12) and folate in relation to the development of Alzheimer’s disease. Neurology. 2001;56:1188–1194. doi: 10.1212/wnl.56.9.1188. [DOI] [PubMed] [Google Scholar]
- 54.Dewachter L, Van Leuven F. Secretases as targets for the treatment of Alzheimer’s disease: the prospects. Lancet Neurol. 2002;1:409–416. doi: 10.1016/s1474-4422(02)00188-6. [DOI] [PubMed] [Google Scholar]
- 55.John V, et al. Human beta-secretase (BACE) and BACE inhibitors. J Med Chem. 2003;46:4625–4630. doi: 10.1021/jm030247h. [DOI] [PubMed] [Google Scholar]
- 56.Roberds SL, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- 57.Ritchie CW, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 58.Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 59.McLaurin J, et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 2002;8:1263–1269. doi: 10.1038/nm790. [DOI] [PubMed] [Google Scholar]
- 60.Kotilinek LA, et al. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hock C, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 62.Hoozemans JJ, et al. Non-steroidal anti-inflammatory drugs and cyclooxygenase in Alzheimer’s disease. Curr Drug Targets. 2003;4:461–468. doi: 10.2174/1389450033490902. [DOI] [PubMed] [Google Scholar]
- 63.Resnick SM, Henderson VW. Hormone therapy and risk of Alzheimer disease. JAMA. 2002;288:2170–2172. doi: 10.1001/jama.288.17.2170. [DOI] [PubMed] [Google Scholar]
- 64.Buxbaum JD, et al. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 65.Asai M, et al. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301:231–235. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 66.Leissring MA, et al. A physiologic signaling role for the gamma -secretase-derived intracellular fragment of APP. Proc Natl Acad Sci U S A. 2002;99:4697–4702. doi: 10.1073/pnas.072033799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu DC, et al. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 68.Yu G, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 69.Francis R, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 70.Takasugi N, et al. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- 71.Shen J, et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 72.Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimberly WT, et al. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 74.Yankner BA, et al. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 75.Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 77.Chapman PF, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 78.Koistinaho M, et al. Specific spatial learning deficits become severe with age in beta -amyloid precursor protein transgenic mice that harbor diffuse beta -amyloid deposits but do not form plaques. Proc Natl Acad Sci USA. 2001;98:14675–14680. doi: 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kamenetz F, et al. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 80.Gong Y, et al. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klein WL, Krafft GA, Finch CE. Targeting small Aβ oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 82.Oster-Granite ML, et al. Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci. 1996;16:6732–6741. doi: 10.1523/JNEUROSCI.16-21-06732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oddo S, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 84.Mucke L, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chan SL, Furukawa K, Mattson MP. Presenilins and APP in neuritic and synaptic plasticity: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 2:167–196. doi: 10.1385/NMM:2:2:167. [DOI] [PubMed] [Google Scholar]
- 86.Stahl R, et al. Assessment of axonal degeneration on Alzheimer’s disease with diffusion tensor MRI. Radiologe. 2003;43:566–575. doi: 10.1007/s00117-003-0925-4. [DOI] [PubMed] [Google Scholar]
- 87.Buxbaum JD, et al. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mandelkow EM, et al. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 89.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 90.Masliah E, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 91.Yao PJ. Synaptic frailty and clathrin-mediated synaptic vesicle trafficking in Alzheimer’s disease. Trends Neurosci. 2004;27:24–29. doi: 10.1016/j.tins.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 92.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 93.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 94.Eckert A, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem Pharmacol. 2003;66:1627–1634. doi: 10.1016/s0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]
- 95.McPhie DL, et al. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci. 2003;23:6914–6927. doi: 10.1523/JNEUROSCI.23-17-06914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 97.Lee MS, et al. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 98.Kruman II, et al. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–561. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- 99.Van Leeuwen FW, et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- 100.Song S, et al. Essential role of E2-25K/Hip-2 in mediating amyloid-beta neurotoxicity. Mol Cell. 2003;12:553–563. doi: 10.1016/j.molcel.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 101.Pedersen WA, Wan R, Zhang P, Mattson MP. Urocortin, but not urocortin II, protects cultured hippocampal neurons from oxidative and excitotoxic cell death via corticotropin-releasing hormone receptor type I. J Neurosci. 2002;22:404–412. doi: 10.1523/JNEUROSCI.22-02-00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]