

Abstract
Aberrant activation of the Src family of tyrosine kinases has been implicated in the development and progression of colorectal cancer (CRC). As a result, Src inhibitors are now being studied as possible therapeutic agents to treat metastatic disease. In this review, we discuss the effects of aberrant Src activation in CRC, Src as a target of single-agent drug therapy, and Src as a target of combination therapy with epidermal growth factor receptor inhibition and cytotoxic chemotherapy. The greatest potential for clinically relevant benefit most likely lies in combination regimens. Further evaluation with biomarkers will continue to define the molecular phenotype of patients with CRC who will benefit the most from Src-based therapy.
Keywords: Bosutinib, Dasatinib, EGFR, Saracatinib, Src inhibitors, VEGF
Introduction
Colorectal cancer (CRC) represents the third most common cancer in men and women in the United States, and it is the third most frequent cause of cancer-related deaths.1 More than 100,000 new diagnoses of CRC were made in the United States in 2008, and nearly 50,000 deaths were attributed to this disease during this same year. Unfortunately, many of these patients had advanced or metastatic disease at the time of diagnosis and required systemic chemotherapy.
Advances in chemotherapy, surgery, and radiation therapy have improved outcomes for patients with colon and rectal cancer.2 Unfortunately, mortality rates continue to be high, with 5-year survival rates of approximately 11% in the metastatic setting, and new therapies are needed.1 There have been many advances in targeted therapies for CRC, including vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGF; EGFR) antibodies. Given sufficient time, metastatic CRC becomes resistant to therapy, and further research continues to investigate mechanisms of resistance. The tyrosine kinase (TK) Src is one candidate molecule that could hold promise in the treatment of patients with CRC. This review will highlight the role of Src in carcinogenesis and the potential clinical application of Src inhibition in CRC.
The Src Family of Tyrosine Kinases
The nonreceptor protein TK Src is a 60-kD protein that is a member of a 9-gene family, including Src, Blk, Fgr, Fyn, Hcy, Lck, Lyn, Yes, and Yrk, that plays a major role in the regulation of proliferation, differentiation, migration, adhesion, angiogenesis, invasion, and immune function.3–6 The src oncogene is considered the archetypal member of this family. Research on the Rous sarcoma virus revealed a gene (v-src) that was responsible for these malignancies in 1970.7 Further research demonstrated that the gene was derived from a normal cellular gene, and study of src has provided insights into the malignant process, as it is one of the oldest and most investigated proto-oncogenes.8
The structure of Src is defined by a unique NH2-terminal region, 2 conserved Src homology domains (SH2 and SH3), and a protein TK domain (Figure 1).9,10 The NH2-terminal region contains the myristoylation site that is important for membrane localization. Regulation of Src is dependent on a C-terminal TK (Y527, corresponding to human Y530) that can lead to a less active conformation when phosphorylated by C-terminal Src kinase (Csk).5 Csk has also been shown to be downregulated early in carcinogenesis.11 Autophosphorylation in the kinase domain at Y416 (corresponding to human Y419) alters the conformation and increases the intrinsic kinase activity.12 Src plays an integral role in multiple cellular processes through its interaction with structural and signaling proteins through its SH2 and SH3 domains, including invasion, migration, proliferation, angiogenesis, and apoptosis (Figure 2).4 Src is activated by binding to growth factor receptors and integrins; cellular stress, including increased reactive oxygen species (ROS); and alterations in phosphatase activity.
Figure 1. Structure of Src in Its Active Conformation With ATP-Analogue Ligand10.
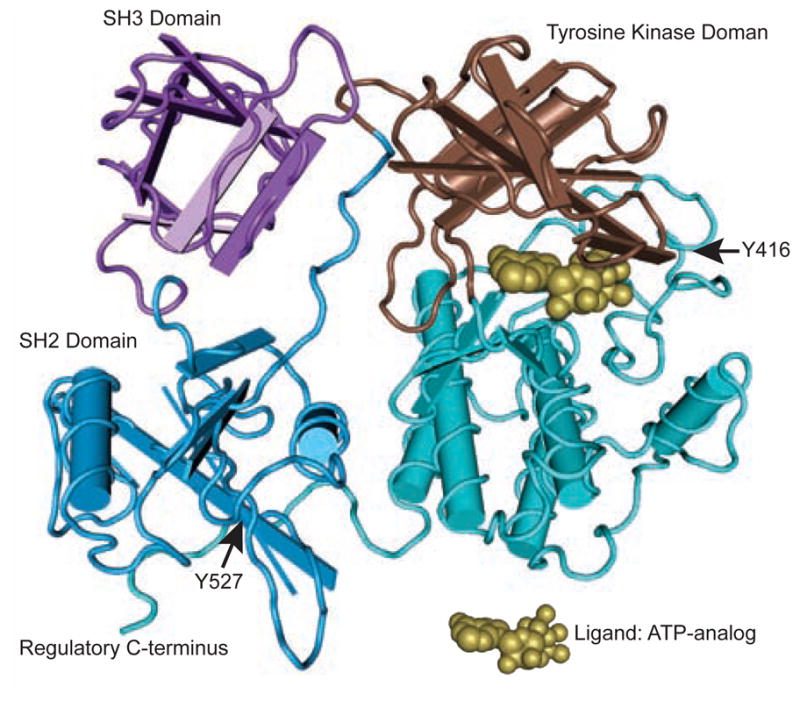
Rendered in Cn3D based on a structure from Xu et al.9 Kopetz, S: Targeting Src and Epidermal Growth Factor Receptor in Colorectal Cancer: Rationale and Progress into the Clinic. Gastrointest Cancer Res 1:S37–S41, 2007. Reprinted with the permission of the International Society of Gastrointestinal Oncology.
Figure 2. Src Plays a Role in Signaling Through a Variety of Membrane-Bound Receptors as Well as in Responding to Intracellular Oxidative Stress4.

The multiple effectors of Src include the PI3K/Akt, Ras/Raf/MAPK, STAT3/STAT5B, and p130 pathways.
Abbreviations: EGFR = epidermal growth factor receptor; MAPK = mitogen-activated protein kinase; PI3K = phosphatidylinositol 3-kinase
Aberrant Src activation has been described in multiple cancers, including colorectal, ovarian, breast, lung, liver, prostate, and pancreatic cancers.13,14 In particular, gastrointestinal cancers show an increase in Src activity as the disease progresses, and chemoresistance in these cells appears to be related to an increase in motility, invasiveness, and detachment as a result of an increased activation of Src.15,16 Activation is likely a consequence of genetic and epigenetic alterations in tumor cells, through increased transcription.6,14 Rare activating mutations of Src have been reported but not duplicated in larger series.17
Src in Colon Cancer
Src is of particular interest in colon cancer because colon carcinomas can both overexpress Src and underexpress the negative-regulatory Src TK protein, which can both lead to higher levels of Src activation.18 Previous research has shown that Src expression is increased in approximately 80% of CRC specimens compared with normal colonic epithelium,19 and colorectal metastases also demonstrate increased activity compared with primary colon tumors.20,21 Src activity has been shown to be 5- to 8-fold higher than in normal colonic mucosal cells, and the activity of Src in primary colon carcinomas was 5- to 7-fold higher than normal colonic mucosa adjacent to the tumor.22 Talamonti and colleagues studied the activation and activity of Src in colonic polyps, primary lesions, and liver metastases relative to normal colonic mucosa.15 Significant increases in TK activity were seen in colonic polyps of high malignant potential. Further increases were observed in activity and level in primary tumors. However, the greatest increases in activity and protein levels were observed in liver metastases, and metastatic lesions were also found to be significantly increased relative to their corresponding primary tumor. Furthermore, increased Src activity has been shown to be an independent indicator of poor clinical prognosis in all stages of colon cancer.14 In one study, increased Src activity was elevated more than 2-fold in tumors when compared with normal mucosa, and a significant correlation between Src activity and a decreased disease-free survival rate was found.23
Overexpression of Src in CRC has been shown to influence cell motility and enhance the ability of cells to spread on a substrate.24 In addition, an EGF-induced increase in Src kinase activity has been shown to stimulate the movement of carcinoma cells into basement membranes, suggesting a role for Src in the invasive behavior of colonic carcinoma cells induced by EGF.25 Overexpression of normal c-Src has also been shown to enhance primary tumor growth in poorly metastatic colon cancer cells.26 Clones producing 4- to 10-fold more c-Src than controls were found to enhance primary tumor growth but were insufficient to induce a metastatic phenotype.
Vascular endothelial growth factor is the predominant proangiogenic cytokine in colon cancer. VEGF stimulation results in enhanced cellular migration, but preclinical studies have found that this could be blocked by pharmacologic inhibition of VEGF receptor (VEGFR)-1 or Src kinase.27 This suggests that VEGFR-1 promotes migration of tumor cells through a Src-dependent pathway. VEGF-induced angiogenesis can also be inhibited by treatment with a retrovirus that encodes an interfering mutant of Src, and overexpression of this mutant has been shown to induce apoptotic death, indicating that VEGF-induced activation of Src is essential for endothelial cell survival and angiogenesis.28 Ectopic expression of activated Src in pancreatic cancer cell lines significantly increased the production of the proangiogenic factor interleukin (IL)-8.29 Conversely, inhibition of endogenous Src kinase activity caused significant decreases in IL-8 production. Src has also been shown to activate STAT signaling pathways, especially STAT3, which regulate increased expression of downstream target genes and also lead to angiogenesis and tumor growth.30–32 However, recent research has also revealed that sustained Src inhibition has only resulted in transient inhibition of STAT3.31,33
Src as a Target of Drug Therapy
Because Src activation has been implicated in a large percentage of common solid tumor types, Src has become a recent target for drug therapy (Figure 3).34 As the mechanisms of activation continue to be elucidated, drug-development targeting Src activation has become more successful. The ubiquitous overexpression of Src and its minimal deletion phenotype indicate that Src inhibition might not have significant toxicity and might have an inhibitory effect in the growth of human colon cancer cells.6 Drugs that have shown the ability to inhibit Src family kinases are currently being evaluated and developed for use in a variety of solid tumors, including dasatinib (BMS-354825), saracatinib (AZD0530), bosutinib (SKI-606), KX2-391, and XL228.35–40
Figure 3. Preclinical Evidence and Potential Clinical Benefit From the Various Mechanisms Attributed to Src Inhibition34.

In colorectal cancer, the greatest benefit will likely be as part of combination therapy as a chemotherapy sensitizer or through antiangiogenic effects. Previously published in Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res 2007; 13:7232-6.
Src Inhibitors as Single Agents
Src inhibitors as single-agent therapies continue to undergo investigation in CRC. As described previously, preclinical data suggest that overexpression of Src in CRC does not directly stimulate cell growth but instead appears to increase cellular motility and invasiveness. Furthermore, most preclinical reports suggest that Src inhibitors affect proliferation in only a small subset of cell lines or animal tumor models.24,41,42 Given these results, single-agent Src inhibitors would be expected to have only a modest benefit in the regression of colorectal tumors. Indeed, Src monotherapy has demonstrated minimal activity in patient-derived xenograft models. Bosutinib, a competitive inhibitor of both Src and Abl TKs, was studied in human pancreatic cancer xenografts, and of the 15 patient tumors, only 3 were found to be sensitive to the inhibitor.43 Similarly, in a study of the Src inhibitor saracatinib in human pancreatic xenografts, only 3 of 16 tumors were found to be sensitive to the drug.44
Results of single-agent Src inhibition in humans have likewise not shown clear activity. In a phase I study of bosutinib, a competitive inhibitor of both Src and Abl TKs, the best reported responses were stable disease (SD) in a variety of tumor types, including breast, colon, and non small-cell lung cancers.45 However, 1 patient with gemcitabine-refractory pancreatic cancer had disease control on single-agent bosutinib for over 1 year. A phase II study of saracatinib in patients with previously treated advanced CRC completed enrollment, but preliminary results failed to demonstrate any activity in 10 evaluated patients, leading to termination of the study.46
Recently, the results of 2 phase I trials involving Src inhibitors have been presented. XL228, a protein kinase inhibitor targeting insulin-like growth factor-1 receptor, the Aurora kinases, fibroblast growth factor receptors 1-3, ABL, and Src family kinases, was evaluated in a phase I single-agent study in 36 patients.40 XL228 was generally well tolerated, and evidence of clinical activity included 1 patient with non small-cell lung cancer with an unconfirmed partial response and 9 additional patients with SD. KX2-391, a non adenosine triphosphate (ATP) competitive substrate pocket directed Src inhibitor, has also been studied in a single-agent phase I trial of 32 patients.47 Seven patients were noted to have prolonged SD for ≥ 4 months, with significant decreases in the biomarkers of a patient with prostate cancer and pancreatic cancer. Common side effects included neutropenia, thrombocytopenia, and fatigue. Drug-specific side effects included transaminitis, vomiting, diarrhea, and pleural effusions. Given the mechanism of action and properties of Src activation, the lack of robust clinical activity is not entirely surprising. Although single-agent trials continue to undergo investigation, Src inhibitors might have greater clinical efficacy in a combined therapy setting with biologic or chemotherapeutic agents (Figure 3).34
Src and Epidermal Growth Factor Receptor
Although overexpression or activation of Src does not significantly increase tumor cell proliferation, cotransfection of Src and EGFR leads to increased proliferation, invasiveness, and tumorigenesis in mice.48,49 This appears to be accomplished in part by direct binding of Src to EGFR and phosphorylation of EGFR by Src.50,51 This then leads to a conformational change in EGFR that promotes access to the ATP binding pocket of EGFR.48 This pathway allows EGFR TK to be activated in the absence of receptor ligand binding.52,53 Although several receptors have been shown to play a role in EGFR transactivation, Src appears to be a common effector in this pathway. Transactivation leads to phosphorylation at unique sites of EGFR that can lead to signal transduction through alternate pathways, such as STAT5B, as well as augmented signaling through mitogen-activated protein kinase (MAPK).54,55 This unique activation of the EGFR pathway by Src suggests that some degree of EGFR activation can occur despite the use of an EGFR-directed antibody such as cetuximab. In CRC cell lines, Src activation correlates with EGFR overexpression, and EGFR has been shown to be activated in response to cisplatin in various types of cells through Src.56–58 Src may enable EGFR to evade degradation by promoting the destruction of Cbl, a kinase responsible for the ubiquitination and degradation of multiple receptors, including EGFR.59
Src could also play a role in EGFR antibody resistance. Src activation has been shown to be increased in a CRC cell line with cetuximab resistance.60 Resistant cells were characterized by markedly lower protein levels of EGFR, an increased association of EGFR with Cbl, and a high level of Src-416 phosphorylation both at baseline and on EGF stimulation. Interestingly, Src inhibition was shown to reverse the resistance to cetuximab-induced apoptosis without affecting the levels of EGFR in the cells.61 A separate model of EGFR inhibitor resistance implicated Src activation as well, with increased signaling through the phosphatidylinositol 3-kinase (PI3K)/Akt pathway.62 Cetuximab-resistant cells with high Src activation were found to have increased EGFR activation of HER3 and PI3K/Akt. These cells were also found to be resensitized to cetuximab when treated with the Src kinase inhibitor, dasatinib. This is consistent with other studies showing that combined treatment with Src inhibitors and EGFR inhibitors can be synergistic in vitro.63 Currently, several trials are exploring the combination of EGFR inhibitors and Src inhibitors. A phase I study combining cetuximab and dasatinib was recently presented, although any results specific to CRC are not yet available because most patients had head and neck cancers.64 Common side effects included headache and dyspnea. A Phase Ib trial of combination therapy using dasatinib, cetuximab, fluorouracil, and oxaliplatin is ongoing at M. D. Anderson Cancer Center (MDACC) in patients with metastatic CRC, and a neoadjuvant study of dasatinib and cetuximab in patients with resectable liver metastases with a pharmacodynamic endpoint is being lead by a Vanderbilt group.
Src and Cytotoxic Therapies
Src activation has been implicated in resistance to chemotherapy.65–70 As a result, recent research has tested the synergistic cytotoxicity of combining Src inhibitors with traditional chemotherapy agents. In pancreatic adenocarcinoma cell lines, Src expression and kinase activity in cell lines directly correlated with gemcitabine chemoresistance.65,67 When an Src TK inhibitor was combined with gemcitabine in a gemcitabine-resistant cell line, the combination substantially decreased tumor growth and inhibited metastasis in vivo.65 Src TK has also been found to be overexpressed in both mouse and human ovarian cancer cells.69,71 Interestingly, Src inhibition can sensitize ovarian cancer cells to chemotherapeutic agents such as docetaxel and cisplatin.70,72 In vitro and in vivo studies of Src inhibition revealed enhanced cytotoxicity of docetaxel in both chemosensitive and chemoresistant ovarian cancer cell lines. Src inhibition has also been shown to resensitize paclitaxel-resistant cells to the cytotoxic effects of paclitaxel possibly by decreasing the critical intracellular concentration at which paclitaxel induces tubulin stabilization and binding.69 Recent evidence has also shown that cisplatin-induced cell death can be inhibited by activated Src induced tyrosine phosphorylation of a gap junction protein and a decrease in gap junction communication suggesting that cell-cell communication can modulate chemosensitivity in part through Src.73 Similar findings have been demonstrated in colon cancer models.
Griffiths et al evaluated the expression of kinase-defective mutants of c-Src in human metastatic colon cancer cells.74 A human metastatic colon cancer cell line was transfected with 2 distinct kinase-defective mutants of Src. The response of these cells to oxaliplatin and SN38 (the active metabolite of irinotecan) was compared with the response seen in vector control cells in terms of cell cycle arrest and apoptosis. Both kinase-defective forms of Src sensitized cells to apoptosis induced by oxaliplatin but not by SN38. Cells with Src point mutations in SH3 or SH2 domains that inhibit Src functions were similarly sensitive to oxaliplatin, suggesting that reduction in kinase activity was responsible for the altered sensitivity. However, in this one cell line, inhibition of Src kinase activity in vector control cells did not alter the oxaliplatin response over 72 hours. The data suggested that longer-term suppression of Src kinase activity might be required to sensitize colon cancer cells to oxaliplatin-induced apoptosis.
More recent data in colon cancer models have demonstrated that Src activity was constitutively increased in oxaliplatin-resistant cell lines and oxaliplatin-induced chronic Src activation in a mouse model of colorectal liver metastases.75 Further research has shown that in colon carcinoma cells, oxaliplatin appears to acutely activate Src through an ROS-dependent mechanism and that pretreatment with antioxidants can inhibit oxaliplatin-induced Src activation.75 A combination of oxaliplatin with an Src inhibitor led to synergistic cytotoxicity only in cell lines with acute Src activation after oxaliplatin administration, suggesting that a combinatorial benefit is seen in only a subset of cancers. The same study revealed that the combination of dasatinib and oxaliplatin resulted in significantly smaller tumors compared with single-agent treatment, corresponding with reduced proliferation and angiogenesis.
Because combination therapy with Src inhibitors will most likely yield the most benefit in selected patients, further trials combining Src inhibitors with cytotoxic agents are ongoing. A combination of dasatinib and gemcitabine in refractory breast cancer and pancreatic cancer is currently enrolling patients at MDACC, and a phase I trial of dasatinib, gemcitabine, and cetuximab in predominantly pancreatic patients is currently enrolling at Duke University. In CRC, a phase II trial of dasatinib and FOLFOX (5-fluorouracil/leucovorin/oxaliplatin), with or without cetuximab (depending on KRAS status), is currently enrolling patients. It is hoped that this study will help define the benefit of Src inhibition in KRAS subsets.
Biomarkers for Src Inhibitors
As stated previously, Src has many different substrates, and Src inhibitors can affect a variety of protein kinases. Therefore, the identification and validation of predictive biomarkers will be essential to the development and application of clinically relevant Src inhibitors. In a study of saracatinib in human pancreatic cancer xenografts, 1 gene pair (LRRC19 and IGFBP2) was found to have a high predictive power for saracatinib sensitivity, suggesting a potential for this gene pair to act as a biomarker for pancreatic tumor sensitivity.44 In breast cancer cell lines, elevated levels of moesin, Yes-associated protein-1, and caveolin-1 (CAV-1) were associated with dasatinib-induced growth inhibition.76 In pancreatic cancer xenografts, sensitivity to bosutinib was also associated with elevated expression of CAV-1, a known substrate of Src.43 CAV-1 overexpression and secretion has been linked to Src activation.77 Because bosutinib was shown to be effective in reducing tumor burden in cells that expressed elevated levels of CAV-1, it is possible that circulating CAV-1 levels could be used as a predictive biomarker. Although multiple biomarkers have shown promise for predicting response, further studies will be needed to validate these potential biomarkers in order to appropriately select the subset of patients that might derive the most benefit from Src inhibition.
Conclusion
The role of the proto-oncogene Src and its family of kinases has been elucidated over many years, and it has been shown to play an integral role in CRC, including tumor cell proliferation, angiogenesis, and chemoresistance. Preclinical and clinical data continue to support the potential utility of Src inhibitors in the treatment of patients with CRC. Although current trials suggest that the utility of Src inhibitors in the single-agent setting might provide only limited clinical benefit, the greatest potential for clinically relevant effects most likely lies in combination regimens with targeted therapy, such as cetuximab, or other cytotoxic chemotherapeutic agents, such as oxaliplatin. Further evaluation, including evaluating biomarkers, will help determine the molecular phenotype of patients with CRC who would benefit the most from Src inhibitor based combination therapy. As more Src inhibitors enter the clinical arena, implementation of scientifically rational combination therapy might produce significant advances in the treatment and management of patients with CRC.
Acknowledgments
Funding source: NIH CA136980 (Dr. Kopetz).
Footnotes
Disclosures
Dr. Kopetz has received research funding from Bristol-Myers Squibb. Dr. Lieu has no relevant relationships to disclose.
References
- 1.Jemal A, Siegel R, Ward E, et al. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27:3677–83. doi: 10.1200/JCO.2008.20.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor SJ, Shalloway D. Src and the control of cell division. Bioessays. 1996;18:9–11. doi: 10.1002/bies.950180105. [DOI] [PubMed] [Google Scholar]
- 4.Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989–98. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- 5.Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7:777–85. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- 6.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 7.Martin GS. Rous sarcoma virus: a function required for the maintenance of the transformed state. Nature. 1970;227:1021–3. doi: 10.1038/2271021a0. [DOI] [PubMed] [Google Scholar]
- 8.Stehelin D, Varmus HE, Bishop JM, et al. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–3. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 9.Xu H, Yu Y, Marciniak D, et al. Epidermal growth factor receptor (EGFR)-related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol Cancer Ther. 2005;4:435–42. doi: 10.1158/1535-7163.MCT-04-0280. [DOI] [PubMed] [Google Scholar]
- 10.Kopetz S. Targeting Src and epidermal growth factor receptor in colorectal cancer: rationale and progress into the clinic. Gastrointest Cancer Res. 2007;1:S37–41. [PMC free article] [PubMed] [Google Scholar]
- 11.Kunte DP, Wali RK, Koetsier JL, et al. Down-regulation of the tumor suppressor gene C-terminal Src kinase: an early event during premalignant colonic epithelial hyperproliferation. FEBS Lett. 2005;579:3497–502. doi: 10.1016/j.febslet.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 12.Roskoski R., Jr Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 14.Dehm SM, Bonham K. SRC gene expression in human cancer: the role of transcriptional activation. Biochem Cell Biol. 2004;82:263–74. doi: 10.1139/o03-077. [DOI] [PubMed] [Google Scholar]
- 15.Talamonti MS, Roh MS, Curley SA, et al. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91:53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah AN, Gallick GE. Src, chemoresistance and epithelial to mesenchymal transition: are they related? Anticancer Drugs. 2007;18:371–5. doi: 10.1097/CAD.0b013e32801265d7. [DOI] [PubMed] [Google Scholar]
- 17.Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–90. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- 18.Cam WR, Masaki T, Shiratori Y, et al. Reduced C-terminal Src kinase activity is correlated inversely with pp60 activity in colorectal carcinoma. Cancer. 2001;92:61–70. doi: 10.1002/1097-0142(20010701)92:1<61::aid-cncr1292>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 19.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 20.Bolen JB, Veillette A, Schwartz AM, et al. Activation of pp60c-src protein kinase activity in human colon carcinoma. Proc Natl Acad Sci USA. 1987;84:2251–5. doi: 10.1073/pnas.84.8.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Termuhlen PM, Curley SA, Talamonti MS, et al. Site-specific differences in pp60c-src activity in human colorectal metastases. J Surg Res. 1993;54:293–8. doi: 10.1006/jsre.1993.1046. [DOI] [PubMed] [Google Scholar]
- 22.Cartwright CA, Kamps MP, Meisler AI, et al. pp60c-src activation in human colon carcinoma. J Clin Invest. 1989;83:2025–33. doi: 10.1172/JCI114113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aligayer H, Boyd DD, Heiss MM, et al. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer. 2002;94:344–51. doi: 10.1002/cncr.10221. [DOI] [PubMed] [Google Scholar]
- 24.Jones RJ, Avizienyte E, Wyke AW, et al. Elevated c-Src is linked to altered cell-matrix adhesion rather than proliferation in KM12C human colorectal cancer cells. Br J Cancer. 2002;87:1128–35. doi: 10.1038/sj.bjc.6600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunton VG, Ozanne BW, Paraskeva C, et al. A role for epidermal growth factor receptor, c-Src and focal adhesion kinase in an in vitro model for the progression of colon cancer. Oncogene. 1997;14:283–93. doi: 10.1038/sj.onc.1200827. [DOI] [PubMed] [Google Scholar]
- 26.Irby R, Mao WG, Coppola D, et al. Overexpression of normal c-Src in poorly metastatic human colon cancer cells enhances primary tumor growth but not metastatic potential. Cell Growth Differ. 1997;8:1287–95. [PubMed] [Google Scholar]
- 27.Lesslie DP, Summy JM, Parikh NU, et al. Vascular endothelial growth factor receptor-1 mediates migration of human colorectal carcinoma cells by activation of Src family kinases. Br J Cancer. 2006;94:1710–7. doi: 10.1038/sj.bjc.6603143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–6. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 29.Trevino JG, Summy JM, Gray MJ, et al. Expression and activity of Src regulate interleukin-8 expression in pancreatic adenocarcinoma cells: implications for angiogenesis. Cancer Res. 2005;65:7214–22. doi: 10.1158/0008-5472.CAN-04-3858. [DOI] [PubMed] [Google Scholar]
- 30.Kijima T, Niwa H, Steinman RA, et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13:355–62. [PubMed] [Google Scholar]
- 31.Yu CL, Meyer DJ, Campbell GS, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–3. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 32.Bromberg JF, Horvath CM, Besser D, et al. Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18:2553–8. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sen B, Saigal B, Parikh N, et al. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–65. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13:7232–6. doi: 10.1158/1078-0432.CCR-07-1902. [DOI] [PubMed] [Google Scholar]
- 35.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 36.Green T, Fennell M, Whittaker R, et al. Preclinical activity of AZD0530, a novel, oral, potent, and selective inhibitor of the Src family kinases. Paper presented at: the 2004 EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; September 28-October 1, 2004; Geneva, Switzerland. Poster 361. [Google Scholar]
- 37.Cooper J, Mita M, Curtright J, et al. A phase I study examining weekly dosing and pharmacokinetics (PK) of a novel spectrum selective kinase inhibitor, XL999, in patients with advanced solid malignancies (ASM) J Clin Oncol. 2006;24(18 suppl):603s. (abstract 13024) [Google Scholar]
- 38.Coluccia AM, Benati D, Dekhil H, et al. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 2006;66:2279–86. doi: 10.1158/0008-5472.CAN-05-2057. [DOI] [PubMed] [Google Scholar]
- 39.Bu Y, Gao L, Smolinski M. KX01 (KX2-391), a Src-family kinase inhibitor targeting the peptide-binding domain, suppresses oncogenic proliferation in vitro and in vivo. Paper presented at: the 99th AACR Annual Meeting; April 12–16, 2008; San Diego, CA. Abstract 4983. [Google Scholar]
- 40.Smith D, Britten C, Clary L, et al. A phase I study of XL228, a potent IGF1R/AURORA/SRC inhibitor, in patients with solid tumors or hematologic malignancies. J Clin Oncol. 2009;27(15 suppl):149s. (abstract 3512) [Google Scholar]
- 41.Johnson FM, Saigal B, Talpaz M, et al. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11:6924–32. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 42.Boyer B, Bourgeois Y, Poupon MF. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene. 2002;21:2347–56. doi: 10.1038/sj.onc.1205298. [DOI] [PubMed] [Google Scholar]
- 43.Messersmith WA, Rajeshkumar NV, Tan AC, et al. Efficacy and pharmacodynamic effects of bosutinib (SKI-606), a Src/Abl inhibitor, in freshly generated human pancreas cancer xenografts. Mol Cancer Ther. 2009;8:1484–93. doi: 10.1158/1535-7163.MCT-09-0075. [DOI] [PubMed] [Google Scholar]
- 44.Rajeshkumar NV, Tan AC, De Oliveira E, et al. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009;15:4138–46. doi: 10.1158/1078-0432.CCR-08-3021. [DOI] [PubMed] [Google Scholar]
- 45.Messersmith WA, Krishnamurthi S, Hewes BA, et al. Bosutinib (SKI-606), a dual Src/Abl tyrosine kinase inhibitor: preliminary results from a phase 1 study in patients with advanced malignant solid tumors. J Clin Oncol. 2007;25(18 suppl):150s. (abstract 3552) [Google Scholar]
- 46.Eng C, Kopetz S, Morris J, et al. Phase II study of the novel oral Src-kinase inhibitor, AZD0530, in previously treated advanced colorectal cancer patients. Paper presented at: the 99th AACR Annual Meeting; April 12–16, 2008; San Diego, CA. Abstract LB-76. [Google Scholar]
- 47.Adjei A, Cohen R, Kurzrock R, et al. Results of a phase I trial of KX2-391, a novel non-ATP competitive substrate-pocket directed SRC inhibitor, in patients with advanced malignancies. J Clin Oncol. 2009;27(15 suppl):148s. doi: 10.1007/s10637-013-9929-8. (abstract 3511) [DOI] [PubMed] [Google Scholar]
- 48.Tice DA, Biscardi JS, Nickles AL, et al. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci USA. 1999;96:1415–20. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maa MC, Leu TH, McCarley DJ, et al. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA. 1995;92:6981–5. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stover DR, Becker M, Liebetanz J, et al. Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85. J Biol Chem. 1995;270:15591–7. doi: 10.1074/jbc.270.26.15591. [DOI] [PubMed] [Google Scholar]
- 51.Biscardi JS, Maa M-C, Tice DA, et al. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–43. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 52.Carpenter G. Employment of the epidermal growth factor receptor in growth factor-independent signaling pathways. J Cell Biol. 1999;146:697–702. doi: 10.1083/jcb.146.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwin F, Wiepz GJ, Singh R, et al. A historical perspective of the EGF receptor and related systems. Methods Mol Biol. 2006;327:1–24. doi: 10.1385/1-59745-012-x:1. [DOI] [PubMed] [Google Scholar]
- 54.Kloth MT, Laughlin KK, Biscardi JS, et al. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J Biol Chem. 2003;278:1671–9. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- 55.Biscardi JS, Belsches AP, Parsons SJ. Characterization of human epidermal growth factor receptor and c-Src interactions in human breast tumor cells. Mol Carcinog. 1998;21:261–72. doi: 10.1002/(sici)1098-2744(199804)21:4<261::aid-mc5>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 56.Osherov N, Levitzki A. Epidermal-growth-factor-dependent activation of the src-family kinases. Eur J Biochem. 1994;225:1047–53. doi: 10.1111/j.1432-1033.1994.1047b.x. [DOI] [PubMed] [Google Scholar]
- 57.Benhar M, Engelberg D, Levitzki A. Cisplatin-induced activation of the EGF receptor. Oncogene. 2002;21:8723–31. doi: 10.1038/sj.onc.1205980. [DOI] [PubMed] [Google Scholar]
- 58.Boerner JL, Biscardi JS, Silva CM, et al. The role of EGF and transactivating agonists in EGFR/c-Src synergy. Paper presented at: the 96th Annual AACR Meeting; April 16–20, 2005; Anaheim, CA. Abstract 5238. [Google Scholar]
- 59.Bao J, Gur G, Yarden Y. Src promotes destruction of c-Cbl: Implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci USA. 2003;100:2438–43. doi: 10.1073/pnas.0437945100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu Y, Li X, Liang K, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67:8240–7. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 61.Lu Y, Li X, Liang K, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance to the anti-EGFR monoclonal antibody cetuximab. Paper presented at: the 97th Annual AACR Meeting; April 14–18, 2007; Los Angeles, CA. Abstract 4082. [Google Scholar]
- 62.Wheeler DL, Lida M, Kruser TJ, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009;8:696–703. doi: 10.4161/cbt.8.8.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kopetz S, Wu J, Davies M, et al. Synergistic activity of Src and EGFR inhibitors in colon cancer. Paper presented at: the 97th Annual AACR Meeting; April 14–18, 2007; Los Angeles, CA. Abstract 4079. [Google Scholar]
- 64.Feinstein T, Agrawal S, Stoller R, et al. Phase I and pharmacokinetic (PK) study of dasatinib (D) and cetuximab (C) in patients (pts) with advanced solid malignancies. J Clin Oncol. 2009;27(15 suppl):156s. (abstract 3540) [Google Scholar]
- 65.Duxbury MS, Ito H, Zinner MJ, et al. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res. 2004;10:2307–18. doi: 10.1158/1078-0432.ccr-1183-3. [DOI] [PubMed] [Google Scholar]
- 66.Lesslie DP, III, Parikh NU, Shah A, et al. Combined activity of dasatinib (BMS-354825) and oxaliplatin in an orthotopic model of metastatic colorectal carcinoma. Paper presented at: the 97th Annual AACR Meeting; April 1–5, 2006; Washington, DC. Abstract 4745. [Google Scholar]
- 67.Duxbury MS, Ito H, Zinner MJ, et al. siRNA directed against c-Src enhances pancreatic adenocarcinoma cell gemcitabine chemosensitivity. J Am Coll Surg. 2004;198:953–9. doi: 10.1016/j.jamcollsurg.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 68.Avizienyte E, Brunton VG, Fincham VJ, et al. The SRC-induced mesenchymal state in late-stage colon cancer cells. Cells Tissues Organs. 2005;179:73–80. doi: 10.1159/000084511. [DOI] [PubMed] [Google Scholar]
- 69.George JA, Chen T, Taylor CC. SRC tyrosine kinase and multidrug resistance protein-1 inhibitions act independently but cooperatively to restore paclitaxel sensitivity to paclitaxel-resistant ovarian cancer cells. Cancer Res. 2005;65:10381–8. doi: 10.1158/0008-5472.CAN-05-1822. [DOI] [PubMed] [Google Scholar]
- 70.Pengetnze Y, Steed M, Roby KF, et al. Src tyrosine kinase promotes survival and resistance to chemotherapeutics in a mouse ovarian cancer cell line. Biochem Biophys Res Commun. 2003;309:377–83. doi: 10.1016/j.bbrc.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 71.Wiener JR, Windham TC, Estrella VC, et al. Activated Src protein tyrosine kinase is overexpressed in late-stage human ovarian cancers. Gynecol Oncol. 2003;88:73–9. doi: 10.1006/gyno.2002.6851. [DOI] [PubMed] [Google Scholar]
- 72.Han LY, Landen CN, Trevino JG, et al. Antiangiogenic and antitumor effects of Src inhibition in ovarian carcinoma. Cancer Res. 2006;66:8633–9. doi: 10.1158/0008-5472.CAN-06-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peterson-Roth E, Brdlik CM, Glazer PM. Src-induced cisplatin resistance mediated by cell-to-cell communication. Cancer Res. 2009;69:3619–24. doi: 10.1158/0008-5472.CAN-08-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Griffiths GJ, Koh MY, Brunton VG, et al. Expression of kinase-defective mutants of c-Src in human metastatic colon cancer cells decreases Bcl-xL and increases oxaliplatin- and Fas-induced apoptosis. J Biol Chem. 2004;279:46113–21. doi: 10.1074/jbc.M408550200. [DOI] [PubMed] [Google Scholar]
- 75.Kopetz S, Lesslie DP, Dallas NA, et al. Synergistic activity of the Src family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer Res. 2009;69:3842–9. doi: 10.1158/0008-5472.CAN-08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Finn RS, Dering J, Ginther C, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007;105:319–26. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- 77.Goetz J, Lajoie P, Wiseman S, et al. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–35. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]