

Abstract
Context
Despite recent progress in describing the common neural circuitry of emotion and stress processing, the bases of individual variation are less well understood. Genetic variants that underlie psychiatric disease have proved particularly difficult to elucidate. Functional genetic variation of neuropeptide Y (NPY) was recently identified as a source of individual differences in emotion. Low NPY levels have been reported in major depressive disorder (MDD).
Objective
To determine whether low-expression NPY genotypes are associated with negative emotional processing at three levels of analysis.
Design
Cross-sectional, case-control.
Setting
Academic medical center.
Participants
Forty-four individuals with MDD and 137 healthy controls; 152 (84%) were classified by NPY genotype as low, intermediate, or high, according to previously established haplotype-based expression data.
Main Outcome Measures
Healthy subjects participated in functional magnetic resonance imaging while viewing negative (versus neutral) words (n=58), and rated positive and negative affect during a pain-stress challenge (n=78). Genotype distribution was compared between 113 control and 39 MDD subjects.
Results
Among healthy individuals, negatively valenced words activated medial prefrontal cortex. Activation within this region was inversely related to genotype-predicted NPY expression (p=0.029). Whole-brain regression of responses to negative words showed that rostral anterior cingulate cortex activated in the low-expression group and deactivated in the high-expression group (p<0.05). During the stress challenge, individuals with low-expression NPY genotypes reported more negative affective experience before and after pain (p=0.002). Low-expression NPY genotypes were over-represented in MDD after controlling for age and sex (p=0.004). Population stratification did not account for the results.
Conclusions
These findings support a model in which NPY genetic variation predisposes certain individuals to low NPY expression, thereby increasing neural responsivity to negative stimuli within key affective circuit elements, including medial prefrontal and anterior cingulate cortices. These genetically influenced neural response patterns appear to mediate risk for some forms of MDD.
Introduction
The neural substrates of emotion have been intensely studied in recent years. These studies have identified key brain structures and circuits that underlie affective processing in humans and other mammals, including the prefrontal cortex (PFC), the anterior cingulate cortex (ACC), and the amygdala.1-3 While much progress has been made in describing the common circuit elements that underlie emotion across individuals, the bases of individual differences in affective processing have received less attention. Among humans, such individual differences are of great importance because they are central to conceptualizations of personality and temperament, and they contribute to risk for psychiatric illness. The wide inter-individual variation in human affective functioning is partly heritable, with roughly half of the observed variance in emotional traits attributable to genetic factors.4 Thus, identification of genetic variations that influence affective processing may provide a window into the neurobiology that underlies individual differences in emotion and risk for affective disorders.
A promising candidate gene that has received increasing attention is the gene for neuropeptide Y (NPY). The NPY gene encodes a prepro-peptide which is cleaved to NPY, a 36-amino-acid neurotransmitter that is evolutionarily conserved, widely distributed in the brain, and expressed at high concentrations.5-8 NPY is co-released with other neurotransmitters by a variety of neuronal cell types, including GABAergic interneurons in the cerebral cortex.9 Experiments in animal models have indicated that stress increases expression and release of NPY in the amygdala, and that NPY reduces anxiety-like behavior.10 NPY also modulates central pain processes in animal models.11, 12 While pain stimuli have been well characterized as universal stressors by physical and emotional responses,13 NPY's role in pain-related emotional reactivity is not well understood.
Several lines of evidence suggest that variation in NPY expression may be important for emotional processing and affective disorders in humans. Plasma NPY concentrations have been positively associated with resilience to psychological stress.14-17 Conversely, low NPY in plasma, cerebrospinal fluid, and postmortem tissue has been variably associated with mood disorders.18-25 Variation in NPY expression appears to be driven in part by variation in the NPY gene.22,26 In particular, at least one functional locus was identified within human NPY haplotypes that predicted expression in lymphoblastoid cell lines, plasma, and brain.26 Individuals with low-expression genotypes exhibited greater hemodynamic responses in the amygdala when presented with threat-related stimuli, lower endogenous opioid release during a pain stressor, and greater trait anxiety.26 Furthermore, a 2004 report linked a single-nucleotide polymorphism in the NPY gene with treatment-resistant MDD.22
These findings suggest a model in which genetic variation in the NPY gene predisposes some individuals to low NPY expression within key stress-regulatory neural circuits. Reduced capacity for NPY expression, in turn, would lead to differential processing of stimuli with negative affective valence and potentially increase the risk of developing affective disorders. We examined the predictive validity of this model at three levels. First, we used functional magnetic resonance imaging (fMRI) and an emotional processing task to test the hypothesis that healthy individuals with low-expression NPY genotypes would exhibit greater cortical activation in response to negative stimuli. Second, we tested the hypothesis that healthy individuals with low-expression NPY genotypes would report more negative affective experiences during stress. Because pain is a potent, universal stressor that is readily manipulated experimentally, we used moderate levels of sustained pain as a stress challenge. Finally, we tested our hypothesis that low-expression NPY genotypes are over-represented among patients with MDD.
Methods
Neuroimaging
One-hundred-eleven healthy adults completed an fMRI study of passive affective processing. After screening for quality control (described in Supplemental Methods) useable data were available for 93 subjects (mean ± SD age, 29 ± 9 years; 52% male). Task effects were determined in the sample of 93 individuals. Of the 70 who participated in genotyping, 58 were classified by NPY genotype and 12 were unclassified according to a previously established haplotype classification scheme (see Table 1 and Genotyping below). Sampling and recruitment is described in Major Depression Association. All subjects in the fMRI experiment were right-handed and fluent English speakers. They were not taking exogenous hormones or medications with central nervous system activity, and they were instructed to abstain from all psychoactive substances for 24 hours prior to the study. Written informed consent was obtained and all procedures were approved by the Institutional Review Board at the University of Michigan.
Table 1. NPY classification in study sub-samples a.
| Sub-sample | NPY classification | Classified total | Unclassified | ||
|---|---|---|---|---|---|
| low | med | high | |||
| fMRI | 8 | 35 | 15 | 58 | 12 |
| Pain-stress challenge | 15 | 47 | 16 | 78 | 18 |
| MDD association | |||||
| Healthy subjects | 22 | 68 | 23 | 113 | 24 |
| MDD subjects | 15 | 19 | 5 | 39 | 5 |
| All subjects | 37 | 87 | 28 | 152 | 29 |
Number of participants in each sub-sample are shown by NPY expression level (low, medium, high), as predicted by NPY genotype.
As described previously,27 subjects performed an affective word task during which they silently read emotionally-valenced words. The blood oxygenation level dependent (BOLD) signal was measured in the whole brain using a GE Signa 3-Tesla MRI scanner with a standard RF coil and T2*-weighted pulse sequence. Images were spatially normalized to standardized space (Montreal Neurological Institute, MNI) and smoothed with a 6-mm Gaussian kernel. Spatial coordinates are reported in MNI space. See Supplemental Methods for further details.
BOLD responses were modeled with SPM2 (Wellcome Department of Cognitive Neurology, University College London, UK) using a general linear model and canonical hemodynamic response function. Statistical analysis proceeded in two stages. At the first level, activation maps were derived for individual subjects, including task-related covariates of interest and nuisance covariates (head translation and rotation). At the second level, a random effects analysis was employed to determine group effects, resulting in statistical parametric (t or F) maps. Statistical tests were applied to the two primary contrasts of interest, negative–neutral words and positive–neutral words, since these isolated affective processing and controlled for non-specific lexical and visual processing. Where those contrasts showed significant effects, we also explored responses to word stimuli relative to rest periods (i.e., negative–rest and neutral–rest) to aid interpretation. A mask excluded the cerebellum and brainstem below the midbrain because these regions were not well represented. The resulting voxel-wise maps (2 × 2 × 2 mm) were thresholded with two-sided uncorrected p < 0.001 and extent k > 55 voxels (440 mm3), which protected against overall type I error at p < 0.05 according to Monte Carlo simulations with AlphaSim.28 All reported p- and z-values are two-sided.
For analyses in regions of interest, the average percent change in BOLD signal within the region was computed. We used ordinal logistic regression with NPY genotype group (low, medium, high) as the dependent variable and percent signal change as a covariate (SPSS 17.0, SPSS Inc., Chicago IL). Parameter estimates β (ordered log odds) and 95%-confidence intervals (95%-CI) are reported. We tested our a priori hypothesis of an NPY genotype effect in a single region (medial PFC), identified as the single cluster activated by this task (negative–neutral words). This hypothesis was based on (1) prior reports that low-expression NPY genotypes are associated with greater amygdala activation specifically to negative (versus neutral) stimuli26, 29 and (2) the proposed role of this region in emotion processing1-3 and depression.30-34 The task also produced deactivations in other regions (neutral–positive, 2 clusters; neutral–negative, 4 clusters; eTable). To characterize the regional and valence-related specificity of the NPY effect, these clusters were also tested for an effect of genotype using a Bonferroni correction based on the number of clusters per contrast to account for multiple comparisons.
Pain-stress challenge
Ninety-six healthy adults (mean ± SD age, 25 ± 4 years; 66% male) participated in a pain-stress challenge described previously.35, 36 Sampling and recruitment is described in Major Depression Association. Seventy-eight of the 96 subjects were classified by NPY genotype and 18 were unclassified (Table 1). Fifty-one of these participants also completed the fMRI affective word task. Each individual underwent a standardized pain paradigm in which hypertonic saline was infused intramuscularly into the masseter muscle, resulting in deep sustained muscle pain for 20 minutes at a level that was individually calibrated to a level ∼40% of “the most pain imaginable.” Subjects provided affective ratings at baseline and immediately after the pain protocol. Written informed consent was obtained and all procedures were approved by the Institutional Review Board at the University of Michigan.
Participants rated affective experience before and after pain with the 60-item Positive and Negative Affective Schedule (PANAS)37, 38 which includes two main pseudo-independent subscales, Negative Affect and Positive Affect. At both time points, the Positive Affect subscale was approximately normally distributed, but the Negative Affect subscale was severely skewed toward low values. For that reason, we analyzed PANAS responses in two ways. First, we used a composite measure (the difference of Positive Affect and Negative Affect) which was readily interpreted, normally distributed, and appropriate for hypothesis testing using repeated-measures ANOVA and Tukey post-hoc tests (SPSS 17.0). Five individuals who were missing baseline data were excluded from that analysis. Second, we used nonparametric Spearman correlation to test for associations between NPY genotype and individual PANAS subscales before and after pain.
Major depression association
We genotyped 44 individuals with MDD who were recruited for two separate studies in the Department of Psychiatry at the University of Michigan39, 40 (39 classified by NPY genotype, 5 unclassified, Table 1). Participants were recruited through local advertisement for neuroimaging studies of MDD. Recruitment criteria were identical between the two studies except that one recruited women only,39 whereas the other recruited both sexes.40 Major medical illness and other axis I diagnoses were excluded, except generalized anxiety disorder, social anxiety disorder, and specific phobia. Subjects were diagnosed with MDD and a current moderate-to-severe depressive episode using the Structured Clinical Interview for DSM-IV41 administered by an experienced psychiatric research nurse, and diagnosis was confirmed with a clinical interview by a psychiatrist. The healthy comparison sample consisted of 137 healthy controls (113 classified by NPY genotype, 24 unclassified, Table 1). Participants were recruited through local advertisement for neuroimaging studies of MDD or pain processing.35, 36, 40 Subjects were screened to exclude major medical illness, psychiatric disorder, or substance use disorder. Written informed consent was obtained and procedures were approved by the Institutional Review Board at the University of Michigan.
We tested a single a priori hypothesis that low-expression NPY genotypes would be over-represented in the MDD sample. Ordinal regression (SPSS 17.0) was employed with NPY genotype group (low, intermediate, high) as the dependent variable and diagnostic group as an independent factor. Sex and age were not well matched between groups and were therefore entered as covariates. Because we tested a single hypothesis using a haplotype-based classification scheme validated in prior work,26 no correction for multiple comparisons was indicated.42, 43 Other association tests were exploratory and aimed at ruling out confounders.
Genotyping
Seven polymorphisms within and near the NPY gene, including six single nucleotide polymorphisms and a two-nucleotide in/del, were genotyped with 5′ nuclease assay, as previously described.26 Each marker was in Hardy-Weinberg equilibrium (all p > 0.3, Pearson χ2 test). Six polymorphisms comprised five major haplotypes, H1–H5 (Table 2). Each subject was assigned to a genotype group (low, intermediate, or high) based on protein and mRNA expression levels previously established in vitro and in vivo (Table 2).26 Because definitive expression data are not available for the two minor haplotypes H4 and H5 (allele frequency 3–5%), individuals carrying those haplotypes (16% of our sample) were not included in genetic analyses (unclassified individuals in Table 1).
Table 2. NPY haplotypes a.
| Markers | Haplotype | ||||
|---|---|---|---|---|---|
| H1 | H2 | H3 | H4 | H5 | |
| rs3037354 | Ins | Del | Ins | Ins | Ins |
| rs17149106 | G | G | G | G | T |
| rs16147 | C | T | T | C | T |
| rs16139 | T | T | T | T | C |
| rs5573 | A | G | G | A | G |
| rs5574 | T | C | C | C | C |
The three major haplotypes (H1, H2, and H3) defined the NPY classification of each subject (low, H1/H1; intermediate, H1/H2, H1/H3, or H3/H3; and high, H2/H2 or H2/H3) based on expression levels previously determined in vitro and in vivo.26
Population stratification was evaluated as a potential confound using ancestry informative markers (AIMs), as described previously.26 In brief, 186 highly informative markers were genotyped using an Illumina Goldengate assay. Factor analysis resulted in a seven-factor solution which yielded ethnic factor scores for each individual. To test for population stratification in the neuroimaging and pain-stress challenge experiments, we performed Spearman correlations between ethnic factor scores and BOLD percent signal change or PANAS composite scores, respectively. For the MDD association study, AIMs were unavailable for 9 healthy controls and 25 MDD patients. Therefore, we estimated Caucasian, African, or Asian ancestry based on a European, African, or Asian factor score > 0.5 when available (n = 118), and used self-reported Caucasian/white, African American, Asian, or other race/ethnicity otherwise (n = 34).
Results
Hemodynamic responses to affective stimuli
From the 93 healthy subjects who completed the fMRI affective word task, 58 were genotyped for NPY and classified as low, intermediate, or high expression. Twelve additional unclassified individuals carried uncommon haplotypes that lack definitive expression data, so they were not included in genotype analyses (Table 1).
For the key contrast of interest, negative-vs-neutral words, this task activated the medial PFC (corrected p < 0.05; n = 93; SPM one-sample t test; peak coordinates = −2,56,22; z = 4.3; cluster size = 2184 mm3; Figure 1A-C). We extracted responses within this task-related cluster and tested it as a region of interest. Neither sex nor age was associated with NPY genotype (p > 0.3, ordinal regression) or percent signal change in medial PFC (p > 0.8, linear regression). Similarly, ancestry informative markers were not associated with NPY genotype or percent signal change (all p > 0.1, Spearman correlations). Consistent with our primary hypothesis, medial PFC responses to negative (versus neutral) words were inversely related to predicted NPY expression level (p = 0.029, β = −2.00, 95%-CI = [−3.80, 0.20], n = 58, ordinal regression; Figure 1D). Comparison to a resting condition indicated that the effect was driven by greater hemodynamic responses to negative words, and a lack of response to neutral words, among the low-expression group (Figure 1E).
Figure 1.
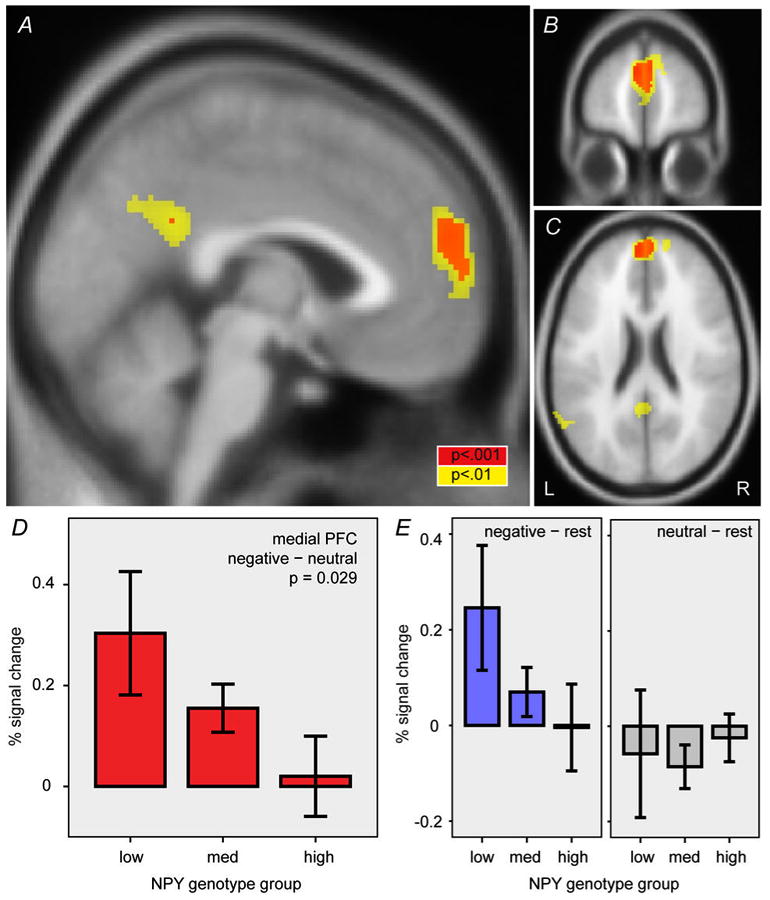
Effect of NPY genotype on medial PFC responses to negative words. Task effect in medial PFC with the negative–neutral word contrast is shown in three sections: (A) sagittal at x = −2, (B) coronal at y = 56, and (C) horizontal at z = 22. Red and yellow areas indicate uncorrected 2-sided p < 0.001 and 0.01, respectively. This cluster was extracted as a region of interest to test for effect of NPY genotype. (D) Effect of NPY genotype group on percent signal change in the medial PFC region of interest shown in A-C (p = 0.029, ordinal regression). (E) Percent signal change for negative–rest and neutral–rest contrasts. Error bars, mean ± standard error. L, left. R, right.
We followed up on this finding by performing a complementary whole-brain linear regression on NPY genotype with the negative–neutral contrast. As shown in Figure 2A-C, this analysis revealed an effect of genotype in the rostral ACC (corrected p < 0.05; peak coordinates = 14,38,0; z = 3.7; cluster size = 592 mm3). The low-expression group showed rostral ACC activation to negative (versus neutral) words, whereas the high-expression group showed deactivation (Figure 2D). Notably, activation of rostral ACC was not evident as a task effect (Figure 1A-C) because responses were oppositely directed in the different genotype groups. Comparison to the resting condition suggested that hemodynamic responses in rostral ACC decreased with negative words among high-expression individuals, and decreased with neutral words among the low-expression group (Figure 2E).
Figure 2.
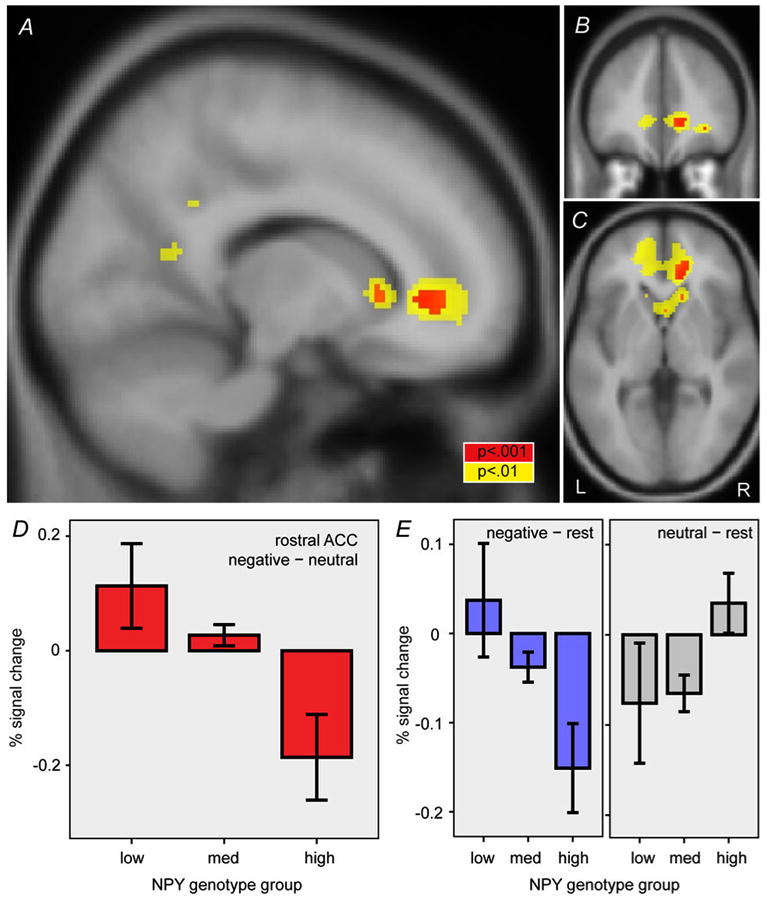
Effect of NPY genotype on rostral ACC responses to negative words. Effect of NPY genotype in right rostral ACC with the negative–neutral word contrast is shown in three sections: (A) parasagittal at x = 14, (B) coronal at y = 38, and (C) horizontal at z = 0. Red and yellow areas indicate uncorrected 2-sided p < 0.001 and 0.01, respectively. (D) Effect of NPY genotype group on percent signal change in the rostral ACC region identified in A-C. (E) Percent signal change for negative–rest and neutral–rest contrasts. Error bars, mean ± standard error. L, left. R, right.
NPY genotype effects were further examined in brain regions where other task effects were found. There was no significant activation for the positive–neutral contrast, but task effects were observed in bilateral parietal and left temporal cortices with the neutral–negative contrast and in left ventrolateral frontal cortex with the neutral–positive contrast (eTable). Percent signal change within these regions was not associated with NPY genotype (p > 0.3, logistic regression, n = 58). Thus, the effect of NPY genotype appeared to be specific to medial frontal cortex and to negative stimuli.
Affective experience during stress
Ninety-six healthy adults who had completed the experimental pain-stress challenge were genotyped for NPY.35, 36 Seventy-eight individuals were classified as low, intermediate, or high NPY expression; 18 additional individuals were unclassified (Table 1).
Self-rated affect was associated with NPY genotype before and after the pain challenge (Figure 3). Neither sex nor age was associated with NPY genotype (both p > 0.3, ordinal regression) or PANAS ratings (p > 0.1, main effect in repeated-measures ANOVA). Similarly, factor weights of ancestry informative markers were not associated with NPY genotype or PANAS ratings (all p > 0.15, Spearman correlations), indicating that population stratification is unlikely to account for the association. Repeated-measures ANOVA on the PANAS composite rating indicated an effect of NPY genotype (p = 0.002, F2,70 = 6.84), an effect of pain (p < 0.001, F1,70 = 13.4), and no genotype-pain interaction (p = 0.16, F2,70 = 1.89). Post-hoc tests demonstrated more negative affect ratings in the low-expression group compared to the other two groups (p = 0.002 for low vs intermediate; p = 0.01 for low vs high; p = 0.99 for intermediate vs high; Tukey HSD test). Examination of subscales before and after pain suggested the effect of NPY genotype was greater on the Negative Affect subscale (p = 0.08, ρ = −0.21, n = 73 before pain; p = 0.02, ρ = −0.26, n = 78 after pain; Spearman correlations) than on the Positive Affect subscale (p = 0.13, ρ = 0.18, n = 73 before pain; p = 0.74, ρ = 0.038, n = 78 after pain; Spearman correlations). Among individuals that participated in both neuroimaging and stress-challenge studies (n = 51), we found no association between PANAS ratings and activation of medial PFC or rostral ACC (p > 0.05, Pearson correlations).
Figure 3.

Effect of NPY genotype on affect self-report during stress. The composite PANAS score, defined as Positive Affect – Negative Affect, is shown as a function of NPY genotype group (mean and 95%-confidence intervals). PANAS ratings were more negative in the low-expression group before (left panel) and after (right panel) a standardized pain-stress challenge (repeated-measures ANOVA, see text).
Association with major depression
Thirty-nine individuals with moderate-to-severe MDD and 113 healthy comparison subjects were classified by NPY genotype (Table 1). Demographic and clinical characteristics are shown in Table 3.
Table 3. Demographic and clinical characteristics of MDD and control samples.
| Characteristic | MDD (n = 39) |
Control (n = 113) |
|---|---|---|
| Female sex, No. (%) | 34 (87) | 44 (39) |
| Age, mean (SD), y | 36 (11) | 27 (7) |
| Race/ethnicity | ||
| Caucasian, No. (%) | 27 (69) | 77 (68) |
| African American, No. (%) | 7 (18) | 20 (18) |
| Asian, No. (%) | 0 (0) | 8 (7) |
| Mixed/other ancestry, No. (%) | 5 (14) | 8 (7) |
| 17-item Hamilton depression scale, mean (SD) | 23 (5) | |
| Atypical features, No. (%) † | 7 (18) | |
| Melancholic features, No. (%) † | 11 (29) | |
| Co-morbid anxiety disorder, No. (%) † | 12 (32) | |
| First-episode (vs recurrent), No. (%) † | 11 (29) | |
| Age of onset, mean (SD), y ‡ | 25 (13) | |
| Duration of episode, mean (SD), mo ‡ | 21 (25) |
Data unavailable for 1 subject
Data unavailable for 4 subjects
Genotype distributions are shown in Figure 4. We confirmed that NPY genotype was not associated with sex or age (p > 0.3, ordinal regression). However, patients in the MDD sample were older (p < 0.001, two-sample t test) and more often female (p < 0.001, Fisher's exact test). We addressed this imbalance by entering age and sex as covariates in the ordinal regression model. An association between MDD diagnosis and NPY genotype was present before adjustment, and it strengthened after adjusting for age and sex (p = 0.004, Table 4).
Figure 4.

Association of NPY genotype with MDD diagnosis. The percent of subjects within each diagnostic group is shown versus NPY genotype group for MDD patients (top panel) and healthy control subjects (bottom panel). Low-expression genotypes are more prevalent in the MDD group (ordinal regression, see text).
Table 4. Ordinal regression analysis of NPY genotype and MDD.
| n a | β b | 95%-CI c | p | |
|---|---|---|---|---|
| All subjects, adjusted for age and sex | 113, 39 | 1.24 | 0.39, 2.09 | 0.004 |
| All subjects, unadjusted | 113, 39 | 0.83 | 0.11, 1.55 | 0.024 |
| All subjects, adjusted for age, sex, and race | 113, 39 | 1.20 | 0.33, 2.07 | 0.007 |
| Female only, adjusted for age | 44, 34 | 1.39 | 0.43, 2.35 | 0.005 |
| Caucasian only, adjusted for age and sex | 77, 27 | 1.14 | 0.12, 2.16 | 0.029 |
| Age- and sex-matched samples | 25, 39 | 0.94 | -0.05, 1.93 | 0.062 |
Number of healthy participants and MDD patients, respectively
Parameter estimate from ordinal regression
Confidence interval for parameter β
Two follow-up analyses were performed to further explore age and sex as potential confounders. Because most patients were female, we tested women only and found the association after adjusting for age (p = 0.005, Table 4). In addition, we performed a restricted analysis of only those healthy controls who had been recruited for the MDD studies, which resulted in a small, well-matched control sample (sex: p = 0.15, Fisher's exact test; age: p = 0.71, t71 = 0.37, two-sample t test) that did not differ from other healthy controls in NPY genotype distribution (p = 0.51, ordinal regression). Within this underpowered sample, we found a trend (p = 0.06, Table 4) toward overrepresentation of low-expression NPY genotypes in the MDD group.
Further control analyses indicated that population stratification (i.e., racial/ethnic stratification) was unlikely to account for the apparent association between NPY genotype and MDD. First, NPY genotype was not associated with Caucasian, African American, or Asian race/ethnicity (p > 0.15, ordinal regression). Second, race/ethnicity did not differ between MDD patients and controls (p = 0.27, χ2 = 3.88, df = 3, Pearson χ2 test). Third, we performed an additional association test between MDD diagnosis and NPY genotype, adjusting for Caucasian, African American, and Asian status, in addition to age and sex, and found the same result (p = 0.007, Table 4). Fourth, because a majority of participants were Caucasian, we verified that the association was present in Caucasians only (p = 0.029, Table 4).
Discussion
Our results implicate genetically driven NPY expression in emotional functioning at three levels of analysis. At the neural circuit level, we found that low-expression NPY genotypes were associated with greater hemodynamic responses in medial PFC and rostral ACC in healthy individuals viewing negative words. At the level of psychological experience, individuals with low-expression NPY genotypes reported more negative affect during a stressor involving sustained, moderate pain over 20 minutes. At the level of syndromal, categorical diagnosis, we found that low-expression NPY genotypes were more prevalent among patients with MDD. These convergent findings support a model in which genetically driven low NPY expression predisposes certain individuals to hyper-responsivity to negative stimuli within key affective circuit elements, including medial PFC, rostral ACC, and (based on prior work26,29) the amygdala. The association of these same low-expression NPY genotypes with negative affect during stress and with MDD suggests that these NPY-associated neural response patterns may mediate risk for at least some forms of depression.
The association we found with activation of medial PFC and rostral ACC builds upon prior neuroimaging studies that have implicated NPY genotype in amygdala function. Using the same haplotype groupings that we employ here, Zhou and colleagues used fMRI with threat-related stimuli (fearful and angry faces) and reported that low-expression NPY genotypes were associated with increased hemodynamic responses in right amygdala and hippocampus.26 Domschke and colleagues used fMRI while subliminally presenting emotional faces to MDD patients.29 Analyzing a single-nucleotide polymorphism in the NPY gene (rs16147, −399T/C), they found that amygdala responses to angry faces (and to a lesser extent, sad faces) were greater among individuals with the CC genotype, which would include the low-expression group in our analyses.29 We detected no task or genotype effects in the amygdala. We attribute this result to our use of a different fMRI task, one that involves reading emotionally-valenced words and that does not generally engage the amygdala.27, 32, 34, 44, 45 Thus, we view our findings as complementary to (rather than in conflict with) previous studies of amygdala responses to threat-related facial stimuli. By using an emotion word task, we demonstrate for the first time that NPY genotype has effects on the function of medial PFC and rostral ACC, core circuit elements that have been multiply implicated in normal emotion processing, regulation of emotion, and MDD pathophysiology.1-3, 30-34 In particular, we found low- and high-expression genotypes were associated with activation and deactivation, respectively, in the rostral ACC. This cortical region has been consistently implicated in normal emotion processing and depression.3, 30, 46 Thus, our fMRI findings add substantially to previously described central effects of NPY genotype, to include key emotional circuits in the frontal cortex. These findings also suggest that NPY expression in frontal cortex5, 19, 23, 24 may have important functional consequences.
Our finding of associations between NPY genotype, affect under stress, and MDD diagnosis are consistent with growing evidence that implicates NPY in both normal emotion regulation and affective disorders.10, 47 Plasma NPY concentration has been positively associated with resilience to psychological stress14-17 and expression of NPY in the central nervous system has been suggested as a general resilience mechanism.48, 49 Conversely, low NPY levels have been implicated in affective illnesses. Low-expression NPY haplotypes were associated with greater trait anxiety and undifferentiated anxiety disorders.26 Low plasma NPY concentrations were found among currently depressed patients with MDD21 but not among patients with remitted MDD.20 Postmortem studies have variably reported low NPY levels in frontal cortex of patients with MDD and bipolar disorder.19, 23, 24 Early studies of cerebrospinal fluid NPY in MDD patients were discrepant,18, 25 but a more recent study reported robust reductions among patients with treatment-resistant MDD.22 Furthermore, the latter study found a greater prevalence of the −399C allele (rs16147) among those same MDD patients.22 Because our low-expression group includes individuals who are −399C/C homozygotes, our study represents a quasi-replication of that finding with a less treatment-resistant sample. Furthermore, our findings from healthy subjects during the pain-stress challenge suggest that NPY genotype influences an individual's affective experience under stress, even before the onset of illness. Taken together, the evidence suggests that genetic predisposition to low NPY expression increases risk for MDD (and possibly other affective disorders) by increasing sensitivity to negative stimuli at the psychological and neural-circuit levels, and possibly at the cell and molecular levels as well.
We tested this model of NPY function in affective processing using a functional genomics strategy that differs from conventional approaches in important ways. Conventional molecular genetic association studies are more susceptible to false positives because the total number of statistical comparisons (and therefore, the extent to which type I error should be corrected) is not always apparent, leading to “hypothesis creep”.42, 43 Furthermore, a nonfunctional locus may be more prone to spurious replication because the direction of the effect is ambiguous.43 We have avoided these pitfalls by testing a single a priori hypothesis using a haplotype-based classification previously validated with in vitro and in vivo NPY expression data.26 This functionally informed strategy increases statistical power by avoiding the multiple-comparison problem, and by targeting genetic variation that has functional impact. This functional genomics approach may also be compared to conventional measurements of peripheral NPY levels. Such measures may approximate the variables of most interest (e.g., synaptic NPY levels), but unlike genotype they are subject to other sources of variability, which could include peripheral sympathetic activation,22 clinical state (depressed versus remission),20 and random measurement error. Thus, our strategy improves on the classic statistical genetics approach by leveraging prior measurements of peripheral and central NPY levels. Our confidence in these results is further strengthened by the coherent directionality of the haplotype-driven effect across three levels of analysis. Nonetheless, independent, replication and meta-analyses of larger pooled samples will be essential to validate these findings.
Several limitations of the present study are noteworthy. First, we have interpreted these findings as supportive of a causative model in which (i) genetically driven variation in NPY expression causes neural hyper-responsiveness in key circuit elements and (ii) hyper-responsive circuits cause negative affect and increase risk of developing MDD. Given the correlative nature of these experiments, however, our findings can only suggest causality, and other models are certainly possible. Experimental interventions in animal models are needed to test causal mechanisms. Second, our subject sample was one of convenience and may not be representative of the general population or of MDD patients encountered in usual clinical practice. For example, our sample was limited to individuals who were willing to volunteer for neuroimaging experiments and genotyping, which could bias certain personality traits of the sample. Third, because definitive expression data was unavailable for minor NPY haplotypes, we were unable to include about 16% of subjects in our analyses. We felt that this limitation was outweighed by the benefits of functionally validated haplotype classification. The role of NPY genotype among those individuals will require characterization of in vivo and in vitro expression data for minor haplotypes. Fourth, about two-thirds of our subjects were of European ancestry, so the extent to which these findings apply to individuals of other genetic backgrounds remains to be seen. Similarly, because our MDD sample was 84% female, we were unable to test for association of low-expression NPY genotype among men. Control analyses indicated that the association with MDD survived (and actually strengthened) after controlling for sex, but sexual dimorphism in the NPY system deserves to be explored. Fifth, the design of this study did not allow us to characterize the degree to which NPY genotype might contribute differentially to risk of MDD versus anxiety. We favor a model of shared risk, but this remains to be tested. Sixth, the sample sizes employed here were limiting in some ways. For example, only 58 subjects were classified in the neuroimaging study, and only 8 had a low-expression genotype. Limited statistical power may have prevented us from detecting brain regions besides mPFC and rACC that are truly modulated by NPY genotype, and parametric statistical tests become less valid for sub-groups that contain smaller numbers of observations.
Our findings may eventually have clinical implications. The heterogeneity of MDD represents a major barrier to improving our understanding of its etiology, pathophysiology, and optimal treatment. Based on the NPY system's established role in anxiety and stress responses in experimental animals, and the increasing evidence for its dysregulation in affective disorders, the NPY system may be an excellent target for MDD subtyping and treatment selection. Along those lines, a recent report suggested that response to antidepressant medication varies with NPY genotype.29 The greatest potential for NPY-based biological markers may lie in guiding development of novel antidepressant agents for the many individuals who fail to respond to currently available treatments.
Supplementary Material
Task effects in the emotion word task (n = 93) a
Acknowledgments
We thank Heng Wang and Wendy Yau for assistance with image processing; Virginia Murphy-Weinberg for study coordination; and the Center for Statistical Consultation and Research at the University of Michigan for advice regarding statistical analysis.
Funded by NIMH (grants P01 MH42251, R25 MH6374, and K23 MH074459), NIDA (grants R01 DA016423 and R01 DA 022520), the NIAAA Intramural Research Program, and the Phil F. Jenkins Research Fund.
The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or preparation, review, or approval of the manuscript. Drs. Mickey and Zubieta had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Disclosures: Dr. Zubieta is a consultant for Eli Lilly & Co. All authors declare no financial conflict of interest.
References
- 1.Dalgleish T. The emotional brain. Nat Rev Neurosci. 2004 Jul;5(7):583–589. doi: 10.1038/nrn1432. [DOI] [PubMed] [Google Scholar]
- 2.Phan KL, Wager T, Taylor SF, Liberzon I. Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. Neuroimage. 2002 Jun;16(2):331–348. doi: 10.1006/nimg.2002.1087. [DOI] [PubMed] [Google Scholar]
- 3.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: The neural basis of normal emotion perception. Biol Psychiatry. 2003 Sep 1;54(5):504–514. doi: 10.1016/s0006-3223(03)00168-9. [DOI] [PubMed] [Google Scholar]
- 4.Bouchard TJ, Jr, McGue M. Genetic and environmental influences on human psychological differences. J Neurobiol. 2003 Jan;54(1):4–45. doi: 10.1002/neu.10160. [DOI] [PubMed] [Google Scholar]
- 5.Adrian TE, Allen JM, Bloom SR, Ghatei MA, Rossor MN, Roberts GW, Crow TJ, Tatemoto K, Polak JM. Neuropeptide Y distribution in human brain. Nature. 1983 Dec 8-14;306(5943):584–586. doi: 10.1038/306584a0. [DOI] [PubMed] [Google Scholar]
- 6.Allen YS, Adrian TE, Allen JM, Tatemoto K, Crow TJ, Bloom SR, Polak JM. Neuropeptide Y distribution in the rat brain. Science. 1983 Aug 26;221(4613):877–879. doi: 10.1126/science.6136091. [DOI] [PubMed] [Google Scholar]
- 7.Tatemoto K, Carlquist M, Mutt V. Neuropeptide Y--a novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature. 1982 Apr 15;296(5858):659–660. doi: 10.1038/296659a0. [DOI] [PubMed] [Google Scholar]
- 8.Gehlert DR. Introduction to the reviews on neuropeptide Y. Neuropeptides. 2004 Aug;38(4):135–140. doi: 10.1016/j.npep.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Kask A, Harro J, von Horsten S, Redrobe JP, Dumont Y, Quirion R. The neurocircuitry and receptor subtypes mediating anxiolytic-like effects of neuropeptide Y. Neurosci Biobehav Rev. 2002 May;26(3):259–283. doi: 10.1016/s0149-7634(01)00066-5. [DOI] [PubMed] [Google Scholar]
- 10.Heilig M. The NPY system in stress, anxiety and depression. Neuropeptides. 2004 Aug;38(4):213–224. doi: 10.1016/j.npep.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Smith PA, Moran TD, Abdulla F, Tumber KK, Taylor BK. Spinal mechanisms of NPY analgesia. Peptides. 2007 Feb;28(2):464–474. doi: 10.1016/j.peptides.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 12.Brumovsky P, Shi TS, Landry M, Villar MJ, Hokfelt T. Neuropeptide tyrosine and pain. Trends Pharmacol Sci. 2007 Feb;28(2):93–102. doi: 10.1016/j.tips.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Craig AD. A new view of pain as a homeostatic emotion. Trends Neurosci. 2003 Jun;26(6):303–307. doi: 10.1016/s0166-2236(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 14.Morgan CA, 3rd, Rasmusson AM, Wang S, Hoyt G, Hauger RL, Hazlett G. Neuropeptide-Y, cortisol, and subjective distress in humans exposed to acute stress: replication and extension of previous report. Biol Psychiatry. 2002 Jul 15;52(2):136–142. doi: 10.1016/s0006-3223(02)01319-7. [DOI] [PubMed] [Google Scholar]
- 15.Morgan CA, 3rd, Rasmusson AM, Winters B, Hauger RL, Morgan J, Hazlett G, Southwick S. Trauma exposure rather than posttraumatic stress disorder is associated with reduced baseline plasma neuropeptide-Y levels. Biol Psychiatry. 2003 Nov 15;54(10):1087–1091. doi: 10.1016/s0006-3223(03)00433-5. [DOI] [PubMed] [Google Scholar]
- 16.Morgan CA, 3rd, Wang S, Southwick SM, Rasmusson A, Hazlett G, Hauger RL, Charney DS. Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol Psychiatry. 2000 May 15;47(10):902–909. doi: 10.1016/s0006-3223(99)00239-5. [DOI] [PubMed] [Google Scholar]
- 17.Yehuda R, Brand S, Yang RK. Plasma neuropeptide Y concentrations in combat exposed veterans: relationship to trauma exposure, recovery from PTSD, and coping. Biol Psychiatry. 2006 Apr 1;59(7):660–663. doi: 10.1016/j.biopsych.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 18.Berrettini WH, Doran AR, Kelsoe J, Roy A, Pickar D. Cerebrospinal fluid neuropeptide Y in depression and schizophrenia. Neuropsychopharmacology. 1987 Dec;1(1):81–83. doi: 10.1016/0893-133x(87)90013-3. [DOI] [PubMed] [Google Scholar]
- 19.Caberlotto L, Hurd YL. Reduced neuropeptide Y mRNA expression in the prefrontal cortex of subjects with bipolar disorder. Neuroreport. 1999 Jun 3;10(8):1747–1750. doi: 10.1097/00001756-199906030-00022. [DOI] [PubMed] [Google Scholar]
- 20.Czermak C, Hauger R, Drevets WC, Luckenbaugh DA, Geraci M, Charney DS, Neumeister A. Plasma NPY concentrations during tryptophan and sham depletion in medication-free patients with remitted depression. J Affect Disord. 2008 Oct;110(3):277–281. doi: 10.1016/j.jad.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto H, Onishi H, Koide S, Kai T, Yamagami S. Plasma neuropeptide Y in patients with major depressive disorder. Neurosci Lett. 1996 Sep 20;216(1):57–60. doi: 10.1016/0304-3940(96)13008-1. [DOI] [PubMed] [Google Scholar]
- 22.Heilig M, Zachrisson O, Thorsell A, Ehnvall A, Mottagui-Tabar S, Sjogren M, Asberg M, Ekman R, Wahlestedt C, Agren H. Decreased cerebrospinal fluid neuropeptide Y (NPY) in patients with treatment refractory unipolar major depression: preliminary evidence for association with preproNPY gene polymorphism. J Psychiatr Res. 2004 Mar-Apr;38(2):113–121. doi: 10.1016/s0022-3956(03)00101-8. [DOI] [PubMed] [Google Scholar]
- 23.Ordway GA, Stockmeier CA, Meltzer HY, Overholser JC, Jaconetta S, Widdowson PS. Neuropeptide Y in frontal cortex is not altered in major depression. J Neurochem. 1995 Oct;65(4):1646–1650. doi: 10.1046/j.1471-4159.1995.65041646.x. [DOI] [PubMed] [Google Scholar]
- 24.Widdowson PS, Ordway GA, Halaris AE. Reduced neuropeptide Y concentrations in suicide brain. J Neurochem. 1992 Jul;59(1):73–80. doi: 10.1111/j.1471-4159.1992.tb08877.x. [DOI] [PubMed] [Google Scholar]
- 25.Widerlov E, Lindstrom LH, Wahlestedt C, Ekman R. Neuropeptide Y and peptide YY as possible cerebrospinal fluid markers for major depression and schizophrenia, respectively. J Psychiatr Res. 1988;22(1):69–79. doi: 10.1016/0022-3956(88)90030-1. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Z, Zhu G, Hariri AR, Enoch MA, Scott D, Sinha R, Virkkunen M, Mash DC, Lipsky RH, Hu XZ, Hodgkinson CA, Xu K, Buzas B, Yuan Q, Shen PH, Ferrell RE, Manuck SB, Brown SM, Hauger RL, Stohler CS, Zubieta JK, Goldman D. Genetic variation in human NPY expression affects stress response and emotion. Nature. 2008 Apr 24;452(7190):997–1001. doi: 10.1038/nature06858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heitzeg MM, Nigg JT, Yau WY, Zubieta JK, Zucker RA. Affective circuitry and risk for alcoholism in late adolescence: differences in frontostriatal responses between vulnerable and resilient children of alcoholic parents. Alcohol Clin Exp Res. 2008 Mar;32(3):414–426. doi: 10.1111/j.1530-0277.2007.00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward BD. AlphaSim. [Accessed January, 2010]; http://afni.nimh.nih.gov.
- 29.Domschke K, Dannlowski U, Hohoff C, Ohrmann P, Bauer J, Kugel H, Zwanzger P, Heindel W, Deckert J, Arolt V, Suslow T, Baune BT. Neuropeptide Y (NPY) gene: Impact on emotional processing and treatment response in anxious depression. Eur Neuropsychopharmacol. 2009 Oct 23; doi: 10.1016/j.euroneuro.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 2003 Sep 1;54(5):515–528. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 31.Price JL, Drevets WC. Neurocircuitry of Mood Disorders. Neuropsychopharmacology. 2009 Aug 19; doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshimura S, Okamoto Y, Onoda K, Matsunaga M, Ueda K, Suzuki S, Shigetoyamawaki Rostral anterior cingulate cortex activity mediates the relationship between the depressive symptoms and the medial prefrontal cortex activity. J Affect Disord. 2009 Apr;122(1-2):76–85. doi: 10.1016/j.jad.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 33.Elliott R, Rubinsztein JS, Sahakian BJ, Dolan RJ. The neural basis of mood-congruent processing biases in depression. Arch Gen Psychiatry. 2002 Jul;59(7):597–604. doi: 10.1001/archpsyc.59.7.597. [DOI] [PubMed] [Google Scholar]
- 34.Hsu DT, Langenecker SA, Kennedy SE, Zubieta JK, Heitzeg MM. fMRI BOLD responses to negative stimuli in the prefrontal cortex are dependent on levels of recent negative life stress in major depressive disorder. Psychiatry Res. 2010 Sep 30;183(3):202–208. doi: 10.1016/j.pscychresns.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001 Jul 13;293(5528):311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]
- 36.Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. mu-opioid receptor-mediated antinociceptive responses differ in men and women. J Neurosci. 2002 Jun 15;22(12):5100–5107. doi: 10.1523/JNEUROSCI.22-12-05100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watson D, Clark LA. PANAS-X Manual for the Positive and Negative Affect Schedule - Expanded Form. University of Iowa; 1994. [Google Scholar]
- 38.Watson D, Clark LA, Tellegen A. Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol. 1988 Jun;54(6):1063–1070. doi: 10.1037//0022-3514.54.6.1063. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy SE, Koeppe RA, Young EA, Zubieta JK. Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch Gen Psychiatry. 2006 Nov;63(11):1199–1208. doi: 10.1001/archpsyc.63.11.1199. [DOI] [PubMed] [Google Scholar]
- 40.Mickey BJ, Ducci F, Hodgkinson CA, Langenecker SA, Goldman D, Zubieta JK. Monoamine oxidase A genotype predicts human serotonin 1A receptor availability in vivo. J Neurosci. 2008 Oct 29;28(44):11354–11359. doi: 10.1523/JNEUROSCI.2391-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders. New York: Biometric Research Dept, New York Psychiatric Institute; 1995. [Google Scholar]
- 42.Burmeister M, McInnis MG, Zollner S. Psychiatric genetics: progress amid controversy. Nat Rev Genet. 2008 Jul;9(7):527–540. doi: 10.1038/nrg2381. [DOI] [PubMed] [Google Scholar]
- 43.Sullivan PF. Spurious genetic associations. Biol Psychiatry. 2007 May 15;61(10):1121–1126. doi: 10.1016/j.biopsych.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Elliott R, Rubinsztein JS, Sahakian BJ, Dolan RJ. Selective attention to emotional stimuli in a verbal go/no-go task: an fMRI study. Neuroreport. 2000 Jun 5;11(8):1739–1744. doi: 10.1097/00001756-200006050-00028. [DOI] [PubMed] [Google Scholar]
- 45.Epstein J, Pan H, Kocsis JH, Yang Y, Butler T, Chusid J, Hochberg H, Murrough J, Strohmayer E, Stern E, Silbersweig DA. Lack of ventral striatal response to positive stimuli in depressed versus normal subjects. Am J Psychiatry. 2006 Oct;163(10):1784–1790. doi: 10.1176/ajp.2006.163.10.1784. [DOI] [PubMed] [Google Scholar]
- 46.Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008 Sep;213(1-2):93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sajdyk TJ, Shekhar A, Gehlert DR. Interactions between NPY and CRF in the amygdala to regulate emotionality. Neuropeptides. 2004 Aug;38(4):225–234. doi: 10.1016/j.npep.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 48.Charney DS. Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry. 2004 Feb;161(2):195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- 49.Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci. 2009 Jun;10(6):446–457. doi: 10.1038/nrn2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradley MM, Lang PJ. Affective norms for English words (ANEW): Stimuli, instruction manual and affective ratings. Gainesville, FL: The Center for Research in Psychophysiology, University of Florida; 1999. Technical report C-1. [Google Scholar]
- 51.Noll DC. Rapid MR image acquisition in the presence of background gradients. IEEE International Symposium on Biomedical Imaging; Washington, DC. 2002. [Google Scholar]
- 52.Meyer CR, Boes JL, Kim B, Bland PH, Zasadny KR, Kison PV, Koral K, Frey KA, Wahl RL. Demonstration of accuracy and clinical versatility of mutual information for automatic multimodality image fusion using affine and thin-plate spline warped geometric deformations. Med Image Anal. 1997 Apr;1(3):195–206. doi: 10.1016/s1361-8415(97)85010-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Task effects in the emotion word task (n = 93) a