

Abstract
Background
Roche 454 pyrosequencing has become a method of choice for generating transcriptome data from non-model organisms. Once the tens to hundreds of thousands of short (250-450 base) reads have been produced, it is important to correctly assemble these to estimate the sequence of all the transcripts. Most transcriptome assembly projects use only one program for assembling 454 pyrosequencing reads, but there is no evidence that the programs used to date are optimal. We have carried out a systematic comparison of five assemblers (CAP3, MIRA, Newbler, SeqMan and CLC) to establish best practices for transcriptome assemblies, using a new dataset from the parasitic nematode Litomosoides sigmodontis.
Results
Although no single assembler performed best on all our criteria, Newbler 2.5 gave longer contigs, better alignments to some reference sequences, and was fast and easy to use. SeqMan assemblies performed best on the criterion of recapitulating known transcripts, and had more novel sequence than the other assemblers, but generated an excess of small, redundant contigs. The remaining assemblers all performed almost as well, with the exception of Newbler 2.3 (the version currently used by most assembly projects), which generated assemblies that had significantly lower total length. As different assemblers use different underlying algorithms to generate contigs, we also explored merging of assemblies and found that the merged datasets not only aligned better to reference sequences than individual assemblies, but were also more consistent in the number and size of contigs.
Conclusions
Transcriptome assemblies are smaller than genome assemblies and thus should be more computationally tractable, but are often harder because individual contigs can have highly variable read coverage. Comparing single assemblers, Newbler 2.5 performed best on our trial data set, but other assemblers were closely comparable. Combining differently optimal assemblies from different programs however gave a more credible final product, and this strategy is recommended.
Background
Transcriptome sequencing projects for non-model organisms are popular because they cost less and are more computationally tractable than full genome sequencing projects, but still yield sufficient information to meet the requirements of many research programs. Traditionally, transcriptome projects have been based on Sanger dideoxy-sequenced expressed sequence tags (ESTs), but, because second-generation sequencing technologies provide much higher throughput than Sanger sequencing at a lower cost per base, these new technologies are increasingly used. For model organisms where a wealth of genomic information is available, the massively parallel, short-read (35-100 base) sequencing technologies (Illumina SOLEXA and ABI SOLiD) are most frequently used, as transcriptome reads can be mapped to the reference genome or transcriptome. However, most published non-model organism projects have used the Roche 454 pyrosequencing platform [1-36], because the longer reads generated (currently about 400 bases) are more amenable to de novo assembly and annotation.
A transcriptome project progresses through phases of data acquisition, assembly of the sequence reads to define putative transcripts, and then annotation and exploitation of the assembled data. The transcriptome assembly problem is not simple. Individual reads can have errors and polymorphisms that complicate recognition of overlaps, and individual transcripts (in non-normalised data) can have several orders of magnitude variation in abundance, and thus in effective coverage. Previous analyses of transcriptome data generated by Roche 454 pyrosequencing have almost always used just one software program for assembly (Table 1). Only two studies report the results of more than assembler. The first [1] briefly compared the length of assembled consensuses from Newbler [37], CAP3 [38], and stackPACK [39] on an Arabidopsis thaliana transcriptome dataset, whereas the second [11] demonstrated the comprehensive est2assembly pipeline on a phylogenetically diverse sample of insects, but only compared results for Newbler and MIRA [40]. Neither study provides a systematic comparison of assemblers, and so here we present such a comparison to assist the growing number of transcriptome projects using Roche 454 pyrosequencing data.
Table 1.
Assemblers previously used for de novo assembly of 454 pyrosequencing transcriptome projects
| Assembler | Organism |
|---|---|
| Newbler | Arabidopsis thaliana [1,2]; Eucalyptus grandis [3]; Castanea dentata and Castanea mollissima [4]; Sarcophaga crassipalpis [5]; Acropora millepora [6]; Palomero toluqueño [7]; Eschscholzia californica and Persea americana [2]; Vitis vinifera [8]; Rhagoletis pomonella [9]; Heliconius spp. [10]; Euphydryas aurinia, Manduca sexta, Chrysomela tremulae, Papilio dardanus, Heliconius melpomene, Heliconius erato, and Melitaea cinxia [11]; Panax quinquefolius [12]; Sclerotium rolfsii [13]; Laternula elliptica [14] |
| CAP3 | Zea mays [15,16]; Arabidopsis thaliana [1]; Ambystoma mexicanum [17]; Human breast cancer [18]; Artemisia annua [19]; Solanum arcanum [20]; Epimedium sagittatum [21]; Haemonchus contortus [22]; Laodelphax striatellus [23]; Coleochaete orbicularis and Spirogyra pratensis [24]; Bugula neritina [25]; |
| MIRA | Centaurea solstitialis [26]; Chrysomela tremulae [27,11]; Pandinus imperator [28]; Zygaena filipendulae [29]; Manduca sexta [30,11]; Euphydryas aurinia, Papilio dardanus, Heliconius melpomene, Heliconius erato, and Melitaea cinxia [11]; |
| TGICL | Pythium ultimum [31]; Zoarces viviparous [32]; Medicago truncatula [33] |
| SeqMan | Melitaea cinxia [34]; Cochliomyia hominivorax [35]; Pinus contorta [36] |
| stackPACK | Arabidopsis thaliana [1] |
Here we compare the performance of five assemblers: Newbler, CAP3, MIRA, SeqMan [41], and CLC's Assembly Cell [42] (Table 2). These assemblers differ in the algorithms used (most use variations of the Overlap-Layout-Consensus (OLC) strategy, while CLC uses de Bruijn graph path finding) and how they treat individual reads (whether a read is indivisible, or can be split and ultimately be placed in different contigs). We tested two versions of Newbler because we found the frequently-used, public release version (Newbler Version 2.3, hereafter referred to as Newbler 2.3) to have several undesirable features (see below) and thus contacted the developers to discuss these. They provided a pre-release version (Newbler Version 2.5 p1, hereafter referred to as Newbler 2.5) that addresses some of the issues identified. We did not include TGICL [43] or stackPACK [39] because both are essentially wrappers for the CAP3 assembler. Although Velvet [44] and ABySS [45] are popular assemblers for second-generation sequence data and can use Roche 454 pyrosequencing reads, they are primarily assemblers for genome sequence data from short-read platforms that rely on the high and even coverage depths afforded by these massively parallel technologies. In preliminary experiments on our data we were only able to generate very short contigs using short-read assemblers and so we have not compared Velvet, Oases (the transcriptome-specific version of Velvet, [46]), and ABySS.
Table 2.
Features of assembly programmes compared in this study
| Assembler | Type† | Splits reads* | Author | Cost | Source available | URL |
|---|---|---|---|---|---|---|
| CAP3 | OLC† | No | X Huang and A Madan [38] | Free for use at non-profit organizations | No | http://seq.cs.iastate.edu/ |
| CLC Assembly Cell 3.0 | de Bruijn graph | Yes | CLC | Request quote or trial license | No | http://www.clcbio.com/ |
| MIRA 3.0 | OLC | No | Bastien Chevreux [40] | Free | Yes, GPL | http://sourceforge.net/projects/mira-assembler/ |
| Newbler 2.3 and Newbler 2.5 | OLC | Yes | Roche 454 [37] | Free for academic use | No | http://454.com/products-solutions/analysis-tools/gs-de-novo-assembler.asp |
| SeqMan NGen 2.1 | OLC | No | DNAStar [41] | Request quote or trial license | No | http://www.dnastar.com/t-products-seqman-ngen.aspx |
* i.e. data from one read can appear in multiple contigs
† OLC: Overlap-Layout-Consensus
Our target organism, Litomosoides sigmodontis, is a model filarial nematode, closely related to the causative agents of human filariases (Brugia malayi, Wuchereria bancrofti and Onchocerca volvulus). It originally derives from cotton rat hosts, but can be maintained in laboratory rodents (mice and gerbils) and is thus a tractable experimental system in which to investigate the dynamics of immune response induction and modulation, and test vaccine and drug candidates [47]. We expect L. sigmodontis to have a transcriptome similar to that of the filarial nematode B. malayi and the more distantly related rhabditid nematode Caenorhabditis elegans, with ~18,000 to 21,000 protein-coding genes generating ~30,000 different transcripts with mean length ~1.2 kb. The transcriptome project is part of a larger investigation into L. sigmodontis genomics, and detailed analyses of the content and biology of the transcriptome data will be published elsewhere.
Results
L. sigmodontis transcriptome data
We used a Roche 454 FLX instrument to generate 'standard chemistry' (mean read length ~220 bases) and 'Titanium chemistry' (mean read length ~350 bases) reads from cDNA libraries from three different lifecycle stages of L. sigmodontis: microfilaria (equivalent to the first stage larva of C. elegans and other nematodes), adult males, and adult females (Table 3, see Methods for more details on library preparation, read pre-processing, and assembly parameters). Reads were trimmed for adapters leaving a total of 741,387 reads with 205,065,666 bases used in all assembly experiments (trimmed read length histograms in Additional file 1, Figure S1).
Table 3.
The Litomosoides sigmodontis transcriptome dataset read statistics
| L. sigmodontis lifecycle stage | Technology | Number of reads | Number of raw bases | Number of trimmed reads | Number of trimmed bases | Mean length of trimmed reads | Median length of trimmed reads |
|---|---|---|---|---|---|---|---|
| Microfilaria (first stage larvae) | Titanium | 366,813 | 203,227,223 | 351,387 | 118,039,337 | 335.92 | 374 |
| Adult female | Standard | 180,271 | 48,434,306 | 176,454 | 38,352,888 | 217.35 | 236 |
| Adult male | Standard | 216,940 | 59,231,575 | 213,546 | 48,673,441 | 227.93 | 245 |
| Total | Titanium + Standard | 764,024 | 310,893,104 | 741,387 | 205,065,666 | 276.60 | 257 |
Comparison of assemblers
For each assembler, we used the default parameters recommended for transcriptome assembly (details are given in Methods). After assembly, contigs less than 100 bases in length and singletons were discarded for subsequent analyses. We compared the assemblies using the following standard metrics: total number of reads used in the assembly, number of contigs > 100 bases generated, N50 length of contigs (the smallest contig size in which half the assembly is represented), maximum contig length, summed contig length, and approximate time taken to perform analysis (Table 4). We include the N50 as a measure even though it is not strictly appropriate for transcriptome assemblies (where we expect the median contig length to be in the region of 1.2 kb). We also assessed assembly integrity and completeness by comparison to four reference datasets.
Table 4.
Basic assembly metrics
| CAP3 | CLC | MIRA | Newbler 2.3 | Newbler 2.5 | SeqMan | |
|---|---|---|---|---|---|---|
| Number of contigs† | 24727 | 22746 | 35827 | 12019 | 21734 | 29969 |
| Total Bases | 16733217 | 14875522 | 21339704 | 14456476 | 20066883 | 21355682 |
| Number of contigs (> = 1 kbp) | 4403 | 4174 | 4770 | 6320 | 7661 | 6082 |
| Total Bases (in contigs > = 1 kbp) | 6461079 | 6255785 | 7027775 | 10810962 | 13691429 | 9296011 |
| Max contig length | 4011 | 4368 | 5784 | 5872 | 6228 | 6263 |
| Mean contig length | 677 | 654 | 596 | 1203 | 923 | 713 |
| N50 | 806 | 850 | 708 | 1487 | 1448 | 880 |
| Number of contigs in N50 | 6533 | 5459 | 9148 | 3406 | 4649 | 7555 |
| Reads used (SSAHA2) | 670425 | 679152 | 672036 | 616672 | 667597 | 681974 |
| Multi-hit reads (SSAHA2) | 271648 | 118334 | 392884 | 249210 | 352887 | 322409 |
| Reads used (CLC) | 690889 | 691818 | 696527 | 600132 | 681831 | 711726 |
| Multi-hit reads (CLC) | 91951 | 24485 | 162365 | 213670 | 262178 | 128631 |
| Time taken | 1 day* | 4 minutes * | 3 days * | 2 hours * | 45 minutes * | 6 hours ** |
† only contigs > 100 bases were assessed
* on a dual quad-core 3 GHz Xeon workstation with 32 GB RAM
** on a dual core 2.53 GHz Mac mini server with 4 GB RAM
Numbers of reads used in assembly
The optimal assembler will use all the reads given, and will deliver assemblies with unambiguous mappings of reads to contigs, and thus putative transcripts. Each assembler has a different way of reporting the number of reads utilised. For example, MIRA reported only 2,170 singletons but classified 115,688 unassembled reads as 'debris'. Similarly, Newbler 2.3 and Newbler 2.5 generate separate lists of singletons, repetitive reads, and 'outliers' (problematic reads such as chimaeras). CAP3 and SeqMan only report assembled and unassembled reads. CLC does not track reads at all and maps reads back to the assembly to estimate where they might belong. Therefore, to compare the assemblies, we mapped all the reads back to each assembly using SSAHA2 [48] and the CLC reference aligner (which is part of the Assembly Cell suite [42]), as recommended in a study comparing short-read aligners [49]. SSAHA2's default settings for Roche 454 reads are clearly tuned for sensitivity, because the number of reads with multiple matches is very high compared to the CLC aligner. For both aligners, the highest numbers of reads were mapped back to the SeqMan assembly, while fewest were mapped to the Newbler 2.3 assembly. CAP3, CLC, and MIRA were comparable in terms of the number of reads used. CLC had the fewest reads with multiple matches by far, indicating that it was the least redundant of the six assemblies.
Number, mean length, and summed length of contigs
The optimal assembler will produce the longest summed length of contigs, while avoiding over-assembly of reads into in silico chimaeras, and avoiding the production of near-identical, largely overlapping contigs from allelic copies or error-rich data. It will also produce a transcriptome estimate with a mean and variance in contig length similar to that expected from the whole transcriptome. Newbler 2.3 generated an assembly with the largest N50 and the longest mean contig length (Table 4), but it also produced the smallest assembly span of the six programmes tested. Inspection of contig assemblies using the next-generation sequence assembly visualisation software Tablet [50] showed that Newbler 2.3 was discarding read overlap information in deriving the assembled contig sequence. The overall assembly sizes of CAP3, CLC and Newbler 2.3 are comparable to each other (between 14.4 and 16.7 Mb), as are the assembly sizes of MIRA, Newbler 2.5 and SeqMan (between 20.0 and 21.3 Mb). Newbler 2.5 has an assembly that is 39% larger than the Newbler 2.3 assembly, and seems to have solved the previous version's problem of discarding sequence data. Of all the six assemblers, Newbler 2.5 produced the highest number of contigs longer than 1 kb, and the most bases in contigs longer than 1 kb (these larger overall contig sizes are represented in Figure 1 as the assembly with the steepest initial slope). MIRA and SeqMan both generated comparable assembly spans, but with at least 8,000 more contigs than Newbler 2.5, indicating that they have shorter contigs overall.
Figure 1.
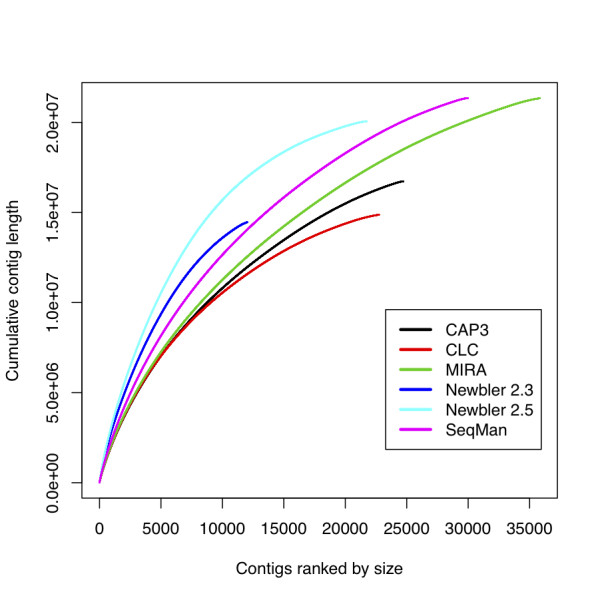
Cumulative contig lengths generated by different assembly programs. For each of six assemblies, contigs longer than 100 bases were ordered by length, and the cumulative length of all contigs shorter than or equal to a given contig was plotted. The total length of the assembly and the number of contigs present in the assembly define the end point of each curve, while the initial slope of each curve reflects the proportion of longer contigs.
Speed of analyses
The optimal assembler will complete analyses in a short period of time, and the time taken will scale well with increasing data volumes. While speed itself is not an overriding optimality criterion, rapid analyses will permit robust and extensive exploration of parameter space. In addition, some algorithms can make efficient use of multi-threaded processors or cluster computing, effectively reducing their wall-clock run time. The speed metric is not linked to assembly quality. CLC was astonishingly fast (taking only a few minutes to assemble 741,387 reads) compared to MIRA (which took 3 days in 'accurate' mode on the same hardware). This speed comes at a significant cost however, as CLC does not track read placement in the assembly. Tracking of read placement in an assembly is a very valuable feature, as it allows inspection of the data underpinning suspect assemblies, and thus CLC sacrifices speed for verifiability.
Assembly redundancy, shared and novel bases
In comparison to other assemblies, the optimal assembly will include the largest proportion of the unique bases present in the sum of all assemblies. Thus an assembler that produced a large assembly that included many contigs that were redundant could be worse that an assembly that was shorter but only included unique bases. To determine if the differing assembly sizes were due to novel sequences in each assembly, or just due to repetitive and redundant assemblies, we used BLAT [51] with default parameters to pairwise align all six assemblies (Figure 2).
Figure 2.

Novel sequence in pair-wise comparisons between assemblies produced by different assemblers. For each assembly, we calculated the number of bases in the other assemblies that were not present in the focal assembly.
These pairwise comparisons show that the SeqMan assembly had the most novel bases when compared to the others, and the others five assemblies had the fewest novel bases when compared to SeqMan. At the other end of the spectrum, the Newbler 2.3 assembly had the fewest novel bases and almost all its bases were seen in the other assemblies. For example, the SeqMan assembly had over 5 Mb of novel sequence compared to the assembly from Newbler 2.3, and the latter had only 23,366 novel bases in the reciprocal comparison. CAP3, CLC, MIRA, and Newbler 2.5 were all very similar in that they generated assemblies that had about 1-2 Mb of novel sequence compared to each other.
SeqMan, MIRA and Newbler 2.5 all generated assemblies in excess of 20 Mb, 25-50% larger than the 14-16 Mb assemblies produced by CAP3, CLC, and Newbler 2.3. However, these additional contigs and bases did not represent an excess of novel sequence, except in the case of SeqMan, suggesting that MIRA and Newbler 2.5 produce some redundant contigs (identical except for real polymorphisms or sequencing error). CLC generated the least redundant assembly, a direct consequence of using a de Bruijn graph algorithm with small fragments (21-mers) that collapses most repeats.
Alignments to reference sequences
Another optimality criterion for a novel de novo assembled transcriptome is how well it recapitulates previously determined sequences for the target species, and how well it represents sequences from related organisms. The best assembler will return contigs that match previous data well, and will deliver a high coverage of the conserved proteome of related taxa. We used four comparator datasets:
(i) a set of 2699 Sanger dideoxy sequenced ESTs from L. sigmodontis (Mark Blaxter, unpublished), assembled into 1602 clusters spanning 0.9 Mb (see http://www.nematodes.org/nembase4/species_info.php?species=LSC). We expect the Roche 454 pyrosequencing assemblies to faithfully reflect these longer reads and clusters;
(ii) the proteome predicted from the genome sequence of B. malayi [52]. We expect the B. malayi RefSeq protiens (11,472 GenBank entries, totalling 4.3 M amino acid residues) to be a good model for the proteome of L. sigmodontis, and thus proportional coverage of this proteome should reflect assembly quality;
(iii) gene families conserved across the phylum Nematoda derived from MCL-TRIBE [53] analysis of 62 species assembled in NEMBASE4 database (Mark Blaxter and Ben Elsworth, unpublished; see http://www.nematodes.org/nembase4/tribe_tree.php). We selected 3,681 tribes, containing 120,926 EST clusters, that had representatives in species spanning the inferred base of the phylum. They represent an estimate of the conserved core of genes that may be present in all nematode proteomes, and again proportional coverage should reflect assembly quality.
(iv) a set of 9,782 C. elegans proteins, grouped into 3,731 euKaryotic Ortholog Groups (KOGs), that are known to have orthologs in the proteomes of six other eukaryotic genomes [54]. We expect mRNAs for these conserved proteins to be present in the L. sigmodontis data, and thus proportional coverage should reflect assembly quality.
These datasets are not independent, but each offers a different assessment of assembly quality. Tables 5, 6, 7 and 8 show the results of aligning the contigs from each assembly to each of these four datasets. In all four cases, the best assembler was either Newbler 2.5 or SeqMan. Newbler 2.3 had the lowest number of significant matches to known reference and related sequences. However, to identify which assembler had the largest number of well-assembled long contigs, we did a second assessment of each dataset where only those hits that covered at least 80% of a target sequence were considered. With this restriction, CLC had the fewest hits in all cases except hits to conserved nematode proteins (Table 7), suggesting that the longer contigs from the CLC assembly were not as accurate.
Table 5.
BLAT hits to 1602 Litomosoides sigmodontis EST clusters
| CAP3 | CLC | MIRA | Newbler 2.3 | Newbler 2.5 | SeqMan | |
|---|---|---|---|---|---|---|
| % of ESTs hit by BLAT | 87.8 | 90.3 | 89.6 | 81.6* | 89.6 | 90.8 |
| (% of bases covered) | (78.2) | (80.0) | (80.1) | (71.3)* | (78.7) | (82.0) |
| % of ESTs hit by BLAT where each hit covered at least 80% of the target EST sequence | 59.9 | 51.9 | 62.5 | 59.4 | 63.9 | 65.4 |
| (% of bases covered) | (59.1) | (50.5)* | (61.1) | (59.4) | (63.7) | (64.3) |
* indicates a value significantly lower than the others, using a Huber M-estimator.
Table 6.
BLASTX hits to 11,472 Brugia malayi proteins
| CAP3 | CLC | MIRA | Newbler 2.3 | Newbler 2.5 | SeqMan | |
|---|---|---|---|---|---|---|
| % of proteins hit | 76.7 | 78.4 | 77.3 | 68.9* | 77.9 | 78.6 |
| (% of bases covered) | (60.4) | (62.4) | (59.7) | (51.8)* | (61.5) | (63.0) |
| % of protein hit by individual HSPs that cover 80% of target protein | 27.0 | 26.0 | 26.5 | 29.2 | 32.4 | 28.7 |
| (% of bases covered) | (16.8) | (16.1) | (16.4) | (19.9) | (22.3) | (18.0) |
Note: E-value cutoff 1e-5
* indicates a value significantly lower than the others (p < 0.01), using a Huber M-estimator.
Table 7.
BLASTX hits to 3,681 tribes containing 120,926 conserved nematode proteins
| CAP3 | CLC | MIRA | Newbler 2.3 | Newbler 2.5 | SeqMan | |
|---|---|---|---|---|---|---|
| % of unique tribes hit | 91.7 | 92.2 | 91.4 | 87.0* | 92.0 | 92.4 |
| % of unique tribes hit where individual HSPs covered 80% of target protein | 81.7 | 81.1 | 78.9 | 77.1* | 82.1 | 81.6 |
Note: E-value cutoff 1e-5
* indicates a value significantly lower than the others (p < 0.01), using a Huber M-estimator.
Table 8.
BLASTX hits to 3,731 KOGs containing 9,782 C. elegans proteins
| CAP3 | CLC | MIRA | Newbler 2.3 | Newbler 2.5 | SeqMan | |
|---|---|---|---|---|---|---|
| % of unique KOGs hit | 89.2 | 89.7 | 88.6 | 83.7* | 89.7 | 90.1 |
| % of unique KOGs hit where individual HSPs covered 80% of target protein | 30.2 | 27.1 | 28.6 | 33.0 | 35.7 | 30.4 |
Note: E-value cutoff 1e-5
* indicates a value significantly lower than the others (p < 0.01), using a Huber M-estimator.
Merging assemblies to improve credibility
As each assembler uses a different algorithm to derive final contigs, and each of these algorithms may model different portions of the true transcriptome with different accuracies, we reasoned that combining assemblies might identify the subset of the assemblies that had high credibility, as it was found by both (or multiple) approaches. We checked if pairs of assemblies performed better than individual assemblies under the various optimality criteria proposed above, particularly in their recapitulation of the reference data. We combined two assemblies at a time by treating their (first-order) contigs as pseudo-reads and assembled these using a traditional OLC assembler (CAP3 with default settings). Only second-order contigs that contained first-order contigs from both constituent assemblies were considered for further analysis because two independent assemblers had agreed on the consensus sequence in that contig: we term these "robust contigs".
Whereas previously the six assemblies had generated from 12,000 to 36,000 contigs and spanned 14.5 Mb to 21.4 Mb (Table 4), we found that merging assemblies resulted in a much narrower range of contig numbers and total span (Table 9). The N50 and mean contig sizes also increased, approaching that predicted for a complete transcriptome.
Table 9.
Secondary assemblies by merging pairs of initial assemblies using CAP3 with default settings
| Assembly 1 | Assembly 2 | Number of "Reads" (contigs) in Assembly 1 | Bases in Assembly 1 | Number of "Reads" (contigs) in Assembly 2 | Bases in Assembly 2 | Number of second-order contigs with "reads" from both assemblies | Bases in second-order contigs with "reads" from both assemblies |
|---|---|---|---|---|---|---|---|
| MIRA | SeqMan | 35827 | 21339704 | 29969 | 21355682 | 18068 | 16293192 |
| MIRA | Newbler 2.5 | 35827 | 21339704 | 21734 | 20066883 | 15951 | 15866051 |
| Newbler 2.5 | SeqMan | 21734 | 20066883 | 29969 | 21355682 | 15783 | 15701053 |
| CLC | Newbler 2.5 | 22746 | 14875522 | 21734 | 20066883 | 15778 | 15825663 |
| CAP3 | MIRA | 24727 | 16733217 | 35827 | 21339704 | 15688 | 14243534 |
| CLC | SeqMan | 22746 | 14875522 | 29969 | 21355682 | 15504 | 14679975 |
| CAP3 | SeqMan | 24727 | 16733217 | 29969 | 21355682 | 15387 | 14824287 |
| CLC | MIRA | 22746 | 14875522 | 35827 | 21339704 | 15334 | 14357031 |
| CAP3 | Newbler 2.5 | 24727 | 16733217 | 21734 | 20066883 | 14275 | 14830304 |
| CAP3 | CLC | 24727 | 16733217 | 22746 | 14875522 | 14149 | 13753398 |
| Newbler 2.3 | Newbler 2.5 | 12019 | 14456476 | 21734 | 20066883 | 9733 | 13252303 |
| MIRA | Newbler 2.3 | 35827 | 21339704 | 12019 | 14456476 | 9380 | 11731374 |
| CLC | Newbler 2.3 | 22746 | 14875522 | 12019 | 14456476 | 8884 | 12318589 |
| CAP3 | Newbler 2.3 | 24727 | 16733217 | 12019 | 14456476 | 8484 | 11426423 |
| Newbler 2.3 | SeqMan | 12019 | 14456476 | 29969 | 21355682 | 8274 | 11452990 |
These robust contigs also performed better in the tests assessing representation of reference datasets (Table 10 and Table 11). This was especially true for matches filtered to include only those covering at least 80% of the reference sequence. The co-assemblies of [CLC and MIRA] and [MIRA and Newbler 2.5] matched the most EST clusters. The [CLC and Newbler 2.5] combined assembly matched the most B. malayi peptides.
Table 10.
Alignments to 1602 EST clusters where > 80% of the EST was covered by a match, by pairs of assemblies merged using an OLC assembler
| Assembly pair | % EST clusters hit | % EST bases covered | |
|---|---|---|---|
| CLC | MIRA | 65.4 | 65.1 |
| MIRA | Newbler 2.5 | 65.4 | 65.3 |
| MIRA | SeqMan | 64.9 | 64.6 |
| CLC | Newbler 2.5 | 64.7 | 64.6 |
| Newbler 2.5 | SeqMan | 63.2 | 62.9 |
| CAP3 | Newbler 2.5 | 63.0 | 62.6 |
| CAP3 | CLC | 62.9 | 62.4 |
| CLC | SeqMan | 62.9 | 62.5 |
| CLC | Newbler 2.3 | 62.4 | 62.5 |
| CAP3 | MIRA | 62.2 | 61.9 |
| CAP3 | SeqMan | 61.7 | 61.3 |
| MIRA | Newbler 2.3 | 61.7 | 61.9 |
| Newbler 2.3 | Newbler 2.5 | 61.4 | 61.5 |
| Newbler 2.3 | SeqMan | 60.0 | 60.2 |
| CAP3 | Newbler 2.3 | 59.7 | 59.6 |
Table 11.
Alignments to 11,472 B. malayi peptides using BLASTX (e value < 1e-5) where > 80% of the protein was covered by a match, by pairs of assemblies merged using an OLC assembler
| Assembly Pair | % Peptides hit | % Peptide bases covered | |
|---|---|---|---|
| CLC | Newbler 2.5 | 38.2 | 28.3 |
| MIRA | Newbler 2.5 | 37.3 | 27.4 |
| CLC | MIRA | 37.1 | 27.1 |
| CLC | Newbler 2.3 | 36.5 | 27.4 |
| Newbler 2.5 | SeqMan | 36.5 | 26.8 |
| CAP3 | Newbler 2.5 | 36.4 | 26.9 |
| CAP3 | CLC | 36.1 | 26.3 |
| CLC | SeqMan | 35.9 | 26.1 |
| MIRA | SeqMan | 35.7 | 25.7 |
| Newbler 2.3 | Newbler 2.5 | 35.3 | 26.3 |
| CAP3 | SeqMan | 35.1 | 25.2 |
| Newbler 2.3 | SeqMan | 34.4 | 25.6 |
| CAP3 | MIRA | 34.0 | 24.2 |
| CAP3 | Newbler 2.3 | 34.0 | 25.1 |
| MIRA | Newbler 2.3 | 33.3 | 24.0 |
Although there was no consistent pattern indicating a clear superiority of any pair of assemblies (other than the exclusion of the Newbler 2.3 assembly), these merged assemblies show that it is possible to get more long, robust contigs that align better to reference sequences by combining the outputs of two different assemblies. Co-assembling three primary assemblies at a time and considering only the contigs that had 'reads' from all three assemblies gave smaller total contig numbers than pairwise co-assemblies. The number of stringent matches to reference sequences went up by less than 1% in all cases (see Additional File 2, Tables S2-S7), suggesting that the additional effort involved in generating and combining these assemblies may not be worthwhile.
Summary
We compared six assemblers by aligning their assembly contigs to four reference sequence sets. The Newbler 2.3 assembly scored worst in each comparison, probably because it had the smallest span of all the assemblies. Under stricter comparison parameters, the CLC assembly was poorer, suggesting that this assembly is more fragmented than the others. Although no single assembler was optimal in every case, Newbler 2.5 and SeqMan had the best alignments to related reference sequences overall. However, SeqMan generated over 8000 contigs extra for approximately the same total number of assembled bases (21.4 Mb versus 20.1 Mb).
We were able to improve the assembly by merging two assemblies at a time using a traditional OLC assembler (CAP3). This approach generated more high quality alignments to our reference sets.
Discussion and Conclusions
We compared six programmes (Table 2) for the task of de novo assembly of transcriptome data from an organism with little or no previous genomic resources. We tested each using default or minimally adjusted parameters, as has been the common practice in previous 454 pyrosequencing transcriptome studies (Table 1). It may be that it is possible to generate better assemblies for particular datasets by exploring the parameter space for each assembler. However, as sequencing becomes cheaper and more accessible, the vast majority of 454 transcriptome assemblies will probably be done by researchers who are new to assembly and just want something that works with a few sensible default parameters. Of the six de novo transcriptome assemblers tested, Newbler 2.5 had the best contig length metrics for our data, and, along with SeqMan, had the best alignments to related reference sequences.
Each assembler has certain advantages and disadvantages that are presented in more detail in the Supplementary Materials (Additional file 1). Versions 2.3 and 2.5 of Newbler were the only assemblers that explicitly attempted to reconstruct and group alternative transcripts and isoforms. If only one assembler had to be used because of time or resource constraints, we would currently recommend Newbler 2.5 overall. However, none of the other assemblers are deprecated apart from Newbler 2.3. We strongly recommend redoing transcriptome assemblies that were performed with Newbler 2.3 or earlier versions, if the goal is to get as complete an assembly as possible.
It is possible that different raw data sets may be better assembled by different programs (e.g. MIRA may be better for normalized transcriptome sequence data, and CAP3 for paired transcriptome data), and thus researchers should perhaps cross-compare the best available ones on their data using the optimality criteria used here.
Merging assemblies performed with different programs is a frequently used approach in genome assembly projects, especially those that employ multiple sequencing technologies. Application of this strategy to the problem of de novo transcriptome assembly appears particularly useful. While a merged assembly may simply sum the errors made by both programs, filtering the resultant second stage contigs on the basis of their containing first stage contigs from both of the starting sets generates a transcriptome assembly that is on average longer and that better represents known or assumed reference sequences than do either of the starting contig sets. A preferred assembly strategy is thus to perform initial assemblies with multiple high-quality assemblers and then to merge these using a traditional OLC assembler such as CAP3. Second-order contigs that have support from multiple first-order assemblies are much more likely to be accurate.
Need for new assemblers
All the de novo assemblers in this study, with the exception of CLC, use the OLC assembly strategy. CLC uses de Bruijn graphs. As read lengths and throughput increase and sequencing costs come down, an average transcriptome assembly project for a non-model organism may comprise several million 800 base reads. Under the OLC paradigm, the computational time for assembling more reads rises exponentially with data complexity because the number of pairwise comparisons to detect overlaps will increase, and the layout graphs will be harder to resolve into a consensus sequence. The CLC de novo assembler is clearly a step in the right direction because its de Bruijn graph algorithm achieves reasonable results in very little time on large datasets. Other de Bruijn graph assemblers such as Oases [46] and ABySS [55] are promising but are currently not suitable for most non-normalised 454 pyrosequencing de novo transcriptome assembly projects because many transcripts have very low coverage that cannot be assembled reliably. Perhaps the way forward is to use the de Bruijn graph method for transcripts with high coverage and the OLC method for transcripts with low coverage. We have shown that an assembly-merging strategy delivers robust contigs from intermediate assemblies produced by current programs, and this strategy is likely to be of utility in deriving the best assemblies from future programs as well.
Methods
Library preparation
The L. sigmodontis samples were prepared by Stella Konstantinou, Eleana Theophilou and Simon Babayan, and sequenced by the GenePool, Edinburgh. Briefly, total RNA from three L. sigmodontis samples (adult male, adult female, microfilaria) was converted to double stranded cDNA using Evrogen's MINT cDNA synthesis kit. First strand cDNA was synthesised using reverse transcriptase (RT) from a 3'-primer with oligo(dT) sequence that annealed to the poly-A stretch of RNA and synthesised cDNA until the 5' end of the mRNA. Finally, double stranded cDNA synthesis was performed using PCR amplification, and the final product contained the same MINT adapter sequence at both 3' and 5' ends. The cDNA was fragmented, size-selected, library-prepped, and sequenced according to standard Roche-454 FLX and Titanium protocols. Sequence files have been deposited into the Sequence Read Archive (SRA) with accession number ERA011678 http://www.ebi.ac.uk/ena/data/view/ERA011678.
Read pre-processing
We chose to remove MINT adapters from the sequences ourselves rather than use the built-in adapter removal tools in MIRA, Newbler, SeqMan, and CLC, because each assembler pre-processes reads slightly differently and CAP3 does not remove adapters at all. The Roche 454 instrument outputs sequences in Standard Flowgram Format (SFF) files that come with quality and clipping information. To ensure that each assembler got exactly the same input, we changed the trim points directly in the SFF file to exclude adapter sequence rather than first convert the SFF files to fasta and quality files. The 29 base MINT adapter sequence was identified using BLASTN [56] from NCBI's new blast+ suite (Version 2.2.23) and by masking all matches with a bit-score greater than an empirically determined threshold of 25. The longest unmasked portion of the sequence was used to set new trim points in the SFF file.
Most EST pipelines remove poly(A/T) regions from reads because aligners would not be able to find good matches back to genomes that do not have the poly(A/T) sequence, and assemblers might try to align on these sequences. The former reason was not a factor for us as this was an unsequenced organism, but we did a trial run both with and without the poly(A/T) sequence and found no misassemblies on these regions by any of the assemblers. Newbler and MIRA account for such sequences when run in the -cdna and EST modes respectively. SeqMan and CAP3 remove low quality ends (homopolymer runs of poly(A/T) sequence have lower qualities in 454 sequencing), and CLC uses a de Bruijn graph which would not allow misassemblies because it would not be able to resolve the branching structure in a poly(A/T) region. Poly(A/T) sequence might also be indistinguishable from genomic sequences in some cases and therefore should not be removed. We found that the total amount of assembled sequence was about 10% lower for assemblies without poly(A/T) sequence. Therefore, we did not trim the poly(A/T) regions in the sequences. An additional advantage of leaving the poly(A/T) sequence in is that assembled contigs with a poly-A at the end (or poly-T at the start) indicate that the assembly end point is almost certainly correct. In our sequences, a poly(A/T) sequence typically had an N about half-way through because a degenerate oligo-dT primer was used to extract the poly-A tails from the cDNA. Any sequences with more than one N were discarded.
Assembly parameters
For each assembler, the version and parameters used are described in this section. CLC, Newbler, and SeqMan used the trimmed SFF files directly. CAP3 and MIRA used fasta and quality files extracted from the SFF files using Roche 454's sffinfo utility. All the assemblers returned a fasta file with contigs, and, except CLC, they all returned an ACE file with read placement information for each contig. All assemblers were run on an 8-core 3.0 GHz Linux workstation with 32 GB of memory, with the exception of SeqMan NGen, which currently runs only on Mac and Windows operating systems. SeqMan NGen was run on a dual core 2.53 GHz Mac mini server with 4 GB of memory.
The CAP3 binary for 64-bit Intel Linux systems was downloaded on June 20, 2010 from http://seq.cs.iastate.edu/cap3.html. The version was not specified in this download, and we did not change any of the default settings.
The CLC Assembly Cell command line application (version 3.02) has only one assembly-specific parameter, the minimum contig length reported, which we set to 100. It converts Roche 454 .sff files to fasta format, strips out all read identifiers, performs the assembly, and returns only a fasta file with contigs. As it does not keep track of reads, we had to use the CLC aligner, also known as the Reference Assembly application, to perform the additional step of mapping all the input reads back to the assembly contigs using default settings.
MIRA version 3.0.0 (production version) was used with the recommended quick switches for a Roche 454 EST assembly: -job = denovo,est,accurate,454. We used the -notraceinfo option because the traceinfo file provides clipping information, whereas we had already used the clipping information to create trimmed fasta and quality files. We also used -GE:not = 8 which is a general option for specifying the number of threads that should be used for steps that can use multiple cores. MIRA's final contig output file includes singletons but we looked for and removed all contigs with the "_s" prefix ("_c" indicates contigs with more than one read).
The latest releases of Roche 454's Newbler (version 2.3, 091027_1459, and version 2.5, newbler v2.5p1-internal-10Jun23-1) provide a "-cdna" option to assemble transcriptomes. In transcriptome assembly, the assembler frequently fails without the -cdna option because it expects approximately even coverage in genome assembly mode. The only other parameters we used were "-ace" (to generate an ACE file at the end) and "-cpu 8" to use all available cores. Like most other OLC assemblers, Newbler stops extending contigs when it cannot resolve branches in the overlap layout graph. Unlike other assemblers, Newbler then tries to create isotigs out of contigs that are consistently connected by a subset of reads. Each isotig corresponds to an alternative transcript, and any contigs or isotigs that share any read overlaps are put into the same isogroup. We noticed that several contig fragments in the Newbler 2.3 ACE file were not reported in the final assembly fasta file (454Isotigs.fna). Initially, the Newbler developers said that these contigs are not parts of actual transcripts and are therefore not reported, but we traced a few of these fragments and found that they were made up of well aligned, overlapping reads that had been split into contigs and then discarded. The developers have since modified Newbler and allowed us to try the pre-release version on our data that addresses this issue, using the -urt flag that allows contigs to be bridged across regions with only single-depth coverage.
DNAStar's SeqMan Ngen version 2 for Mac has a GUI that calls an underlying script. We chose the de novo transcriptome assembly option for 454 unpaired reads. When we tried a run with default settings and no quality or vector trimming, the assembly completed without any error messages but reported only 891 contigs. Subsequently we tried an assembly with quality trimming enabled which completed in 6 hours and reported a 21 Mb assembly.
Assembly read alignment
To get assembly stats on the number of reads used, we used SSAHA2 [48] and CLC's aligner to map all the input reads back to each assembly's contigs (as recommended in a comparison of next-generation aligners [49]). SSAHA2 was used with the -454 option, and the clc_ref_assemble_long binary from CLC's Assembly Cell distribution version 3.00.44070-beta2 was used with default settings.
Abbreviations
OLC: Overlap Layout Consensus; EST: Expressed Sequence Tag; kb: kilo basepairs; Mb: mega basepairs; MB: mega bytes; GB: giga bytes; KOG: euKaryotic Orthologous Group; HSP: High Scoring Pair, a query sequence can have only one hit to a target sequence in a blast database, but each hit can be made up of many HSPs
Competing interests
The authors declare that they have no competing interests. Although we have used commercial software in this study supplied by the manufacturers, they had no influence on the experiments performed nor on the results reported here.
Authors' contributions
SK and MB conceived and designed the project. Analyses were carried out by SK and reviewed by MB. SK and MB wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Supplementary Materials. Figure S1: Trimmed read length histograms. A section describing the advantages and disadvantages of each assembler.
Results of merging three assemblies. Assembly metrics as a result of merging three first-order assemblies. Alignments of merged assemblies to reference sequences.
Contributor Information
Sujai Kumar, Email: sujai.kumar@ed.ac.uk.
Mark L Blaxter, Email: mark.blaxter@ed.ac.uk.
Acknowledgements
We thank Stella Constantinou and Eleana Theophilou, both Masters students in the Blaxter lab, for generation of the cDNA in collaboration with Simon Babayan, and Judith Allen for access to the L. sigmodontis lifecycle. Roche 454 sequencing was carried out by Anna Montazam and Denis Cleven of the GenePool http://genepool.bio.ed.ac.uk. The GenePool bioinformatics staff (especially Stephen Bridgett) assisted with sample quality checking, and with advice and tips on using Newbler. We also thank the Newbler development team for early access to Newbler 2.5, Alexie Papanicolau for advice on comparing assemblies, Ben Elsworth for NEMBASE4 Tribes, and Graham Thomas for helping install and test assemblers. SK holds a University of Edinburgh School of Biological Sciences graduate scholarship. The sequencing data were generated by funding from the EU project SCOOT, PI Judith Allen.
References
- Weber AP, Weber KL, Carr K, Wilkerson C, Ohlrogge JB. Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiology. 2007;144:32–42. doi: 10.1104/pp.107.096677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall PK, Leebens-Mack J, Chanderbali AS, Barakat A, Wolcott E, Liang H, Landherr L, Tomsho LP, Hu Y, Carlson JE, Ma H, Schuster SC, Soltis DE, Soltis PS, Altman N, DePamphilis CW. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics. 2009;10:347. doi: 10.1186/1471-2164-10-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaes E, Drost DR, Farmerie WG, Pappas GJ, Grattapaglia D, Sederoff RR, Kirst M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat A, DiLoreto DS, Zhang Y, Smith C, Baier K, Powell WA, Wheeler N, Sederoff R, Carlson JE. Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biology. 2009;9:51. doi: 10.1186/1471-2229-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn DA, Ragland GJ, Shoemaker DD, Denlinger DL. Gene discovery using massively parallel pyrosequencing to develop ESTs for the flesh fly Sarcophaga crassipalpis. BMC Genomics. 2009;10:234. doi: 10.1186/1471-2164-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, Colbourne JK, Willis BL, Matz MV. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009;10:219. doi: 10.1186/1471-2164-10-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Arreguín JC, Ibarra-Laclette E, Jiménez-Moraila B, Martínez O, Vielle-Calzada JP, Herrera-Estrella L, Herrera-Estrella A. Deep sampling of the Palomero maize transcriptome by a high throughput strategy of pyrosequencing. BMC Genomics. 2009;10:299. doi: 10.1186/1471-2164-10-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellin D, Ferrarini A, Chimento A, Kaiser O, Levenkova N, Bouffard P, Delledonne M. Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC Genomics. 2009;10:555. doi: 10.1186/1471-2164-10-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz D, Robertson HM, Feder JL, Varala K, Hudson ME, Ragland GJ, Hahn DA, Berlocher SH. Sympatric ecological speciation meets pyrosequencing: sampling the transcriptome of the apple maggot Rhagoletis pomonella. BMC Genomics. 2009;10:633. doi: 10.1186/1471-2164-10-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson L, Fai LS, Chamberlain N, Nadeau N, Joron M, Baxter S, Wilkinson P, Papanicolaou A, Kumar S, Kee T, Clark R, Davidson C, Glithero R, Beasley H, Vogel H, Ffrench-Constant R, Jiggins C. Characterization of a hotspot for mimicry: assembly of a butterfly wing transcriptome to genomic sequence at the HmYb/Sb locus. Molecular Ecology. 2010;19:240–254. doi: 10.1111/j.1365-294X.2009.04475.x. [DOI] [PubMed] [Google Scholar]
- Papanicolaou A, Stierli R, Ffrench-Constant RH, Heckel DG. Next generation transcriptomes for next generation genomes using est2assembly. BMC Bioinformatics. 2009;10:447. doi: 10.1186/1471-2105-10-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Li Y, Wu Q, Luo H, Sun Y, Song J, Lui EM, Chen S. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics. 2010;11:262. doi: 10.1186/1471-2164-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid J, Müller-Hagen D, Bekel T, Funk L, Stahl U, Sieber V, Meyer V. Transcriptome sequencing and comparative transcriptome analysis of the scleroglucan producer Sclerotium rolfsii. BMC Genomics. 2010;11:329. doi: 10.1186/1471-2164-11-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MS, Thorne MA, Vieira FA, Cardoso JC, Power DM, Peck LS. Insights into shell deposition in the Antarctic bivalve Laternula elliptica: gene discovery in the mantle transcriptome using 454 pyrosequencing. BMC Genomics. 2010;11:362. doi: 10.1186/1471-2164-11-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrich SJ, Barbazuk WB, Li L, Schnable PS. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Research. 2007;17:69–73. doi: 10.1101/gr.5145806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveland AL, McCarty DR, Koch KE. Transcript profiling by 3'-untranslated region sequencing resolves expression of gene families. Plant Physiology. 2008;146:32–44. doi: 10.1104/pp.107.108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan JR, Epp LG, Putta S, Page RB, Walker JA, Beachy CK, Zhu W, Pao GM, Verma IM, Hunter T, Bryant SV, Gardiner DM, Harkins TT, Voss SR. Microarray and cDNA sequence analysis of transcription during nerve-dependent limb regeneration. BMC Biology. 2009;7:1. doi: 10.1186/1741-7007-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guffanti A, Iacono M, Pelucchi P, Kim N, Soldà G, Croft LJ, Taft RJ, Rizzi E, Askarian-Amiri M, Bonnal RJ, Callari M, Mignone F, Pesole G, Bertalot G, Bernardi LR, Albertini A, Lee C, Mattick JS, Zucchi I, De Bellis G. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163. doi: 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang Y, Zhang Q, Qi Y, Guo D. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genomics. 2009;10:465. doi: 10.1186/1471-2164-10-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund C, Johnson E, Bomhoff M, Descour A. PAVE: program for assembling and viewing ESTs. BMC Genomics. 2009;10:400. doi: 10.1186/1471-2164-10-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng S, Xiao G, Guo J, Fei Z, Xu Y, Roe BA, Wang Y. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics. 2010;11:94. doi: 10.1186/1471-2164-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantacessi C, Campbell BE, Young ND, Jex AR, Hall RS, Presidente PJ, Zawadzki JL, Zhong W, Aleman B, Loukas A, Sternberg PW, Gasser RB. Differences in transcription between free-living and CO2-activated third-stage larvae of Haemonchus contortus. BMC Genomics. 2010;11:266. doi: 10.1186/1471-2164-11-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Guo H, Zheng H, Zhou T, Zhou Y, Wang S, Fang R, Qian W, Chen X. Massively parallel pyrosequencing-based transcriptome analyses of small brown planthopper (Laodelphax striatellus), a vector insect transmitting rice stripe virus (RSV) BMC Genomics. 2010;11:303. doi: 10.1186/1471-2164-11-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timme RE, Delwiche CF. Uncovering the evolutionary origin of plant molecular processes: comparison of Coleochaete (Coleochaetales) and Spirogyra (Zygnematales) transcriptomes. BMC Plant Biology. 2010;10:96. doi: 10.1186/1471-2229-10-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang H, Wong YH, Voolstra C, Ravasi T, Bajic BV, Qian P. Rapid transcriptome and proteome profiling of a non-model marine invertebrate, Bugula neritina. Proteomics. 2010;16:2972–2981. doi: 10.1002/pmic.201000056. [DOI] [PubMed] [Google Scholar]
- Barker MS, Dlugosch KM, Reddy AC, Amyotte SN, Rieseberg LH. SCARF: maximizing next-generation EST assemblies for evolutionary and population genomic analyses. Bioinformatics. 2009;25:535–536. doi: 10.1093/bioinformatics/btp011. [DOI] [PubMed] [Google Scholar]
- Pauchet Y, Wilkinson P, van Munster M, Augustin S, Pauron D, Ffrench-Constant RH. Pyrosequencing of the midgut transcriptome of the poplar leaf beetle Chrysomela tremulae reveals new gene families in Coleoptera. Insect Biochemistry and Molecular Biology. 2009;39:403–13. doi: 10.1016/j.ibmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Roeding F, Borner J, Kube M, Klages S, Reinhardt R, Burmester T. A 454 sequencing approach for large scale phylogenomic analysis of the common emperor scorpion (Pandinus imperator) Molecular Phylogenetics and Evolution. 2009;53:826–34. doi: 10.1016/j.ympev.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Zagrobelny M, Scheibye-Alsing K, Jensen NB, Møller BL, Gorodkin J, Bak S. 454 pyrosequencing based transcriptome analysis of Zygaena filipendulae with focus on genes involved in biosynthesis of cyanogenic glucosides. BMC Genomics. 2009;10:574. doi: 10.1186/1471-2164-10-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauchet Y, Wilkinson P, Vogel H, Nelson DR, Reynolds SE, Heckel DG, Ffrench-Constant RH. Pyrosequencing the Manduca sexta larval midgut transcriptome: messages for digestion, detoxification and defence. Insect Molecular Biology. 2010;19:61–75. doi: 10.1111/j.1365-2583.2009.00936.x. [DOI] [PubMed] [Google Scholar]
- Cheung F, Win J, Lang JM, Hamilton J, Vuong H, Leach JE, Kamoun S, André Lévesque C, Tisserat N, Buell CR. Analysis of the Pythium ultimum transcriptome using Sanger and Pyrosequencing approaches. BMC Genomics. 2008;9:542. doi: 10.1186/1471-2164-9-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansson E, Asker N, Förlin L, Larsson DG. Characterization of the Zoarces viviparus liver transcriptome using massively parallel pyrosequencing. BMC Genomics. 2009;10:345. doi: 10.1186/1471-2164-10-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung F, Haas BJ, Goldberg SM, May GD, Xiao Y, Town CD. Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics. 2006;7:272. doi: 10.1186/1471-2164-7-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Molecular Ecology. 2008;17:1636–47. doi: 10.1111/j.1365-294X.2008.03666.x. [DOI] [PubMed] [Google Scholar]
- Guerrero FD, Dowd SE, Djikeng A, Wiley G, Macmil S, Saldivar L, Najar F, Roe BA. A database of expressed genes from Cochliomyia hominivorax (Diptera: Calliphoridae) Journal of Medical Entomology. 2009;46:1109–16. doi: 10.1603/033.046.0518. [DOI] [PubMed] [Google Scholar]
- Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11:180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Research. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RT, Christoffels AG, Gopalakrishnan C, Burke J, Ptitsyn AA, Broveak TR, Hide WA. A Comprehensive Approach to Clustering of Expressed Human Gene Sequence: The Sequence Tag Alignment and Consensus Knowledge Base. Genome Research. 1999;9:1143–1155. doi: 10.1101/gr.9.11.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB) 99. 1999. pp. 45–56.
- Swindell SR, Plasterer TN. Sequence Data Analysis Guidebook. Vol. 70. Humana Press; 1997. SEQMAN; pp. 75–89. full_text. [Google Scholar]
- CLC bio: CLC Assembly Cell User Manual. http://www.clcbio.com/index.php?id=1393
- Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman S, Birol I. Assembling genomes using short-read sequencing technology. Genome Biology. 2010;11:202. doi: 10.1186/gb-2010-11-1-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oases: De novo transcriptome assembler for very short reads. http://www.ebi.ac.uk/~zerbino/oases/
- Allen JE, Adjei O, Bain O, Hoerauf A, Hoffmann WH, Makepeace BL, Schulz-Key H, Tanya VN, Trees AJ, Wanji S, Taylor DW. Of mice, cattle, and humans: the immunology and treatment of river blindness. PLoS Neglected Tropical Diseases. 2008;2:e217. doi: 10.1371/journal.pntd.0000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Cox A, Mullikin J. SSAHA: a fast search method for large DNA databases. Genome Research. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri N, Schlötterer C. Mapping accuracy of short reads from massively parallel sequencing and the implications for quantitative expression profiling. PLoS One. 2009;4:e6323. doi: 10.1371/journal.pone.0006323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. Tablet--next generation sequence assembly visualization. Bioinformatics. 2010;26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Research. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra D, Angiuoli SV, Creasy T, Amedeo P, Haas B, El-Sayed NM, Wortman JR, Feldblyum T, Tallon L, Schatz M, Shumway M, Koo H, Salzberg SL, Schobel S, Pertea M, Pop M, White O, Barton GJ, Carlow CK, Crawford MJ, Daub J, Dimmic MW, Estes CF, Foster JM, Ganatra M, Gregory WF, Johnson NM, Jin J, Komuniecki R, Korf I, Kumar S, Laney S, Li B, Li W, Lindblom TH, Lustigman S, Ma D, Maina CV, Martin DM, McCarter JP, McReynolds L, Mitreva M, Nutman TB, Parkinson J, Peregrín-Alvarez JM, Poole C, Ren Q, Saunders L, Sluder AE, Smith K, Stanke M, Unnasch TR, Ware J, Wei AD, Weil G, Williams DJ, Zhang Y, Williams SA, Fraser-Liggett C, Slatko B, Blaxter ML, Scott AL. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317:1756–60. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Research. 2002;30:1575–84. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, Horsman DE, Connors JM, Gascoyne RD, Marra MA, Jones SJ. De novo transcriptome assembly with ABySS. Bioinformatics. 2009;25:2872–2877. doi: 10.1093/bioinformatics/btp367. [DOI] [PubMed] [Google Scholar]
- Altschul S, Madden T, Schäffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials. Figure S1: Trimmed read length histograms. A section describing the advantages and disadvantages of each assembler.
Results of merging three assemblies. Assembly metrics as a result of merging three first-order assemblies. Alignments of merged assemblies to reference sequences.