

Abstract
Numerous mutations in E3 ubiquitin ligase parkin were shown to associate with familial Parkinson's disease. Here we show that parkin binds arrestins, versatile regulators of cell signaling. Arrestin-parkin interaction was demonstrated by coimmuno-precipitation of endogenous proteins from brain tissue, and shown to be direct using purified proteins. Parkin binding enhances arrestin interactions with another E3 ubiquitin ligase, Mdm2, apparently by shifting arrestin conformational equilibrium to the basal state preferred by Mdm2. Although Mdm2 was reported to ubiquitinate arrestins, parkin-dependent increase in Mdm2 binding dramatically reduces the ubiquitination of both non-visual arrestins, basal and stimulated by receptor activation, without affecting receptor internalization. Several disease-associated parkin mutations differentially affect the stimulation of Mdm2 binding. All parkin mutants tested effectively suppress arrestin ubiquitination, suggesting that bound parkin shields arrestin lysines targeted by Mdm2. Parkin binding to arrestins along with its effects on arrestin interaction with Mdm2 and ubiquitination is a novel function of this protein with implications for Parkinson's disease pathology.
Keywords: arrestin, parkin, Mdm2, ubiquitin ligase, GPCR, ubiquitination
Identification of several genes associated with the familial form of Parkinsons's disease (PD) provided a tremendous momentum for the studies of the mechanisms of neurodegeneration in this disorder. Parkin is a gene associated with autosomal recessive juvenile parkinsonism (1). Loss-of-function mutations in parkin are the most frequent cause of the autosomal recessive familial PD and are often found in younger patients with apparently sporadic PD (2). Compound heterozygous mutations in parkin are also common and appear to be a susceptibility factor for the late-onset PD (2-5). Some missense parkin mutants may act as gain-of-function or dominant-negative proteins exerting neurotoxic effect (6, 7). Functionally, parkin is an E3 ubiquitin ligase (8). Dysfunctions in the ubiquitin-proteasome system (UBS) appear to play a prominent role in the PD-related neurodegeneration (9). Parkin demonstrates neuroprotective activity in vitro and in vivo (10), although the underlying mechanisms are unclear. Several parkin substrates were tentatively identified, but the authenticity and relevance to neurodegeneration in PD of some substrates remains controversial (11). Recently, parkin has been shown to exert neuroprotective influence via its interaction with signaling proteins rather than through the UBS (12, 13), although these functions are by no means mutually exclusive. A handful of proteins have been described that interact with parkin and modulate its activity (14-17). An interesting feature of the parkin-linked PD is the absence, in most cases, of Lewy bodies (4, 5), suggesting that functional parkin is critical for Lewy body formation. The role of parkin in sporadic PD is supported by its inactivation by stress and its accumulation in insoluble inclusions in the brain of PD patients and rodents treated with dopaminergic neurotoxins (18-23). Improved understanding of parkin functions and mechanisms of parkin-related neurodegeneration will facilitate the development of novel treatments for familial and sporadic PD.
Arrestins are best known for their role in quenching G protein-mediated signaling via G protein-coupled receptors (GPCRs) (24, 25). They also orchestrate receptor internalization via coated pits and redirect signaling to alternative pathways (26, 27). Two non-visual subtypes, arrestin-21 and arrestin-3, are ubiquitously expressed in the brain. Arrestins interact with many partners, assembling multi-protein complexes and redistributing signaling proteins within the cell, thereby directing their activity towards specific targets (26, 28, 29). Arrestins modulate multiple signaling pathways critical for neuronal death and survival (30-34). The expression of specific arrestin subtypes dramatically changes during neuronal development (35, 36), in Parkinson's disease (37, 38), and in response to psychotropic drugs and drugs of abuse (39). Non-visual arrestins were reported to recruit E3 ubiquitin ligases Mdm2 (30) and AIP4 (40), although only the latter interaction was shown to be direct using purified proteins. Arrestin interaction with Nedd4, that ubiquitinates β2-adrenergic receptor (b2AR), remains controversial: arrestin-3 was reported to recruit Nedd4 to the receptor in one study (41), while in the other arrestin-domain containing protein ARRDC3, rather that arrestin-3, was shown to perform this function (42).
Here we identify parkin as arrestin interaction partner, demonstrating co-immunoprecipitation (co-IP) of endogenous proteins from mouse brain and direct binding of purified parkin and arrestins. We found that this interaction suppresses the ubiquitination of both non-visual arrestins without affecting receptor internalization. Our finding that parkin binding selectively enhances the interaction of another ubiquitin ligase, Mdm2, with arrestins suggests that these proteins play an unexpected role in complex functional interplay between different E3 ubiquitin ligases, which is likely involved in Parkinson's pathology.
Experimental Procedures
Antisera and reagents
Rabbit polyclonal antibodies against residues 357-418 of arrestin2 and 350-409 of arrestin3 (a generous gift of Dr. J. L. Benovic) were described previously (43, 44). Rabbit and mouse monoclonal anti-myc, mouse anti-HA, and rabbit anti-parkin antibodies were from Cell Signaling Technology (Beverly, MA). Rabbit and mouse anti-FLAG antibodies were from Sigma (St.Louis, MO). Rat monoclonal high affinity anti-HA antibody was from Roche Molecular Biochemicals, (Indianapolis, IN). Tissue culture media and reagents were from Invitrogen (Carlsbad, CA), restriction endonucleases from New England Biolabs (Ipswich, MA), and all other chemicals were from Sigma (St Louis, MO), unless otherwise specified.
Arrestin, parkin and receptor constructs
Expression constructs for wild type (WT) and mutant arrestins were described previously (29, 45, 46). Plasmids encoding HA- and myc-tagged WT parkin and selected mutants were generously provided by Drs. Ted Dawson (Johns Hopkins University, Baltimore, MD) and Konstanze Winklhofer (Ludwig-Maximilians-University, Munich, Germany), respectively (13, 47, 48). Other mutations were introduced by PCR and confirmed by dideoxy-sequencing. MBP-parkin E. coli expression construct (pMal-Parkin) was a generous gift of Dr. Noriyuki Matsuda (Tokyo Metropolitan Institute of Medical Science, Japan) (49). pMal-MBP was constructed by removing parkin coding sequence and replacing the first parkin codon with a stop codon (between BamHI and EcoRI sites). HA-tagged β2-adrenergic receptor and its chimera with V2 vasopressin receptor C-terminus (50) were generous gifts of Dr. M. G. Caron (Duke University).
Cell culture, transfection, and stimulation
HEK-293A and HeLa cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% FBS and 1% penicillin-streptomycin in a humidified incubator at 37 °C and 5% CO2. Lipofectamin2000 (Invitrogen, Carlsbad, CA) (1:2.5 DNA:lipid) in Opti-MEM was used to transfect cells. DNA amounts in each transfection were kept constant by the addition of empty vector. All experiments were conducted 48 h post-transfection.
Protein purification
WT arrestins-1, -2, and −3 and mutants with unique cysteines on cysteine-less background were purified by sequential heparin- and Q-Sepharose chromatography, as previously described (51). Cysteine-less arrestin-3 (C17S, C60V, C126S, C141L, C151V, C243V, C252V, C270S, and C409S) was created by introducing the same mutations that generated fully functional cysteine-less arrestin-2 (29, 52, 53) and replacing the only additional cysteine (Cys409) in the flexible C-tail with serine. This base mutant bound light-activated phosphorhodopsin as well as WT arrestin-3 (data not shown). MBP-parkin and control MBP were purified on maltose column, following described procedure (49). Purified His-tagged parkin was purchased from BostonBiochem (Boston, MA).
In vitro binding assays with purified proteins
MBP pull-down assay
Purified MBP-Parkin (15 μg), MBP (15 μg) or binding buffer (20 mM Hepes, pH 7.3, 150 mM NaCl) in 50 μl were incubated with 50 μl Amylose resin (50% slurry, BioLabs) at 4°C overnight with gentle agitation, then purified arrestin-3 (5 μg) was added and incubated at 4°C for another 3 h. Suspensions were transferred to centrifuge filters (Ultrafree, 5 μm, Millipore) and washed three times with the binding buffer. Bound proteins were eluted from Amylose resin by 100 μl of buffer containing 100 mM maltose, 20 mM Hepes-Na, pH 7.3, 150 mM NaCl. The eluted samples were analyzed by SDS-PAGE and Western blot.
Direct binding assay based on changes in fluorescence anisotropy
Arrestins containing unique cysteines on cysteine-less background (CL-arrestin-1-F79C, CL-arrestin-2-L33C, and CL-arrestin-3-F88C) were chemically modified with fluorescent label monobromobimane (Toronto Research Chemicals), as described for arrestin-1 (54). Bimane was selected because it has a relatively long lifetime of the active state (∼ 10 ns) (55), which makes it suitable for relatively large proteins and complexes in 45-140 kDa range. Arrestins were irradiated with polarized 380 nm light. G factor was determined and taken into account in calculations based on the following equation r=(IVV − GIVH)/(IVV + 2GIVH) (56). Measured G factor for arrestin-1, -2, and 3 was 0.33-0.38, 0.45-0.49, and 0.35-0.38, respectively. Based on Perrin equation, , where η is solvent viscosity, T is temperature, R is gas constant and V is molecular volume of the fluorescent dye or dye conjugate (56), calculated polarization and anisotropy for a 45 kDa protein with probe life time of 10 ns are ∼0.35 and 0.26, respectively. Average experimentally measured anisotropy of free arrestins was ∼0.32. The anisotropy of 470 nm light emitted by bimane-labeled arrestins (1 μM) in buffer (20 mM Tris, 100mM NaCl, pH 7.5) with and without MBP-Parkin (0.25-10 μM) was measured in a spectrofluorometer (Photon Technology International) equipped with polarizers. The data were fit by the equation:
where Y is the change of anisotropy; [A] is the concentration of bimane-labeled arrestin; [B] is the concentration of MBP-Parkin; Kd is the binding affinity between arrestin and MBP-Parkin.
Immunoprecipitation
Rat brain
Pierce co-immunoprecipitation kit with AminoLink coupled antibody (Thermo Fisher Scientific, Rockford, IL) was used for co-immunoprecipitation (co-IP) of native proteins from the rat brain. Mouse monoclonal anti-parkin PRK8 antibody (Sigma, St. Louis, MO) and control mouse IgG (Vector laboratories, Burlingame, CA) (50 μg) were coupled to the Aminolink resin following manufacturer's instructions. The rat cortex tissue (approximately 10 mg per reaction) was sonicated in Lysis buffer, lysed for 12 h at 4°C, and centrifuged 30 min at 4°C to remove cell debris. The supernatant was pre-cleared with control resin (provided with the kit) for 2 h at 4°C, and co-IP was carried out overnight at 4°C. The resin was washed 5 × 500 μl of Lysis buffer, and co-immunoprecipited proteins were eluted with 50 μl of Elution buffer. Immunoprecipitated parkin was detected by Western blot with rabbit antiparkin antibody (Cell Signaling, Danvers, MA), and arrestin-2 was detected with rabbit polyclonal anti-arrestin antibody described previously (43, 44).
Purified proteins
Anti-parkin (PRK8 antibody) and control antibodies (75 μg) were covalently attached to 0.1 ml of the matrix (Pierce, Rockford, IL) following manufacturer's instructions. Pure His-parkin (6 μg) was mixed with purified arrestin-2 or -3 (6 μg), incubated on ice for 15 min, and then immunoprecipitated overnight (14 h). The columns were washed and the proteins eluted according to manufacturer's instructions. Aliquots of input, parkin co-IP, and control co-IP were run on SDS-PAGE and detected by Western blot with rabbit anti-parkin, anti-arrestin-2, or anti-arrestin-3 antibodies, followed by HRP-conjugated anti-rabbit secondary antibodies and ECL WestPico reagent (Pierce, Rockford, IL).
Cultured cells
Cells were scraped off plates, collected by centrifugation in phosphate-buffered saline and resuspended in the immunoprecipitation buffer (IPB) containing 50 mM Tris.HCl, 2 mM EDTA, 250 mM NaCl, 10% (v/v) glycerol, 0.5% NP-40, 20 mM NaF, 1 mM sodium orthovanadate, and 10 mM N-ethylmaleimide. Benzamidine (2 mM final concentration) and phenylmethylsulfonyl fluoride (1 mM) were added immediately before use. Cells were lysed at 4°C for 1 h and centrifuged to remove the debris. The supernatant was pre-cleared by incubating with 25-30 μl of Protein G Agarose for 1 h at 4°C. Target proteins were then immunoprecipitated by incubating the supernatant overnight at 4°C with appropriate antibodies (1-2 μg per 60 mm Petri dish) and 20-25 μl of Protein G agarose. Beads were washed three times with IPB, and the proteins were eluted by boiling in Laemli SDS buffer for 5 min.
Receptor trafficking
HeLa cells were transfected with the chimeric β2/V2 receptor, either alone or in combination with arrestin-2 or -3, parkin, or both. Cells were serum-starved overnight 48 h post-transfection, and then incubated with isoproterenol (10 μM) for 1, 2 or 4 h in DMEM supplemented with 20 mM HEPES and 1 mM ascorbic acid to induce receptor internalization (control cells were exposed to the medium only). Upon completion of incubation, cells were washed 3 × 500 μl in ice-cold TBS, and incubated with 2nM [3H]CGP-12177 on ice for 3 h to label surface receptors, washed 3 × 500 μl with ice-cold TBS, and the amount of bound ligand was determined by scintillation counting.
Western blotting
The proteins were analyzed by reducing SDS-PAGE and Western blotting onto Immobilon-P (Millipore, Bedford, MA, USA) membrane. The membrane was blocked and then incubated in TBS supplemented with 0.1% Triton X-100 and 1% BSA and appropriate primary antibody. Blots were incubated with secondary antibodies coupled with horseradish peroxidase (Jackson Immunoresearch Laboratories, West Grove, PA, USA) and visualized by SuperSignal enhanced chemiluminescence reagent (Pierce, Rockford, IL, USA). The bands on the film were quantified using Versadoc 4 (Bio-Rad, Hercules, CA).
Statistical analysis
StatView (SAS Institute) software was used for statistical analysis of quantitative data. The data were analyzed by analysis of variance (ANOVA) to determine the effects of nominal independent variables (factors), or by analysis of co-variance (ANCOVA) (when continuous independent variables, or regressors, were involved such as parkin or arrestin-2 concentration) (57). In all cases, p<0.05 was considered significant.
Results
Parkin directly interacts with arrestins
Parkin is an E3 ubiquitin protein ligase (58) shown to interact with multiple partners (11). Arrestins participate in numerous signaling pathways and were reported to bind several E3 ubiquitin ligases (30, 40, 59). Both non-visual arrestins (35, 36) and parkin (11) are expressed at high levels in neurons. Therefore, we tested whether arrestins bind parkin in HEK-293A cells expressing HA-tagged WT parkin and myc-tagged arrestins and found that both non-visual arrestins co-IP with parkin (Fig. 1A). Next, we tested whether endogenous arrestins interact with parkin by co-IP from brain tissue. We found that anti-parkin antibody readily immunoprecipitates endogenous parkin, and that arrestin-2, the prevalent isoform that outnumbers arrestin-3 by ∼10-20-fold in the brain (36), is detectable in the immunoprecipitate (Fig. 1B). Recent demonstration that signaling proteins, including E3 ubiquitin ligase Mdm2, that were believed to bind exclusively non-visual arrestins, also interact with visual subtypes (29, 46, 52, 60, 61), prompted us to test arrestin-1 and -4 (formerly known as rod and cone arrestins, respectively). Parkin demonstrates comparable binding to visual and both non-visual subtypes and arrestin-2/3 chimeras (Fig.1C), suggesting that parkin likely interacts with all arrestins, including visual isoforms expressed at very high levels in photoreceptor cells (62-65).
Fig. 1. Parkin interacts with endogenous and expressed arrestins.
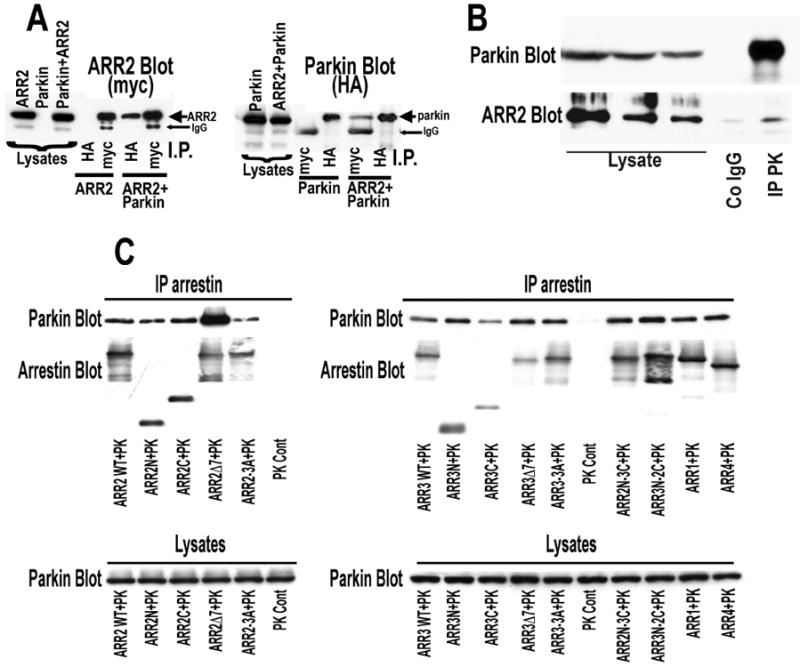
A. HEK293 cells were transfected with myc-tagged arrestin-2 (ARR2), HA-tagged parkin or both arrestin-2 and parkin. The proteins were immunoprecipitated with anti-HA or anti-myc antibodies as described in methods. Left panel: Western blot for myc detecting arrestin2 in cell lysates and IP products. Right panel: Western blot for HA detecting parkin in cell lysates and IP products. B. Endogenous arrestin-2 (prevalent isoform in the brain) immunoprecipitates with endogenous parkin from rat brain. Parkin (upper blot) and arrestin-2 (lower blot) detected in aliquots of rat cortical lysates and sample immunoprecipitated with anti-parkin antibody (IP PK), but not control IgG (Co IgG). C. Parkin interacts with multiple arrestin isoforms and mutants. Left panel: immunoprecipitation (IP) with anti-FLAG antibodies from HEK293 cells expressing myc-tagged parkin alone (control) or with FLAG-tagged WT and indicated mutant forms of arrestin-2. Right panel: IP with anti-FLAG antibodies from HEK293 cells expressing myc-tagged parkin alone (control) or with FLAG-tagged WT and mutant arrestin-3 (ARR3), and arrestin-2/3 chimeras containing arrestin-2 N-domain and arrestin-3 C-domain (ARR2N-3C), or vise versa (ARR3N-2C), arrestin-1 (ARR1), also known as rod arrestin, and arrestin-4 (ARR4) also known as cone arrestin. Even in the presence of protease inhibitor cocktail some WT and mutant arrestins appear to undergo partial proteolysis during IP generating faster running bands that are not observed in lysates.
However, co-IP shows that the proteins are in the same macromolecular complex, but cannot prove that they interact directly, rather than via unidentified intermediaries. This is particularly true for non-visual arrestins shown to co-IP with more than a hundred different proteins (66). Obviously, any of these partners could potentially “bridge” arrestin and parkin, localizing them to the same complex without the two actually touching each other. To test whether parkin binds arrestins directly, we used fully functional purified wild type (WT) arrestins (29, 67, 68) and two forms of parkin, with N-terminal His- and MBP-tag, both of which were shown to be catalytically active (49, 69). Purified arrestin-2 and −3 in the presence of purified His-parkin were retained by columns with covalently attached anti-parkin, but not control antibody (Fig. 2A). In addition, pure arrestin-3 was mixed with equal amounts of MBP-tagged parkin or purified MBP (control) and upon brief incubation on ice loaded onto amylose column. Non-specifically bound proteins were removed by three washes with isotonic binding buffer, and specifically bound proteins were then eluted with 100 mM maltose. Arrestin-3 was retained by the column via MBP-parkin, but not via control MBP (Fig. 2B). Thus, parkin interaction with arrestins is direct and does not require any additional proteins (Fig. 2), and this interaction is observed between endogenous proteins expressed at physiologically relevant levels (Fig. 1B).
Fig. 2. Parkin binds arrestins directly with micromolar affinity.
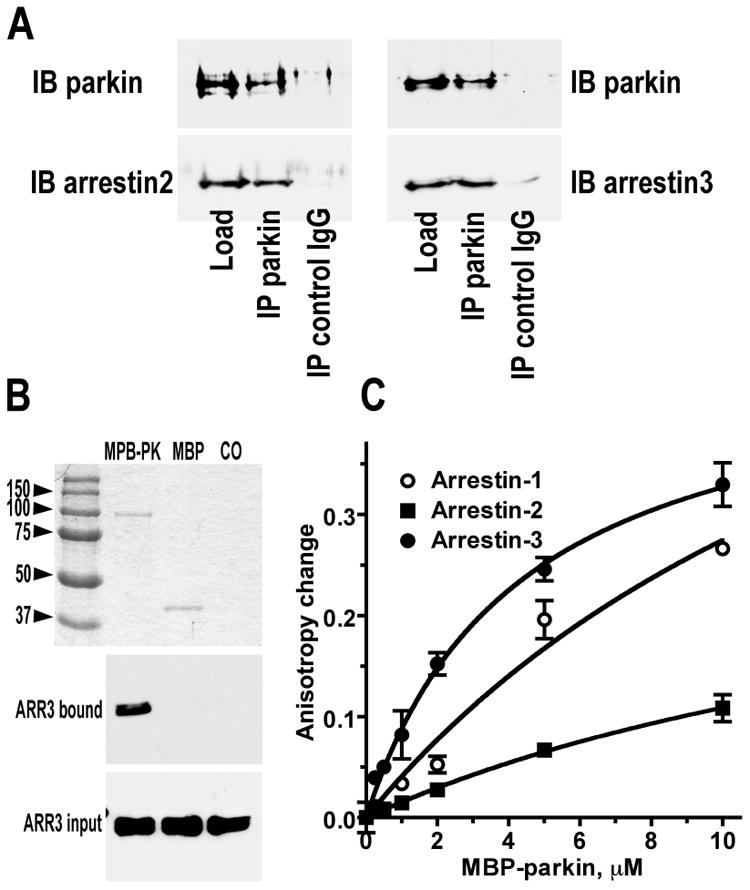
A. Purified His-parkin (6 μg) was mixed with 6 μg of purified arrestin-2 (left) or -3 (right) and immunoprecipitated overnight (14 h) with anti-parkin (IP parkin) and control (IP control IgG) antibodies (50 μg) covalently attached to 50 μl of the matrix. Aliquots of input and retained proteins were subjected to SDS-PAGE and detected by Western blot with rabbit anti-parkin (IB parkin), anti-arrestin-2 (IB arrestin2), or anti-arrestin-3 (IB arrestin3) antibodies. B. Purified MBP-Parkin (15 μg) (MBP-PK), MBP (15 μg) (MBP), or binding buffer (20 mM Hepes, pH 7.3, 150 mM NaCl)(CO) in 50 μl were loaded onto 50 μl Amylose resin and incubated with purified arrestin-3 (5 μg) at 4°C for 3 h. After three washes bound proteins were eluted with 100 μl of buffer containing 100 mM maltose. The eluted samples were analyzed by SDS-PAGE. Top panel shows Coomassie-stained gel with indicated eluted proteins, middle and bottom panels show Western blots of eluted arrestin-3 (ARR3) and aliquots of the input. C. Indicated purified arrestins (1 μM) labeled with bimane at unique cysteines, as described in methods, were irradiated with polarized 380 nm light. Anisotropy of emitted 470 nm light in the absence and presence of indicated concentrations of purified MBP-parkin is shown (means ± SD from ten measurements in representative experiments). The results of three independent experiments yield apparent KD of MBP-parkin binding to arrestin-1, -2, and -3 of 15.54±10.12 μM, 20.07±5.60 μM, and 3.36±1.86 μM, respectively.
Finally, we used yet another interaction assay with pure fluorescently labeled arrestins and MBP-parkin (Fig. 2C). This assay is based on the fact that upon excitation of fluorescent label with polarized light the polarization of the emitted light depends on the tumbling rate of the labeled molecule. Thus, larger complexes that tumble slower show greater anisotropy of emitted light. We labeled fully functional cysteine-less arrestin-1, -2, and -3 base mutants (29, 52, 61, 68, 70-72) with unique cysteines in selected positions with bimane. Bimane has relatively long lifetime of the activated state (∼10 ns) (55), which makes it a suitable reporter for larger proteins and complexes. Labeled arrestins were excited by polarized light and showed anisotropy of emitted light approximating that predicted for ∼45 kDa molecule (56). Anisotropy progressively increased with the addition of increasing concentrations of purified MBP-parkin (∼95 kDa (49)), reflecting the formation of much larger (∼140 kDa) arrestin-MBP-parkin complexes with slower tumbling rate (Fig 2C). In addition to confirming direct interaction by a third independent method, this approach allows one to determine the affinity of MBP-parkin for these arrestin subtypes. The data show that arrestin-3 has significantly higher affinity for parkin than arrestin-1 and -2, with apparent KD of 3.36±1.86 μM, 15.54±10.12 μM, and 20.07±5.60 μM, respectively (means ± SD of three independent experiments) (Fig. 2C). This difference in affinity and consequent rapid dissociation explains why we were unable to trap arrestin-1 and −2 in complex with MBP-parkin on amylose column that retained arrestin-3 (Fig. 2B).
Parkin engages both domains of arrestin proteins
Structurally, arrestins are elongated two-domain molecules (73-77), in which N- and C-domains are independent folding units that can be expressed (60) separately and retain certain functions (29, 60, 78-81). Many non-receptor interaction partners, including Mdm2 (46), tubulin (29), calmodulin (52), and MAP kinases ASK1, MKK4, JNK3, c-Raf1, MEK1, and ERK2 (81) engage both arrestin domains. Therefore, to determine the localization of parkin-binding arrestin elements, we tested its interactions with individually expressed N- and C-domains of arrestin-2 and -3. Both domains of arrestin-2 and -3 efficiently co-IP with parkin (Fig. 1C). Thus, similar to many other binding partners, parkin apparently engages both arrestin domains. Since the movement of the two domains relative to each other is a part of the conformational change in arrestin upon its binding to receptors (29, 82) and microtubules (29, 61), interaction with both domains is likely to make parkin binding sensitive to arrestin conformation. To test this idea, we used two types of previously characterized mutants: conformationally loose arrestin-3A mimicking receptor-bound state (45, 83-86), where the C-tail is detached by triple alanine substitution of residues anchoring it to the N-domain, and receptor binding-deficient Δ7 mutants frozen in the basal state, where the domain movement is blocked by the 7-residue deletion in the inter-domain hinge, preventing its transition into the active state and receptor binding (29, 82). The effect of these mutations was subtype-dependent: 3A somewhat decreased, while Δ7 mutation significantly increased parkin binding to arrestin-2, but neither had a dramatic effect on its interaction with arrestin-3 (Fig. 1C).
The binding sites of microtubules (29, 61) and calmodulin (52) are localized on the concave side of the two arrestin domains, overlapping with the receptor-binding site (53, 68, 87-90), which makes the interaction of these proteins and GPCRs with arrestins mutually exclusive (52, 91). In contrast, the majority of other binding partners interact with both free and receptor-bound arrestins, apparently engaging the non-receptor-binding side of the arrestin molecule (reviewed in (27, 92)). Therefore, we tested whether arrestin binding to the receptor affects its interaction with parkin. Both arrestin-2 and parkin co-immunoprecipitated with the inactive HA-tagged chimeric β2-adrenergic receptor with the C-terminus of vasopressin V2 receptor (b2V2), which was shown to form stable high-affinity complexes with arrestins (50) (Fig. 3A). Comparable amounts of arrestin were co-immunoprecipitated with the receptor in the presence and absence of parkin. In the reverse IP of arrestin-2, both the receptor and parkin were co-immunoprecipitated (Fig. 3A). Co-expression of parkin did not significantly affect the amount of receptor immunoprecipitated via arrestin (Fig. 3A). These data suggest that arrestin binding to the receptor or parkin does not appreciably affect the interaction with the other partner. Thus, parkin likely engages arrestin elements that are not shielded by bound receptor.
Fig. 3. Receptor and parkin do not compete for arrestin binding.
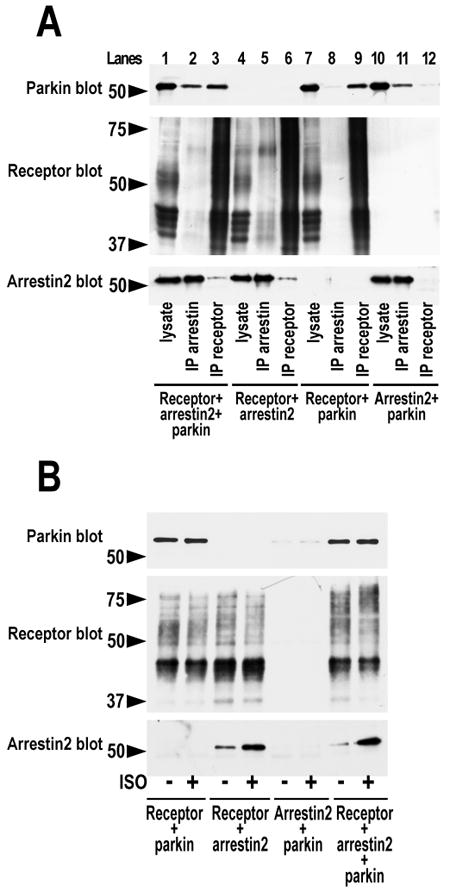
A. HEK293 cells were transfected with indicated combinations of HA-tagged chimeric β2-adrenergic with the C-terminus of V2 vasopressin receptor, FLAG-tagged arrestin-2, and myc-tagged parkin. Receptor or arrestin was immunoprecipitated with anti-HA (IP receptor) or anti-FLAG (IP arrestin) antibodies, respectively. Parkin (upper blot), receptor (middle blot) and arrestin (lower blot) in aliquots of lysates and immunoprecipitated samples were visualized with appropriate antibodies. B. In the same experimental design, before lysis the cells were incubated for 15 min at 37°C in control medium or with 10 μM of β2-adrenergic agonist isoproterenol (ISO). Receptor was immunoprecipitated with anti-HA antibody, and indicated proteins that co-IP with the receptor were visualized with appropriate antibodies. Whereas the amount of arrestin-2 co-immunoprecipitated with the receptor significantly increases upon receptor stimulation, the amount of parkin does not. The positions of molecular weight markers are indicated on the left. The results of a representative experiments out 2-3 performed are shown.
Interestingly, while the amount of receptor-bound arrestin is greatly increased upon receptor stimulation, the amount of associated parkin does not appreciably change (Fig. 3B). Moreover, parkin co-immunoprecipitates with the inactive and activated receptor even in the absence of overexpressed arrestin (Fig. 3A,B). Although one cannot exclude the role of endogenous arrestins in the latter case, these data suggest that parkin may interact with receptor in arrestin-independent fashion, either directly or via an alternative scaffold. The fact that the amount of receptor-associated parkin does not change with a several-fold increase of bound arrestin in the presence of agonist (Fig. 3B) also indicates that parkin may preferentially bind arrestins in their basal conformation, similar to Mdm2 (46, 60).
Parkin suppresses arrestin ubiquitination induced by receptor binding without affecting receptor internalization
Parkin is an E3 ubiquitin ligase, and both arrestin-2 and -3 were shown to undergo transient ubiquitination upon receptor binding (59). Therefore, we tested the effect of arrestin-parkin interaction on this process. Using HA-ubiquitin, we readily detected basal ubiquitination of both arrestin-2 and -3. Unexpectedly, we found that arrestin ubiquitination was dramatically suppressed by parkin co-expression (Fig. 4A,B,C). Using the b2V2 receptor, we confirmed previous report (59) that arrestin ubiquitination is enhanced by its binding to the agonist-activated receptor (Fig. 4A,B). Parkin co-expression reduced basal and receptor activation-dependent ubiquitination of arrestin-2 to about the same extent, by 70-75% (Fig. 4A,C). In contrast, parkin reduced basal ubiquitination of arrestin-3 by only ∼35%, whereas its receptor-dependent ubiquitination was suppressed more than 10-fold (Fig. 4B,C).
Fig. 4. Parkin dramatically reduces basal and receptor-stimulated ubiquitination of arrestins, but does not affect receptor internalization.
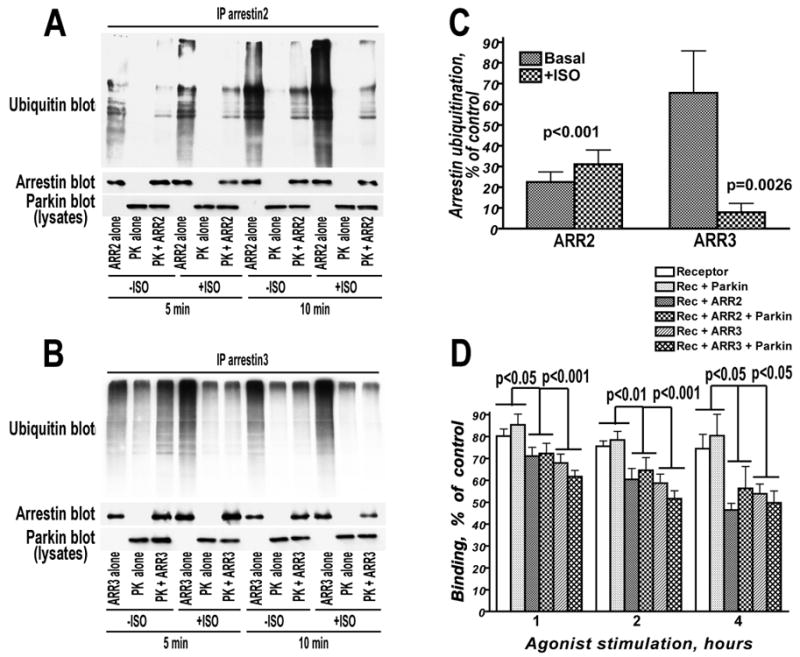
A, B. HEK293 cells expressing untagged chimeric β2-adrenergic receptor with the C-terminus of vasopressin V2 receptor were co-transfected HA-tagged ubiquitin and FLAG-tagged arrestin-2 (A) or arrestin-3 (B) with or without myc-tagged parkin, as indicated. The cells were treated for indicated time with or without 10 μM of β2-adrenergic agonist isoproterenol (ISO). In immunoprecipitated arrestin samples the amount of arrestin (middle blots) and its ubiquitination (upper blot) was determined by Western blot with appropriate antibody. The amounts of expressed parkin in cell lysates are shown in the lower blots. Note that because multiple forms of arrestin with different numbers of ubiquitin moieties are generated, the main band of non-ubiquitinated protein is by far the most prominent in arrestin blots. C. Quantification of parkin-dependent suppression of arrestin ubiquitination (panels A and B). The ratios of the amount of ubiquitinated arrestin determined with and without receptor activation in the presence or absence of parkin, expressed as % of control (no parkin) are shown. Representative results of one experiment (out of four performed) are shown in panels A and B. Statistical analysis of the data was performed by two-way repeated measure ANOVA with PARKIN as within group factor and ISO stimulation as a between group factor. The effect of PARKIN on the arrestin-2 ubiquitination was significant (F(1,6)=135.2, p<0.0001) as was the effect of ISO (F(1,6)=7.9, p=0.031), whereas the interaction was not significant, indicating that the degree of parkin-induced inhibition of arrestin-2 ubiquitination was independent of receptor stimulation. The effects of PARKIN and ISO on the arrestin-3 ubiquitination were also significant (F(1,6)=57.8, p=0.0003, and 6.67, p=0.042), as was PARKIN × ISO interaction (F(1,6)=18.8, p=0.0049), indicating that PARKIN inhibits activation-induced arrestin-3 ubiquitination significantly stronger. D. Parkin effect on the arrestin-dependent receptor trafficking. HeLa cells were transfected with the chimeric β2V2 receptor, alone or in combination with arrestin-2, arrestin-3, parkin, or both, as indicated. Serum-starved cells were incubated with 10 μM isoproterenol for indicated time. Control cells were incubated without agonist. Cells were then washed 3 × 500 μl in ice-cold TBS, and cell surface receptor was determined by measuring specific binding of cell-impermeable antagonist [3H]CGP-12177 (2nM). The graph shows means±S.E.M. of 6 (for 1 and 2 h) or 2 (for 4 h) independent experiments. Note that parkin co-expression does not affect receptor internalization at any time point. The data for each time point were statistically analyzed by two-way ANOVA with ARRESTIN and PARKIN as main factors. The effects of ARRESTIN were significant (F(1,30)=9.89, p=0.0005; 13.47, p<0.0001; and F(1,6)=9.41, p=0.014, for 1, 2, and 4 h, respectively) for all time points, whereas no significant effect of PARKIN or PARKIN × ARRESTIN interaction was detected. There was no significant difference between arrestin-2 and arrestin-3.
Arrestin ubiquitination was proposed to increase its affinity for the receptor, stabilizing the complex and facilitating receptor internalization (59). Therefore, we tested whether a dramatic suppression of receptor activation-induced arrestin ubiquitination by parkin affects receptor trafficking. To achieve maximum sensitivity of the assay, we expressed the b2V2 receptor at the level where co-expression of exogenous arrestin-2 or -3 significantly promoted internalization (Fig. 4D). We found that co-expression of parkin, which dramatically suppressed arrestin ubiquitination (Fig. 4A-C), had no appreciable effect on the b2V2 endocytosis mediated by arrestin-2 or −3 (Fig. 4D). These results suggest that arrestin ubiquitination does not play a significant role in the internalization of b2V2 receptor.
Two mechanisms are consistent with parkin-dependent reduction of arrestin ubiquitination: parkin could either compete with Mdm2, the ubiquitin ligase reported to target arrestins (59), or bound parkin could simply shield arrestin lysines to which Mdm2 attaches the ubiquitin moieties (93). In the first scenario, parkin would reduce Mdm2 association with arrestins, whereas in the second arrestin-Mdm2 interaction would not be affected.
Parkin promotes Mdm2 binding to arrestins
To test whether parkin alters the Mdm2 binding to arrestins, we used cells expressing HA-Mdm2, FLAG-arrestin-2 or -3, and varying concentrations of myc-parkin (Fig. 5). Unexpectedly, we found that parkin enhances arrestin-Mdm2 interaction. Parkin effect was dramatic in case of arrestin-2, but only modest in case of arrestin-3 (Fig. 5A,D). The amount of Mdm2 co-immunoprecipitated with arrestin-2 was increased by parkin up to 3–fold (Fig. 5D). Interestingly, parkin co-expression noticeably increases the level of Mdm2 in cells (Fig. 5A,D). This effect is observed with both non-visual arrestins, and its magnitude appears to correlate with the affinity of arrestin-parkin interaction: in the presence of arrestin-2 and -3 the amount of Mdm2 in cells increases ∼2- and ∼4-fold, respectively (compare Fig. 2C and Fig. 5A,D). Stabilization of cellular Mdm2 suggests that parkin likely promotes its binding to both non-visual arrestins, even though its effect on arrestin-2 was more consistent and therefore easier to detect (Fig. 5D). Since parkin interacts with both arrestin domains (Fig. 1), we tested whether observed differences in parkin effects on Mdm2 binding to arrestin-2 and −3 are mediated by parkin interaction with one particular domain. To this end, we constructed two chimeras with the N-domain of arrestin-2 and C-domain of arrestin-3 (arrestin-2N-3C), and vice versa (arrestin-3N-2C). We found that Mdm2 binding to arrestin-2N-3C was significantly enhanced with increasing parkin expression, whereas its binding to arrestin-3N-2C showed less consistent increase similar to that observed with arrestin-3 (Fig.5A-C,F). Mdm2 binding to separated arrestin-2 N-domain was also enhanced by parkin, although not as stringly as to the full-length arrestin-2, whereas no consistent effect was observed with separate arrestin-2 C-domain (Fig. 5E,F). Because arrestin-2N-3C chimera behaved similarly to the arrestin-2 N-domain, whereas arrestin-3N-2C resembled arrestin-3, we conclude that the origin of the N-domain largely determined the extent of the observed increase in Mdm2 binding.
Fig. 5. Parkin promotes the arrestin interaction with Mdm2.
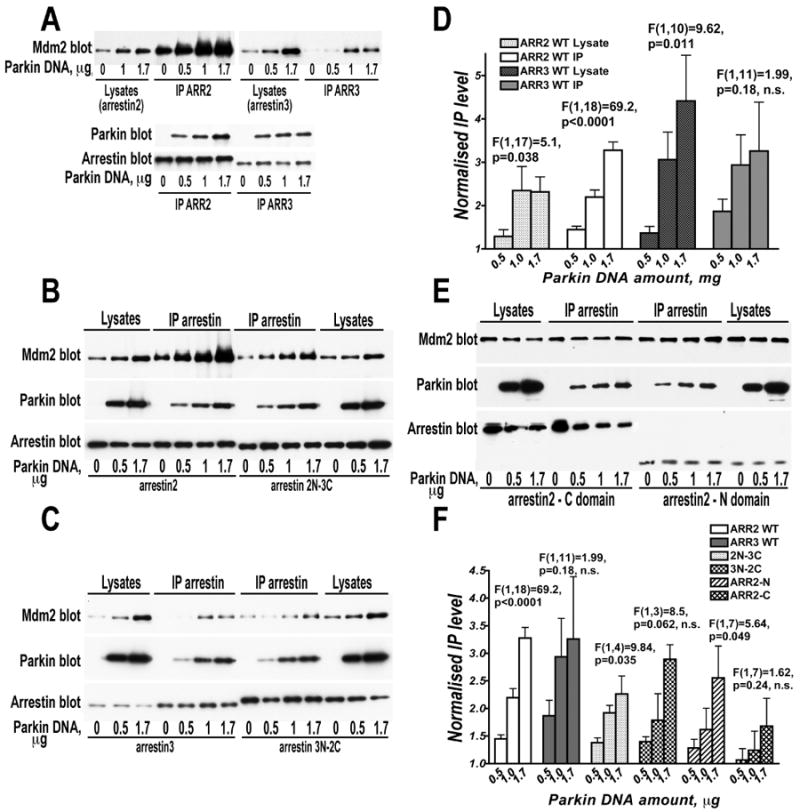
A, B, C, E. Indicated FLAG-tagged arrestins were immunoprecipitated from HEK293 cells co-expressing HA-tagged Mdm2 and varying amounts of WT myc-tagged parkin (the amounts of parkin DNA used for transfection of 60 mm dishes are shown in μg). Upper panels show Western blots for Mdm2; middle panels, parkin; lower panels, arrestins (ARR2, arrestin-2; ARR3, arrestin-3; arrestin-2N-3C, chimera with the N-domain of arrestin-2 and C-domain of arrestin-3; arrestin-3N-2C, reverse chimera; arrestin-2 N- and C-domains included residues 1–180 and 179–418, respectively). Lanes represent equal aliquots of cell lysates or immunoprecipitated samples, as indicated. In panel E, parkin appears to reduce somewhat the expression of the C-domain, but this effect was not studied further. D. Quantification of the effect of parkin concentration on the amount of Mdm2 in cell lysates containing equal amounts of total protein and expressed arrestins, and on the amount of Mdm2 co-immunoprecipitated with equal amount of arrestin-2 or arrestin-3. The data were normalized to the Mdm2 levels in cells or IP samples without parkin. Note that parkin co-expression significantly increases the level of Mdm2 in cells and, to a greater extent, its co-IP with arrestin-2. F. Quantification of parkin effect on the amount of Mdm2 co-immunoprecipitated with equal amount of indicated arrestins. The results of a representative experiments out 2-3 performed are shown. The data were normalized to the Mdm2 level in the IP samples without parkin. The data for each WT and mutant arrestin were analyzed by ANCOVA with Parkin concentration as main factor. The F and p values for each protein are shown on the graphs.
Parkin expression significantly increased the level of Mdm2 in cell lysates, even though the same amount of Mdm2-encoding plasmid was used (Fig. 5A-D). Therefore, increased Mdm2 co-immunoprecipitation with arrestins could be explained either by parkin-dependent increase in its expression, or by stabilization of Mdm2 (which is known to have high turnover rate) by arrestin binding, which is greatly facilitated by parkin. In the first scenario, increasing expression of parkin would be expected to elevate Mdm2 levels, whereas in the second only co-expression of parkin with arrestins would show this effect. To distinguish between these possibilities, we used COS7 cells that express low levels of parkin and both non-visual arrestins, and expressed increasing amounts of parkin in the presence or absence of arrestin, as well as increasing amounts of arrestins with and without parkin, and determined the level of endogenous Mdm2 (Fig. 6). We found that neither arrestin in the absence of parkin, nor parkin in the absence of arrestin appreciably affected Mdm2 level. In contrast, increasing parkin expression in the presense of arrestin, as well as increasing arrestin expression in the presense of parkin progressively increased the level of endogenous Mdm2 in the cytoplasm of COS7 cells (Fig. 6). The simplest interpretation of these data is that arrestin, parkin, and Mdm2 form a ternary complex, in which Mdm2 is protected from normal rapid degradation.
Fig. 6. Endogenous Mdm2 in the cell is stabilized only by simultaneous presense of arrestin and parkin.
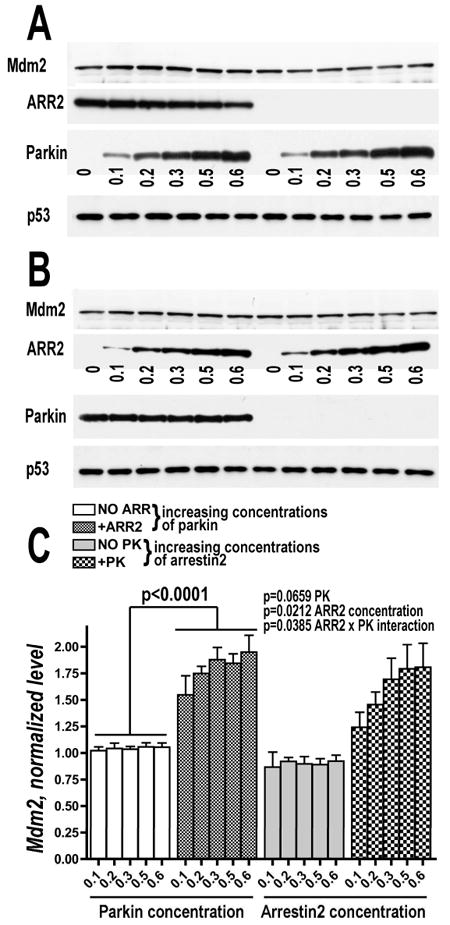
A. Cos7 cells were either co-transfected with a fixed amount of FLAG-arrestin2 and increasing amount of myc-parkin or with increasing amount of myc-parkin alone. Representative Western blots show increasing expression of endogenous Mdm2 (upper panel) in cells co-expressing arrestin-2 (middle panel) and parkin (lower panel) but not in cell expressing parkin alone. The data were normalized to the Mdm2 concentration in cells expressing either no parkin (left two sets of panels) or no arrestin-2 (right two sets of panels). Cytosolic p53 served as loading control. B. A reverse experiment to the one shown in A. Cos7 cells were either co-transfected with a fixed amount of myc-parkin and increasing amount of arrestin-2 or with increasing amount of arrestin-2 alone. Representative Western blots show increasing expression of endogenous Mdm2 in cells co-expressing arrestin-2 and parkin but not in cell expressing parkin alone. C. Quantification of the Western blot data demonstrating increased expression of endogenous Mdm2 in cell co-expressing arrestin-2 and parkin. The data are presented as ratios to the values obtained without expressed parkin (experiment shown in A; left side of the graph) or without arrestin-2 (experiment shown in B; right side of the graph). The data were analyzed by ANCOVA with Arrestin or Parkin as a factor and Parkin or Arrestin concentration as a co-variate. The results of 3 independent experiments are shown.
Arrestins assume several distinct conformations. The main conformational change in arrestins is believed to be the movement of the two domains relative to each other (94). Thus, within arrestin-parkin-Mdm2 ternary complex, one can envision two models of parkin-dependent increase in Mdm2 binding: 1) each of the proteins may directly interact with the other two, so that arrestin-parkin complex provides an additional binding site for Mdm2, via parkin; 2) parkin can shift the conformational equilibrium of arrestin towards the state favorable for Mdm2 binding. Since Mdm2 (60) and parkin (Fig. 1) engage both arrestin domains, in the first scenario parkin-dependent increase of Mdm2 binding to individual arrestin domains should be comparable to that observed with the full-length arrestin. In contrast, if the effect of parkin on Mdm2 binding is mediated solely by the change of arrestin conformational equilibrium, parkin is unlikely to affect Mdm2 interaction with separated arrestin domains. To distinguish between these two possibilities, we tested the effect of increasing parkin concentration on Mdm2 binding to separately expressed N- and C-domains of arrestin-2. The effect of parkin on the Mdm2 recruitment to the N-domain was significant and comparable to that for the 2N-3C arrestin chimera, but smaller than in case of full-length arrestin-2 (Fig. 5E,F). The effect was minimal in case of the C-domain (Fig. 5E,F). Detectable effect on Mdm2 binding to the N-domain of arrestin-2 (Fig. 5E,F) suggests that Mdm2 interaction with arrestin-associated parkin contributes to the effect. However, parkin effects on Mdm2 binding to chimeras and separate N-domain were less robust than on full-length arrestin-2 (Fig. 5). Therefore, we conclude that parkin primarily acts by shifting arrestin conformation to that favorable for Mdm2 binding, and the interaction with the N-domain of arrestin-2 plays key role in parkin-induced increase of Mdm2 binding.
The binding of Mdm2 (46, 60) and parkin (Fig. 1C) is highly sensitive to arrestin conformation: both E3 ubiquitin ligases preferentially interact with arrestin mutants frozen in the basal state. Therefore we tested the effect of arrestin conformation, using two types of conformationally biased arrestins mutants: Δ7 with greatly reduced ability to assume active receptor-binding state (29, 82), and 3A with enhanced conformational flexibility (85) and consequent enhanced binding to different functional forms of their cognate receptors (67, 83, 84, 86, 95). We found that increased Mdm2 binding to the Δ7 mutants of both non-visual arrestins was no longer enhanced by parkin (Fig. 7A,B). In contrast, parkin-dependent increase of Mdm2 binding to arrestin2-3A mutant was at least as robust as with WT arrestin-2 (Fig. 7C). These data are consistent with the idea that parkin increased Mdm2 binding primarily by shifting the conformational equilibrium of arrestins towards the basal state, mimicking the effect of Δ7 mutation. Importantly, in the presense of Δ7 mutants that bind Mdm2 better than parental WT arrestins, parkin did not further increase Mdm2 level in the cell, whereas in the presense of 3A mutants it increased as robustly as with WT arrestins (Fig. 7). Thus, it appears that tight arrestin binding stabilizes Mdm2, whereas parkin predominantly acts by enhancing arrestin-Mdm2 interaction.
Fig. 7. Enhanced Mdm2 recruitment by arrestin mutants frozen in the basal state is not increased by parkin.
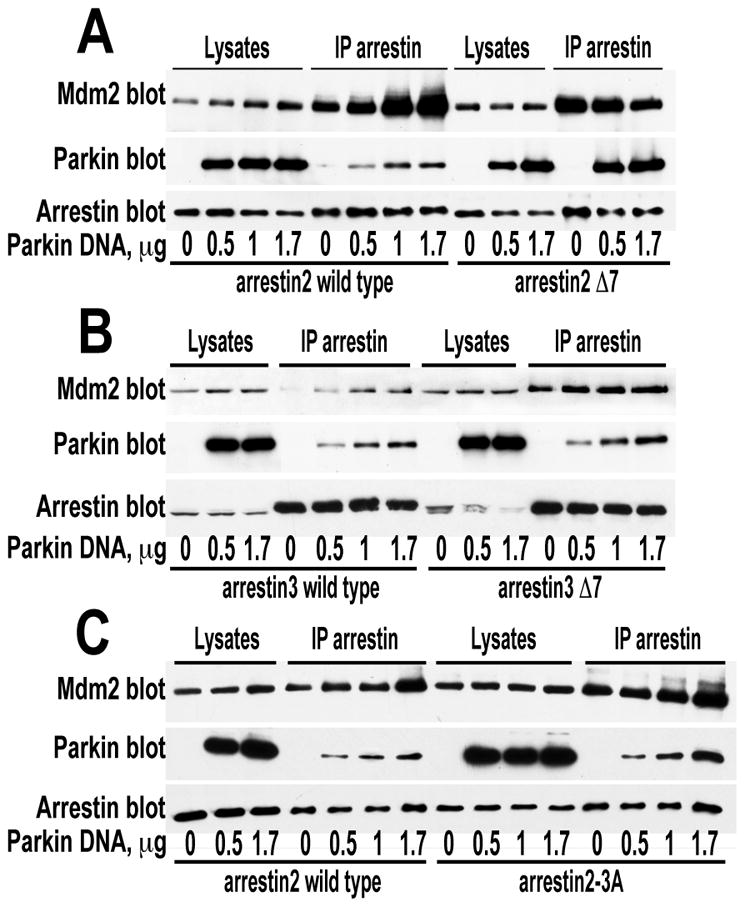
A, B, C. HEK293 cells were co-transfected with HA-Mdm2, indicated FLAG-arrestins, and varying amounts of myc-parkin. Arrestins were immunoprecipitated with anti-FLAg antibodies. Upper blots show Mdm2; middle blots, parkin, lower blots, arrestin. Lanes represent equal aliquots of cell lysates or immunoprecipitated samples, as indicated. Arrestin-2 Δ7 and arrestin-3 Δ7, mutants with seven-residue deletions in the inter-domain hinge, which freezes the protein in the basal conformation and greatly impedes receptor binding; arrestin2-3A, conformationally loose mutant mimicking the active state, that shows enhanced receptor binding. Note that Δ7 mutants of both arrestins bind more Mdm2 than corresponding WT forms, and parkin does not further increase Mdm2 recruitment to these mutants. In contrast, arrestin2-3A mutant shows even more evident enhancement of Mdm2 recruitment by parkin than WT arrestin-2. The results of a representative experiments out 2-3 performed are shown.
Disease-associated parkin mutants bind arrestins and promote arrestin-Mdm2 interaction
Multiple missense mutations in different functional domains of the parkin protein have been identified (96). Mutations in parkin were linked to ∼50% of familial early-onset recessive Parkinson's disease cases (2). Although functional manifestations of individual mutations vary, many parkin mutants displayed altered intracellular distribution and solubility, whereas others showed attenuated enzymatic activity (49, 97, 98). Since arrestin binding is a novel parkin function, we investigated the ability of several disease-associated parkin mutants to interact with arrestins. For these studies we selected representative parkin variants with mutations in different elements of the molecule: R42P (ubiquitin-like domain), R275W (RING finger 1), G430D (RING finger 2), and T415N (connector between the two RING fingers) (Fig. 8A). Two of these mutants, R42P and R275W were shown to retain ubiquitin ligase activity, whereas T415N and G430D were catalytically inactive (49). We found that none of these mutations significantly affected parkin binding to arrestin-2 (Fig. 8B).
Fig. 8. Parkin mutants implicated in the familial Parkinson's disease bind both non-visual arrestins.
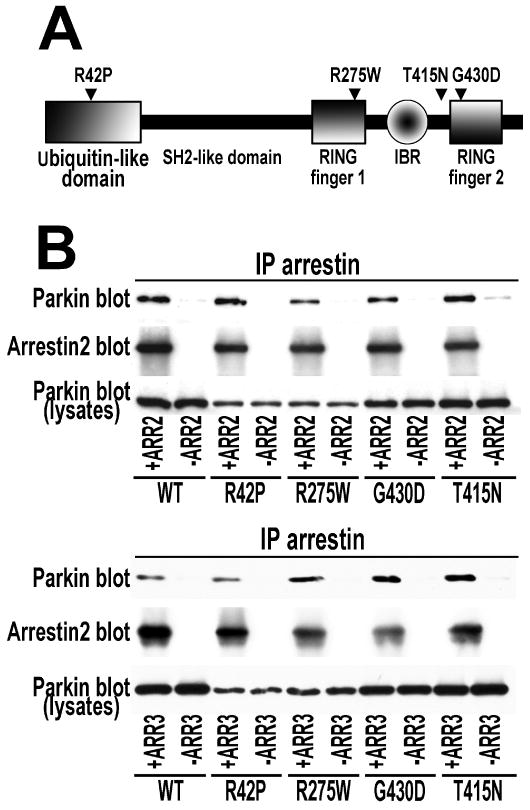
A. Domain structure of parkin showing the positions of mutations tested. B. Myc-tagged WT parkin and indicated mutants were expressed in HEK293 cells alone (-ARR2 and –ARR3 controls) or together with FLAG-tagged WT arrestin-2 (+ARR2) or arrestin-3 (+ARR3). Indicated arrestins were immunoprecipitated with anti-FLAG antibody, and IP samples were analyzed by Western blot for parkin (upper blots) and arrestin (middle blots). Lower blots show parkin expression in cell lysates. The results of representative experiments out 2-3 performed with each parkin-arrestin combination are shown.
Next, we tested whether parkin mutants retain the ability to promote arrestin-Mdm2 interaction and found that three mutants (R42P, R275W, and T415N) increased Mdm2 binding to arrestin-2. R42P and R275W demonstrated a robust effect at the same expression levels as WT parkin, whereas T415N and particularly G430D appeared less potent in this regard (Fig. 9A).
Fig. 9. Parkin mutants reduce receptor-stimulated ubiquitination of arrestins and differentially promote Mdm2 recruitment.
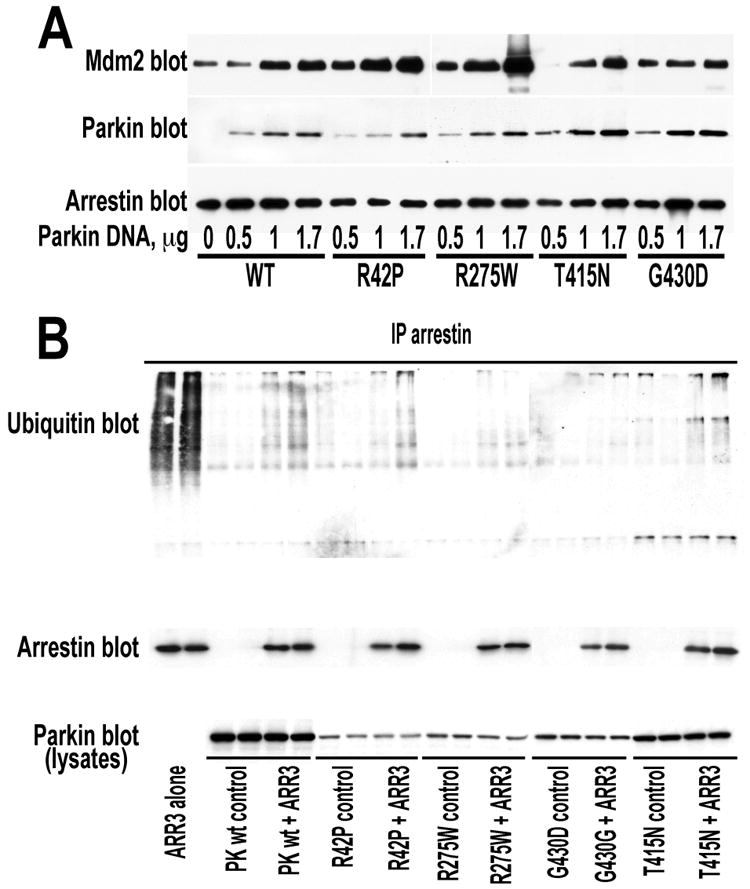
A. HEK293 cells were co-transfected with HA-Mdm2, FLAG-arrestin-2, and varying amounts of myc-tagged WT parkin or indicated mutants. Arrestin was immunoprecipitated with anti-FLAG antibody, and IP samples were analyzed by Western blot for the presence of Mdm2 (upper blot), parkin (middle blot), and arrestin (lower blot). B. HEK293 cells expressing β2V2 receptor chimera, HA-tagged ubiquitin, myc-tagged parkin mutants alone (control) or with FLAG-tagged WT arrestin-3. Arrestin was immunoprecipitated with anti-FLAG antibody, and IP samples were analyzed by Western blot for the presence of ubiquitin (upper blot) and arrestin (middle blot). The expression of WT parkin and indicated mutants in cell lysates is shown in the lower blot. The results of a representative experiments out 2-3 performed with each arrestin-parkin combination are shown.
Since parkin also suppressed arrestin ubiquitination (Fig. 4), we compared the effect of disease-associated mutants and WT parkin on this process. WT parkin and all tested mutants decreased arrestin-3 ubiquitination to a similar extent (Fig. 9B). Apparently, parkin exerts this effect by shielding arrestin lysines targeted by Mdm2. This is consistent with simultaneous increase in Mdm2 binding and decrease in arrestin modification by this ubiquitin ligase.
Discussion
Recent identification of several genes associated with relatively rare (5-10% of all cases) familial forms of Parkinson's disease (99) was widely expected to reveal the mechanisms involved and pave the way to finding a cure. Unfortunately, the functions of proteins encoded by many PD-associated genes are currently either unclear, incomplete, and/or controversial (reviewed in (11, 99)). Comprehensive elucidation of the biological roles of these proteins and functional consequences of mutations linked to the pathology is necessary to translate identification of PD-associated genes into progress in understanding the mechanisms of neurodegeration and devising new therapies.
Parkin is a fairly well-studied PD-associated protein. It is a functional E3 ubiquitin ligase with several identified substrates linked to the death of dopaminergic neurons (58, 98, 100). The effects of several disease-associated mutations on its solubility, subcellular localization, self-ubiquitination and substrate binding have been characterized (49, 97, 98). Parkin interactions with several chaperones and regulatory proteins, as well as its post-translational modifications, were shown to affect its activity (15-17, 20, 21, 101-103). Parkin interactions with arrestins described here reveal previously unappreciated aspect of parkin function. Virtually every cell in vertebrates expresses one or more arrestin subtype, with intracellular concentrations ranging from low nanomolar in some cells and ∼200 nM in neurons (36) to high micromolar and millimolar in cone and rod photoreceptors, respectively (62-65). Arrestins bind an amazing variety of proteins, serving as versatile regulators of cell signaling (26, 94, 104). Although arrestins were first discovered as key players in the homologous desensitization of GPCRs (reviewed in (25, 104)), the emerging common theme in arrestin function is to serve as scaffolds or adaptors, organizing multi-protein signaling complexes and localizing them to specific compartments in the cell, such as coated pits (105), receptor-rich membranes (106, 107), or cytoskeleton (29). Reported parkin localization in post-synaptic densities (101) may well be assisted by its interaction with endogenous arrestins abundant in this receptor-rich compartment (37).
Both arrestin-2 and -3 were reported to interact with many non-receptor binding partners (26, 104). However, most of these interactions were inferred from co-IP, where each non-visual arrestin was shown to bring down more than a hundred different proteins (66). Thus, this method cannot prove direct interaction, as any protein can be recruited to the complex via one or more of arrestin binding partners, rather than via arrestin itself. Even co-IP of endogenous proteins expressed at biologically relevant levels from native tissues or cells (shown in Fig. 1B for parkin and in (59) for Mdm2) has the same caveat. Direct binding can only be proved by the demonstration that purified arrestin interacts with a purified partner, which was done in very few cases. For example, the binding of Mdm2 to arrestin-3, reported in 2001 (59), was never confirmed with two pure proteins in the absence of cell lysate, which obviously contains hundreds of other proteins that could serve as adaptors bridging the two. In fact, among E3 ubiquitin ligases reported to bind arrestins, the proof of direct interaction was only presented for AIP4 (40). Therefore, we used three independent methods with two differentially tagged forms of parkin to reproduce with purified proteins its interaction with three arrestin subtypes to prove that in all cases the binding is direct (Fig. 2).
So far, the affinity of arrestin interactions with only three non-receptor partners was measured: the binding of free arrestin-2 and -3 to clathrin (105), arrestin-2 binding to microtubules (29) and to Ca-liganded calmodulin (52). The assay based on the change of anisotropy of pure fluorescently labeled arrestin-1, -2, and -3 in the presence of increasing concentrations of purified MBP-parkin (Fig. 2C) allows to estimate the affinity of these interactions. The data yielded KDs ranging from 3 to 19 μM. This makes parkin only the fourth non-receptor binding partner and the first ubiquitin ligase with known affinity for arrestins. As reported intracellular concentrations of non-visual arrestins are sub-micromolar (36), at first glance parkin affinity appears fairly low. This affinity means that the interaction is very dynamic and that at any given moment a small fraction of cellular arrestin exists in complex with parkin. Considering that more than a hundred different cellular proteins were reported to bind arrestins (66), and that arrestin molecule is too small to accommodate many partners simultaneously (104), this is exactly what should be expected. The affinity may be modulated by post-translational modifications of arrestins and/or parkin, as well as by the binding of additional partners. For example, our finding that parkin dramatically increases arrestin-2 affinity for Mdm2 (Figs. 5,6) suggests (according to the laws of thermodynamics) that Mdm2 also increases the affinity of arrestin-2 for parkin. As a result, this apparently transient interaction is sufficient to stabilize expressed and endogenous Mdm2 in the cytoplasm (Figs. 5,6) and dramatically reduce the ubiquitination of arrestin-2 and -3 in cells (Figs. 4,9).
Parkin increases the level of co-expressed (Fig. 5) and endogenous (Fig. 6) Mdm2 in the cell. Although higher level of Mdm2 could be the reason for its increased binding to arrestins in the presense of parkin (Figs. 5,6), several lines of evidence are incompatible with this explanation. First, parkin effect on endogenous Mdm2 depends on co-expression of arrestin (Fig. 6), ruling out direct arrestin-independent mechanisms. Second, the increase in Mdm2 by parkin depends on the nature of arrestin: it is robust in the presence of WT and conformationally loose 3A form (Figs. 5-7), but absent in the presence of Δ7 mutants (Fig. 7). Third, parkin suppresses arrestin ubiquitination (Figs. 4, 9), which would be expected to increase if parkin simply elevated Mdm2 level in the cytoplasm bypassing arrestins. The only model compatible with all these observations is that parkin directly binds arrestins (Fig. 2) and increases their affinity for Mdm2, which is stabilized in this ternary complex. Importantly, parkin does not increase the binding to Δ7 mutants with enhanced affinity for Mdm2 (46, 60). Considering that parkin interacts with both arrestin domains, the hypothesis that parkin binding shifts arrestin equilibrium towards the basal conformation similar to that fixed by Δ7 mutation is the simplest model that accounts for these findings. Since Mdm2 was reported to ubiquitinate arrestins (59), simultaneous increased recruitment of Mdm2 to its substrate (Figs. 5,7) and suppression of its modification by parkin (Figs. 4,9) requires an explanation. One possibility is suggested by the fact that arrestin ubiqutination is facilitated by receptor binding (Fig. 4 and (59)), which suggests that Mdm2 prefers active receptor-bound arrestin as a substrate. In this model, parkin-dependent stabilization of the basal arrestin conformation would make arrestin a less suitable substrate for Mdm2, but at the same time a better binding partner. However, parkin does not appreciably reduce arrestin association with the receptor (Fig. 3). Collectively, these data suggest that bound parkin simply shields surface lysines on arrestin that otherwise would be modified by Mdm2. Interestingly, parkin binding was recently found to inhibit ubiquitination of PINK1 (108), another protein implicated in familial Parkinson's disease.
Although ubiquitination was reported to enhance arrestin affinity for receptors (59), we did not detect any effect of parkin expressed at concentrations that dramatically suppress receptor-induced increase in the ubiquitination of arrestin-2 and -3 (Fig. 4A,B,D) on arrestin interaction with the active receptor (Fig. 3B) or on arrestin-dependent facilitation of receptor internalization (Fig. 4C). These data appear to contradict recent report that reduced ubiqitination (by overexpression of the deubiquitinating enzyme USP33) precluded arrestin-3 endosomal recruitment in response to stimulation of the vasopressin V2 receptor (109). One possible reason for this discrepancy is the diffenece in experimental techniques, confocal microscopy versus receptor binding. We measured receptor internalization directly, using the disappearence of the binding sites from the plasma membrane as readout, but did not follow subsequent localization of arrestins. Our data suggest that non-ubiqitinated arrestins support internalization of at least some receptors to the same extent as ubiquitinated arrestins.
At least two out of four parkin mutants associated with familial PD, R42P in ubiquitin-like domain and R275W in the RING1 domain, bind arrestin like WT parkin and robustly enhance the recruitment of Mdm2 (Figs. 8,9). These data are in agreement with previous reports that missense mutations mostly preserve the parkin's ability to interact with its partners (97). However, certain parkin mutations have been shown to disrupt interactions with specific partners: R42P precludes parkin interaction with chaperone 14-3-3b, which suppresses parkin activity (17). C-terminal mutations, including T415N, strongly reduce parkin interaction with E2 enzymes UbcH7 and UbcH8 (58). Since these mutants are deficient in certain other functions (98), by interacting normally with arrestin they can displace WT parkin, explaining apparent dominant-negative effects reported for some mutant forms (6, 7). Interestingly, the mutations in both RING fingers almost completely abolish parkin interactions with one of its bona fide substrates, p38 subunit of aminoacyl-tRNA synthetase (98), even though only intact second RING domain is necessary for its ubiquitin ligase activity (49). Further analysis of different mutant forms of parkin with intact and impaired arrestin binding is necessary to narrow down the interaction site and elucidate possible role of arrestin-parkin interaction in Parkinson's pathology. These studies will enable targeted manipulation of this interaction with a view of constructing arrestin variants that alleviate cytotoxic effects of parkin mutants.
Acknowledgments
The authors are grateful to Drs. Ted Dawson (John Hopkins University) and Konstanze Winklhofer (Ludwig Maximilians University, Germany) for parkin expression constructs, to Drs. M. G. Caron (Duke University) and J. L. Benovic (Thomas Jefferson University) for HA-β2-adrenergic receptor constructs and arrestin-2- and arrestin-3-specific antibodies, respectively, and to Dr. Hassane McHaourab and Mr. Sanjay Mishra (Vanderbilt University) for help with measurements of fluorescence anisotropy.
Funding: Supported in part by NIH grants NS045117 and NS065868 (EVG), EY011500, GM077561, and GM081756 (VVG).
Abbreviations used
- GPCR
G protein-coupled receptor
- Mdm2
murine double minute oncogene (E3 ubiquitin ligase)
- PD
Parkinson's disease
- b2AR
β2-adrenergic receptor
- b2V2
chimeric β2-adrenergic receptor with the C-terminus of vasopressin V2 receptor
- IP
immunoprecipitation
- co-IP
coimmunoprecipitation
- ARRDC3
arrestin domain-containing protein 3
- MBP
maltose-binding protein
- WT
wild type
Footnotes
We use systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in HUGO database).
References
- 1.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:544–545. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 2.Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Brice A French Parkinson's Disease Genetics Study Group, and European Consortium on Genetic Susceptibility in Parkinson's Disease. Association between Early-Onset Parkinson's Disease and Mutations in the Parkin Gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 3.Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, Brooks DJ, Wood NW, Piccini P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–136. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- 4.Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston W. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- 5.Pramstaller PP, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Gonzales-McNeal M, Hilker R, Kramer PL, Klein C. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58:411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- 6.Kyratzi E, Pavlaki M, Kontostavlaki D, Rideout HJ, Stefanis L. Differential effects of Parkin and its mutants on protein aggregation, the ubiquitin-proteasome system, and neuronal cell death in human neuroblastoma cells. J Neurochem. 2007;102:1292–1303. doi: 10.1111/j.1471-4159.2007.04620.x. [DOI] [PubMed] [Google Scholar]
- 7.Sang TK, Chang HY, Lawless GM, Ratnaparkhi A, Mee L, Ackerson LC, Maidment NT, Krantz DE, Jackson GR. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin–protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 9.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 11.Gupta A, Dawson VL, Dawson TM. What causes cell death in Parkinson's disease? Ann Neurol. 2008;64:S3–15. doi: 10.1002/ana.21573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, Brice A, van Bergen En Henegouwen PM, Fon EA. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nature Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- 13.Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D, Tatzelt J, Winklhofer KF. Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avraham E, Rott R, Liani E, Szargel R, Engelender S. Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J Biol Chem. 2007;282:12842–12850. doi: 10.1074/jbc.M608243200. [DOI] [PubMed] [Google Scholar]
- 15.Kalia SK, Lee S, Smith PD, Liu L, Crocker SJ, Thorarinsdottir TE, Glover JR, Fon EA, Park DS, Lozano AM. BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron. 2004;44:931–945. doi: 10.1016/j.neuron.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 16.Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci USA. 2010;107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato S, Chiba T, Sakata E, Kato K, Mizuno Y, Hattori N, Tanaka K. 14-3-3eta is a novel regulator of parkin ubiquitin ligase. EMBO J. 2006;25:211–221. doi: 10.1038/sj.emboj.7600774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nature Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- 19.Winklhofer KF, Henn IH, Kay-Jackson PC, Heller U, Tatzelt J. Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem. 2003;278:47199–47208. doi: 10.1074/jbc.M306769200. [DOI] [PubMed] [Google Scholar]
- 20.Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 22.Wong ES, Tan JM, Wang C, Zhang Z, Tay SP, Zaiden N, Ko HS, Dawson VL, Dawson TM, Lim KL. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J Biol Chem. 2007;282:12310–12318. doi: 10.1074/jbc.M609466200. [DOI] [PubMed] [Google Scholar]
- 23.Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- 24.Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 25.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 26.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Ann Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 27.Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 28.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 29.Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, Nair KS, Slepak VZ, Klug CS, Gurevich VV. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol. 2007;368:375–387. doi: 10.1016/j.jmb.2007.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang P, Gao H, Ni Y, Wang B, Wu Y, Ji L, Qin L, Ma L, Pei G. beta-Arrestin 2 Functions as a G-Protein-coupled Receptor-activated Regulator of Oncoprotein Mdm2. J Biol Chem. 2003;278:6363–6370. doi: 10.1074/jbc.M210350200. [DOI] [PubMed] [Google Scholar]
- 31.Luan B, Zhang Z, Wu Y, Kang J, Pei G. Beta-arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-kappaB activation. EMBO J. 2005;24:4237–4246. doi: 10.1038/sj.emboj.7600882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004;14:303–317. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 33.Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci USA. 2004;101:8603–8607. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins Block G Protein-coupled Receptor-mediated Apoptosis. J Biol Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- 35.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- 36.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed MR, Bychkov E, Gurevich VV, Benovic JL, Gurevich EV. Altered expression and subcellular distribution of GRK subtypes in the dopamine-depleted rat basal ganglia is not normalized by l-DOPA treatment. J Neurochem. 2008;104:1622–1636. doi: 10.1111/j.1471-4159.2007.05104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis. 2005;18:323–335. doi: 10.1016/j.nbd.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Hurlé MA. Changes in the expression of G protein-coupled receptor kinases and beta-arrestin 2 in rat brain during opioid tolerance and supersensitivity. J Neurochem. 2001;77:486–492. doi: 10.1046/j.1471-4159.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 40.Bhandari D, Trejo J, Benovic JL, Marchese A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J Biol Chem. 2007;282:36971–36979. doi: 10.1074/jbc.M705085200. [DOI] [PubMed] [Google Scholar]
- 41.Shenoy SK, Xiao K, Venkataramanan V, Snyder PM, Freedman NJ, Weissman AM. NEDD4 mediates agonist-dependent ubiquitination, lysosomal targeting and degradation of the beta 2 adrenergic receptor. J Biol Chem. 2008;283:22166–22176. doi: 10.1074/jbc.M709668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nabhan JF, Pan H, Lu Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the beta2-adrenergic receptor. EMBO Rep. 2010;11:605–611. doi: 10.1038/embor.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orsini MJ, Benovic JL. Characterization of dominant negative arrestins that inhibit beta2-adrenergic receptor internalization by distinct mechanisms. J Biol Chem. 1998;273:34616–34622. doi: 10.1074/jbc.273.51.34616. [DOI] [PubMed] [Google Scholar]
- 44.Mundell SJ, Loudon RP, Benovic JL. Characterization of G protein-coupled receptor regulation in antisense mRNA-expressing cells with reduced arrestin levels. Biochemistry. 1999;38:8723–8732. doi: 10.1021/bi990361v. [DOI] [PubMed] [Google Scholar]
- 45.Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin × receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- 46.Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their ‘inactive’ conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore DJ, West AB, Dikeman DA, Dawson VL, Dawson TM. Parkin mediates the degradation-independent ubiquitination of Hsp70. J Neurochem. 2008;105:1806–1819. doi: 10.1111/j.1471-4159.2008.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henn IH, Gostner JM, Lackner P, Tatzelt J, Winklhofer KF. Pathogenic mutations inactivate parkin by distinct mechanisms. J Neurochem. 2005;92:114–122. doi: 10.1111/j.1471-4159.2004.02854.x. [DOI] [PubMed] [Google Scholar]
- 49.Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem. 2006;281:3204–3209. doi: 10.1074/jbc.M510393200. [DOI] [PubMed] [Google Scholar]
- 50.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 51.Gurevich VV, Benovic JL. Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 2000;315:422–437. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- 52.Wu N, Hanson SM, Francis DJ, Vishnivetskiy SA, Thibonnier M, Klug CS, Shoham M, Gurevich VV. Arrestin binding to calmodulin: a direct interaction between two ubiquitous signaling proteins. J Mol Biol. 2006;364:955–963. doi: 10.1016/j.jmb.2006.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem. 2011;286 doi: 10.1074/jbc.M110.213835. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sommer ME, Smith WC, Farrens DL. Dynamics of arrestin-rhodopsin interactions: acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent. J Biol Chem. 2006;281:9407–9417. doi: 10.1074/jbc.M510037200. [DOI] [PubMed] [Google Scholar]
- 55.Baudier J, Glasser N, Duportail G. Bimane- and acrylodan-labeled S100 proteins. Role of cysteines-85 alpha and -84 beta in the conformation and calcium binding properties of S100 alpha alpha and S100b (beta beta) proteins. Biochemistry. 1986;25:6934–6941. doi: 10.1021/bi00370a029. [DOI] [PubMed] [Google Scholar]
- 56.Lakowicz JR. Principles of fluorescence spectroscopy. Third. Springer; NY: 2006. [Google Scholar]
- 57.Johnson R. Elementary statistics. seventh. Wadsworth Publishing Co; 1996. [Google Scholar]
- 58.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 59.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 60.Song X, Gurevich EV, Gurevich VV. Cone arrestin binding to JNK3 and Mdm2: conformational preference and localization of interaction sites. J Neurochem. 2007;103:1053–1062. doi: 10.1111/j.1471-4159.2007.04842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV. Each rhodopsin molecule binds its own arrestin. Proc Nat Acad Sci USA. 2007;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh EN, Craft CM. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59:462–474. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, Lefkowitz RJ. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- 68.Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV. A model for the solution structure of the rod arrestin tetramer. Structure. 2008;16:924–934. doi: 10.1016/j.str.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Klug CS, Hubbell WL, Gurevich VV. Structure and function of the visual arrestin oligomer. EMBO J. 2007;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vishnivetskiy SA, Francis DJ, Van Eps N, Kim M, Hanson SM, Klug CS, Hubbell WL, Gurevich VV. The role of arrestin alpha-helix I in receptor binding. J Mol Biol. 2010;395:42–54. doi: 10.1016/j.jmb.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 74.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 75.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3A: evolution of receptor specificity. Journal of Molecular Biology. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 76.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 77.Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 79.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 80.Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 81.Song X, Coffa S, Fu H, Gurevich VV. How does arrestin assemble MAP kinases into a signaling complex? J Biol Chem. 2009;284:685–695. doi: 10.1074/jbc.M806124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vishnivetskiy SA, Hirsch JA, Velez MG, Gurevich YV, Gurevich VV. Transition of arrestin in the active receptor-binding state requires an extended interdomain hinge. J Biol Chem. 2002;277:43961–43968. doi: 10.1074/jbc.M206951200. [DOI] [PubMed] [Google Scholar]
- 83.Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- 84.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 85.Carter JM, Gurevich VV, Prossnitz ER, Engen JR. Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J Mol Biol. 2005;351:865–878. doi: 10.1016/j.jmb.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 86.Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Enhanced Arrestin Facilitates Recovery and Protects Rod Photoreceptors Deficient in Rhodopsin Phosphorylation. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 89.Pulvermuller A, Schroder K, Fischer T, Hofmann KP. Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- 90.Ohguro H, Palczewski K, Walsh KA, Johnson RS. Topographic study of arrestin using differential chemical modifications and hydrogen/deuterium exchange. Protein Sci. 1994:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46 doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gurevich VV, Gurevich EV. Custom-designed proteins as novel therapeutic tools? The case of arrestins. Expert Rev Mol Med. 2010;12:e13. doi: 10.1017/S1462399410001444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shenoy SK, Lefkowitz RJ. Receptor-specific ubiquitination of beta-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J Biol Chem. 2005;280:15315–15324. doi: 10.1074/jbc.M412418200. [DOI] [PubMed] [Google Scholar]
- 94.Gurevich VV, Gurevich EV, Cleghorn WM. Arrestins as multi-functional signaling adaptors. Handb Exp Pharmacol. 2008;186:15–37. doi: 10.1007/978-3-540-72843-6_2. [DOI] [PubMed] [Google Scholar]
- 95.Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG, Gurevich VV. An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J Biol Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- 96.West AB, Maidment NT. Genetics of parkin-linked disease. Hum genet. 2004;114:327–336. doi: 10.1007/s00439-003-1074-6. [DOI] [PubMed] [Google Scholar]
- 97.Hampe C, Ardila-Osorio H, Fournier M, Brice A, Corti O. Biochemical analysis of Parkinson's disease-causing variants of PArkin, an E3 ubiquitin-protein ligase with monoubiquitination capacity. Hum Mol Genet. 2006;15:2059–2075. doi: 10.1093/hmg/ddl131. [DOI] [PubMed] [Google Scholar]
- 98.Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- 99.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 100.Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O, Troncoso J, Johnson B, Saffary R, Goh EL, Song H, Park BJ, Kim MJ, Kim S, Dawson VL, Dawson TM. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci. 2005;25:7968–7978. doi: 10.1523/JNEUROSCI.2172-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- 102.Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA. Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J Biol Chem. 2002;277:486–491. doi: 10.1074/jbc.M109806200. [DOI] [PubMed] [Google Scholar]
- 103.Yamamoto A, Friedlein A, Imai Y, Takahashi R, Kahle PJ, Haass C. Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J Biol Chem. 2005;280:3390–3399. doi: 10.1074/jbc.M407724200. [DOI] [PubMed] [Google Scholar]
- 104.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharm Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 106.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 107.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shiba K, Arai T, Sato S, Kubo S, Ohba Y, Mizuno Y, Hattori N. Parkin stabilizes PINK1 through direct interaction. Biochem Biophys Res Commun. 2009;383:331–335. doi: 10.1016/j.bbrc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 109.Shenoy SK, Modi AS, Shukla AK, Xiao K, Berthouze M, Ahn S, Wilkinson KD, Miller WE, Lefkowitz RJ. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci U S A. 2009;106:6650–6655. doi: 10.1073/pnas.0901083106. [DOI] [PMC free article] [PubMed] [Google Scholar]