

Summary
Myeloproliferative neoplasms (MPN), a group of haematopoietic stem cell (HSC) disorders, are often accompanied by myelofibrosis. We previously identified the fusion of the ETV6 gene to the LYN gene (ETV6-LYN) in idiopathic myelofibrosis with ins(12;8)(p13;q11q21). The introduction of ETV6-LYN into HSCs resulted in fatal MPN with massive myelofibrosis in mice, implicating the rearranged LYN kinase in the pathogenesis of MPN with myelofibrosis. However, the signalling molecules directly downstream from and activated by ETV6-LYN remain unknown. In this study, we demonstrated that the direct activation of STAT5 by ETV6-LYN is crucial for the development of MPN. ETV6-LYN was constitutively active as a kinase through autophosphorylation. ETV6-LYN, but not its kinase-dead mutant, supported cytokine-free proliferation of haematopoietic cells. STAT5 was activated in a JAK2-independent manner in ETV6-LYN-expressing cells. ETV6-LYN interacted with STAT5 and directly activated STAT5 both in vitro and in vivo. Of note, ETV6-LYN did not support the formation of colonies by Stat5-deficient HSCs under cytokine-free conditions and the capacity of ETV6-LYN to induce MPN with myelofibrosis was profoundly attenuated in a Stat5-null background. These findings define STAT5 as a direct target of ETV6-LYN and unveil the LYN-STAT5 axis as a novel pathway to augment proliferative signals in MPN and leukaemia.
Keywords: ETV6-LYN, STAT5, myeloproliferative neoplasm, myelofibrosis
Myeloproliferative neoplasms (MPN) are a heterogeneous group characterized by excessive production of blood cells by haematopoietic precursors and often accompanied by myelofibrosis (Levine and Gilliland, 2008). The V617F somatic mutation in the Janus kinase 2 (JAK2) gene has recently been found in the majority of patients with polycythaemia vera (PV) and more than half of patients with essential thrombocythaemia (ET) and idiopathic myelofibrosis (IMF)(James et al, 2005; Kralovics et al, 2005; Tefferi et al, 2007). The expression of JAK2 V617F causes a PV-like disease with myelofibrosis in a murine bone marrow (BM) transplant model (Lacout et al, 2006; Wernig et al, 2006; Zaleskas et al, 2006). In addition, a gain-of-function MPL W515 mutation was described in nearly 10% of patients with JAK2 V617F-negative IMF (Pardanani et al, 2006; Pikman et al, 2006). However, the mechanism responsible for MPN and the formation of myelofibrosis in patients without JAK2 or MPL mutations is still unclear.
We previously reported the fusion of the ETV6 (TEL) gene to the LYN gene in idiopathic myelofibrosis with ins(12;8)(p13;q11q21) without JAK2 V617F or MPL W515 mutations (Tanaka et al, 2010). ETV6-LYN fusion conferred cytokine-independence to an interleukin-3 (IL-3)-dependent murine haematopoietic line, Ba/F3. Murine haematopoietic stem cells (HSCs) expressing ETV6-LYN formed colonies without exogenous growth factors and induced fatal MPN with massive fibrosis in recipient mice in BM transplant experiments. ETV6 is known to form fusion genes with more than 20 partners including protein tyrosine kinase (PTK) genes (Bohlander 2005). ETV6-PTK fusion genes have been found in MPN and other haematological malignancies (ETV6 and ABL1, ABL2, JAK2, NTRK3, FGFR3, PDGFRB and SYK). ETV6-PTK fusion products are constitutively active as kinases through oligomerization mediated by the PNT domain of ETV6 (Bohlander 2005). ETV6-LYN is the first fusion gene to involve a SRC family kinase gene. LYN kinase is a specific member of the SRC family of non-receptor kinases and an important component in cytokine signal transduction in a variety of cells including haematopoietic cells (Xu et al, 2005). LYN kinase was highly expressed and activated in samples from chronic myeloid leukaemia (CML) patients who progressed to blastic crisis or an accelerated phase during imatinib treatment (Donato et al, 2003). In addition, LYN was constitutively activated in acute myeloid leukaemia (AML) and BCR-ABL1-positive acute lymphoblastic leukaemia (ALL) (Hu et al, 2004; Dos Santos et al, 2008). However, there has been no report of a SRC family gene fused to ETV6 and a possible association with MPN.
In this study, we analysed the signalling pathways activated by ETV6-LYN in haematopoietic cells. We have identified that ETV6-LYN directly activates STAT5, a critical signalling molecule activated downstream of both JAK2 V617F and MPL W515 mutants in MPN. Our findings provide a novel signalling pathway of STAT5 activation that bypasses JAK2, the major kinase of STAT5, and implicate the LYN-STAT5 signalling cascade in the augmentation of proliferative signals in MPN and leukaemia.
Materials and methods
Mice
Stat5+/- mice that had been backcrossed at least eight times onto a C57BL/6 (B6-Ly5.2) background were used (Cui et al, 2004). C57BL/6 mice congenic for the Ly5.2 locus (B6-Ly5.2) were purchased from SLC (Shizuoka, Japan). Mice congenic for the CD45.1 locus (B6-CD45.1) were bred and maintained at Sankyo Lab Service (Tsukuba, Japan). All experiments using mice received approval from The Chiba University Administrative Panel for Animal Care.
Retroviral constructs and retrovirus production
The ETV6-LYN cDNA was subcloned into pMXs-puro and MigR1 (IRES-EGFP) retroviral vectors with or without a FLAG tag (Pear et al, 1998; Kitamura et al, 2003). To produce the recombinant retrovirus, plasmid DNA was transfected into 293gp cells (293 cells containing the gag and pol genes but lacking an envelope gene) along with a VSV-G expression plasmid or 293gpg cells as previously described (Iwama et al, 2004). The kinase-dead mutant of ETV6-LYN (ETV6-LYN KD) was constructed by replacing the C-terminal portion of ETV6-LYN with that of the kinase-dead mutant of LYN (K275A)(Kasahara et al, 2004) and was subcloned into pMXs-puro.
Proliferation assay of Ba/F3 cells
The mouse IL-3-dependent pro-B cell line Ba/F3 (Palacios and Steinmetz, 1985) was cultured in RPMI 1640 medium with 10% fetal calf serum (FCS) in the presence of 2 ng/ml of IL-3 (PeproTech, Rocky Hill, NJ). BaF/3 cells stably expressing ETV6-LYN or ETV6-LYN KD (designated herein as BaF3/ETV6-LYN and BaF3/ETV6-LYN KD, respectively) were established by infecting cells with either the ETV6-LYN or ETV6-LYN KD retrovirus, followed by selection for puromycin-resistance in culture. For proliferation assays, Ba/F3 cells were plated at 5×104/well in 24-microtitre plates in triplicate and cultured with or without 2 ng/ml of IL-3. The number of the cells was counted at 24, 48, and 72 h of culture.
Western blotting and immunoprecipitation
The expression of ETV6-LYN was detected by Western blotting of BaF3/ETV6-LYN cells using a mouse anti-human ETV6 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). To identify the subcellular localization of ETV6-LYN, BaF3/ ETV6-LYN cells were lysed with a buffer [0.5% Nonidet P-40 [P-40], 50 mmol/l Tris-HCl (pH 8.0), 5 mmol/l EDTA (pH 8.0), and 150 mmol/l NaCl] and the soluble fraction was used as the cytoplasmic protein fraction. The remaining fraction was sonicated in a buffer [0.1% NP-40, 0.5 mol/l EDTA (pH 7.4), 300 mmol/l NaCl, and 20 mmol/l sodium pyrophosphate] and the soluble proteins served as the nuclear protein fraction. To detect the tyrosine phosphorylation of ETV6-LYN in BaF3/ETV6-LYN cells, ETV6-LYN was immunoprecipitated from the cell lysate with an anti-ETV6 antibody and tyrosine phosphorylation was detected with an antibody against phosphotyrosine (4G10, Santa Cruz Biotechnology). The blot was stripped of primary antibodies and reprobed with an anti-ETV6 antibody. For detection of the phosphorylation status of JAK2, Erk1/2, p38 MAPK and Akt, BaF3/ETV6-LYN and control cells were lysed with a buffer [1.0% Triton X-100, 50 mmol/L HEPES (pH 7.4), 10% glycerol, 4 mmol/L EDTA (pH 7.4), 150 mmol/L NaCl, and PhosSTOP (Roche Applied Science, Indianapolis, IN)] and sonicated. Equal amounts of whole cell lysate were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and probed with an anti-phospho-JAK2, anti-phospho-Erk1/2, anti-phospho p38 MAPK or anti-phopho-Akt antibody (Cell Signaling Technology, Danvers, MA). For detection of the phosphorylation of JNK, STAT3 and STAT5, the proteins were immunoprecipitated with an anti-JNK, anti-STAT3 or STAT5B antibody (Cell Signaling Technology) and tyrosine phosphorylation was detected with an anti-phospho-JNK for JNK and 4G10 for STAT3 and STAT5.
In vitro kinase assay
Ba/F3 and BaF3/ETV6-LYN cells, deprived of IL-3 for 12 h, were used for the phosphorylation assay. These cells (5 × 106) were lysed with a lysis buffer [1% Triton X-100, 50 mmol/l HEPES (pH 7.4), 10% glycerol, 10 mmol/l sodium pyrophosphate, 100 mmol/l sodium fluoride, 4 mmol/l EDTA, 2 mmol/l sodium orthovanadate, and PhosStop]. STAT5 in the Ba/F3 cell lysate was immunoprecipitated with an anti-STAT5B antibody and ETV6-LYN in the BaF3/ETV6-LYN cell lysate was immunoprecipitated with an anti-ETV6 antibody or an anti-mouse IgG. STAT5 and ETV6-LYN proteins on protein G sepharose beads were mixed in kinase buffer [0.1% Triton X-100, 20 mmol/l HEPES (pH 7.4), 10 mmol/l sodium pyrophosphate, and 4 mmol/l EDTA] and incubated at 37°C or 4°C for 30 min with or without 100 μM ATP. Tyrosine phosphorylation of STAT5 was evaluated by Western blotting with an antiphospho-STAT5 antibody. The in vitro kinase assay was similarly performed with the ETV6-LYN protein translated in vitro using a TNT reticulocyte lysate transcription/translation system (Promega, San Luis Obispo, CA) and GST-STAT5A fusion protein.
Purification of mouse HSCs and colony assay
Mouse HSCs (CD150+c-Kit+Sca-1+lineage marker-: CD150+KSL cells) were purified from E14.5 fetal liver cells of C57BL/6 mice. Mononuclear cells were isolated on Ficoll-Paque™ PLUS (GE Healthcare, Buckinghamshire, UK) and incubated with a mixture of biotin-conjugated monoclonal antibodies against lineage markers including Gr-1, Ter-119, B220, CD4, and CD8α (e-Bioscience, San Diego, CA). Lineage-positive cells were depleted with goat anti-rat IgG microbeads (Miltenyi Biotec GmbH, Germany). The cells were further stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD150, phycoerythrin (PE)-conjugated anti-Sca-1, and allophycocyanin (APC)-conjugated anti-c-Kit antibodies (e-Bioscience). Biotinylated antibodies were detected with streptavidin-APC-Cy7 (BD Biosciences, San Jose, CA). Four-colour analysis and sorting were performed on a desktop cell sorter JSAN (Bay Bioscience, Kobe, Japan). The CD150+KSL cells were infected with the control or ETV6-LYN retrovirus as previously described (Tanaka et al, 2010). Infected cells (50 cells) were plated into 35-mm Petri dishes in 1.1 ml of Methocult M3230 methylcellulose medium (StemCell Technologies Inc., Vancouver, Canada) with or without growth factors [50 ng/ml murine SCF, 20 ng/ml murine IL-3, 2 units/ml human erythropoietin, and 50 ng/ml human thrombopoietin (PeproTech)]. After 10 days of culture, the number of colonies was counted and the colonies were recovered and examined morphologically. The cultures were done in triplicate.
Transplantation of HSCs to irradiated mice
The c-Kit+ E14.5 fetal liver cells were purified using anti-c-Kit magnetic microbeads and LS MACS columns (Miltenyi Biotech, Auburn, CA) and transduced with the control vector (MigR1) or ETV6-LYN as previously described (Takeuchi et al, 2008). The transduced c-Kit+ cells (3×105) were transplanted into lethally irradiated B6-Ly5.1 mice along with 2×105 B6-Ly5.1 BM competitor cells (Ly5.1). The chimerism of donor-derived haematopoiesis was monitored by flow cytometry. Peripheral blood cells were stained with a mixture of monoclonal antibodies that included PE-anti-Gr-1, APC-Cy7-anti-Mac-1, Pacific blue-anti-CD45.2, and PE-Cy7-anti-CD45.1. The proportion of GFP-positive donor cells was evaluated by dividing the number of GFP-positive CD45.2-positive cells by the total number of CD45-positive cells (CD45.1+CD45.2). Peripheral blood white blood cell counts were made determined by an automated cell counter, Celltec α (Nihon Kohden, Tokyo, Japan).
Results
ETV6-LYN supports cytokine-independent proliferation of Ba/F3 cells
To examine the signalling pathways activated by ETV6-LYN in haematopoietic cells, we prepared a kinase-dead ETV6-LYN fusion gene (ETV6-LYN KD), which harbours a lysine to alanine mutation at the ATP-binding site corresponding to K275 of its wild-type counterpart (Fig 1A). ETV6-LYN and ETV6-LYN KD were stably expressed in a mouse IL-3 dependent pro-B cell line, Ba/F3 (BaF3/ETV6-LYN and BaF3/ETV6-LYN KD, respectively).
Fig 1. ETV6-LYN supports cytokine-independent proliferation of Ba/F3 cells.

(A) Schematic representation of ETV6-LYN and its kinase-dead mutant. The kinase-dead mutant of ETV6-LYN (ETV6-LYN KD) was constructed by replacing the C-terminal portion of ETV6-LYN with that of the kinase-dead mutant of LYN (K275A). (B) Detection of ETV6-LYN protein in BaF3/ETV6-LYN cells. The cytoplasmic proteins recovered from Ba/F3 and BaF3/ETV6-LYN cells were separated by SDS-PAGE under reducing (lanes 1 and 2) and non-reducing (lanes 3 and 4) conditions, and then transferred and probed with an anti-Flag antibody. Arrowheads indicate ETV6-LYN oligomers. (C) Detection of tyrosine phosphorylation of ETV6-LYN. ETV6-LYN and ETV6-LYN KD were immunoprecipitated from IL-3-depleted BaF3/ETV6-LYN and BaF3/ETV6-LYN KD cells, respectively, and detected by Western blotting using anti-phosphotyrosine (4G10) and anti-ETV6 antibodies. (D) Detection of kinase activity of ETV6-LYN. In vitro-translated ETV6-LYN and ETV6-LYN KD proteins were subjected to an in vitro kinase assay and detected by Western blotting using anti-phosphotyrosine and anti-ETV6 antibodies. (E) Proliferation of Ba/F3 cells expressing ETV6-LYN or ETV6-LYN KD mutant. Ba/F3 cells were plated at 5×104/well in 24-microtitre plates in triplicate and cultured with or without 2 ng/ml of IL-3. The number of cells was counted at 24, 48, and 72 h of culture. Data are shown as the mean ± SD (n = 3).
ETV6-PTK fusion products are constitutively active as kinases through oligomerization mediated by the PNT domain of ETV6 (Bohlander 2005). A part of ETV6-LYN migrated significantly slowly on an SDS-PAGE gel under non-reducing conditions, showing an approximately 3-fold larger molecular weight compared to that under reducing conditions (75 kDa) (Fig 1B). A faint but even larger band was also detected (Fig 1B). These findings indicate that ETV6-LYN forms oligomers. As expected, ETV6-LYN was highly phosphorylated on tyrosines even in the absence of IL-3. In contrast, ETV6-LYN KD was not phosphorylated at all, indicating that ETV6-LYN KD does not possess kinase activity (Fig 1C). These findings were confirmed by in vitro kinase assays. ETV6-LYN and ETV6-LYN KD were generated by in vitro transcription/translation using reticulocyte lysate and subjected to an in vitro kinase assay. ETV6-LYN but not ETV6-LYN KD was autophosphorylated (Fig 1D). These results indicate that ETV6-LYN forms oligomers and becomes activated through autophosphorylation. In agreement with these findings, ETV6-LYN supported the proliferation of Ba/F3 cells in the absence of IL-3 while ETV6-LYN KD did not (Fig 1E).
Intracellular signalling pathways activated in BaF3/ETV6-LYN cells
We next examined the signalling pathways activated by the ETV6-LYN fusion protein in Ba/F3 cells. Parental Ba/F3 and BaF3/ETV6-LYN KD cells were deprived of IL-3 for 12 h and further incubated with or without IL-3 for 15 min. BaF3/ETV6-LYN cells were also deprived of IL-3 for 12 h. IL-3 activated JAK2, STAT5, and Erk1/2, and Akt to a lesser extent, but not STAT3, in parental Ba/F3 cells. In contrast, STAT5, STAT3, and Akt were constitutively activated in BaF3/ETV6-LYN cells in the absence of IL-3 (Fig 2A). Among these signalling molecules, STAT5 was activated most prominently and this occurred without the activation of JAK2, the major kinase for STAT5 (Benekli et al, 2003; Hennighausen and Robinson, 2008). On the other hand, ETV6-LYN KD failed to activate STAT5 in the absence of IL-3 (Fig 2B), suggesting the involvement of ETV6-LYN in the JAK2-independent activation of STAT5. To understand the relationship between the ETV6-LYN signalling pathway and the canonical cytokine signalling pathway, we treated BaF3/ETV6-LYN cells with tyrosine kinase inhibitors. Dasatinib, but not imatinib, is known to effectively inhibit LYN catalytic activity. As expected, dasatinib but not imatinib inhibited the activation of STAT5 in the absence of IL-3. However, treatment of cells with dasatinib together with IL-3 failed to inhibit STAT5 activation (Fig 2C). These findings indicate that the ETV6-LYN-STAT5 signalling pathway is totally independent of the IL-3 receptor-JAK2-STAT5 signalling pathway.
Fig 2. ETV6-LYN activates STAT5 in a JAK2-independent manner.
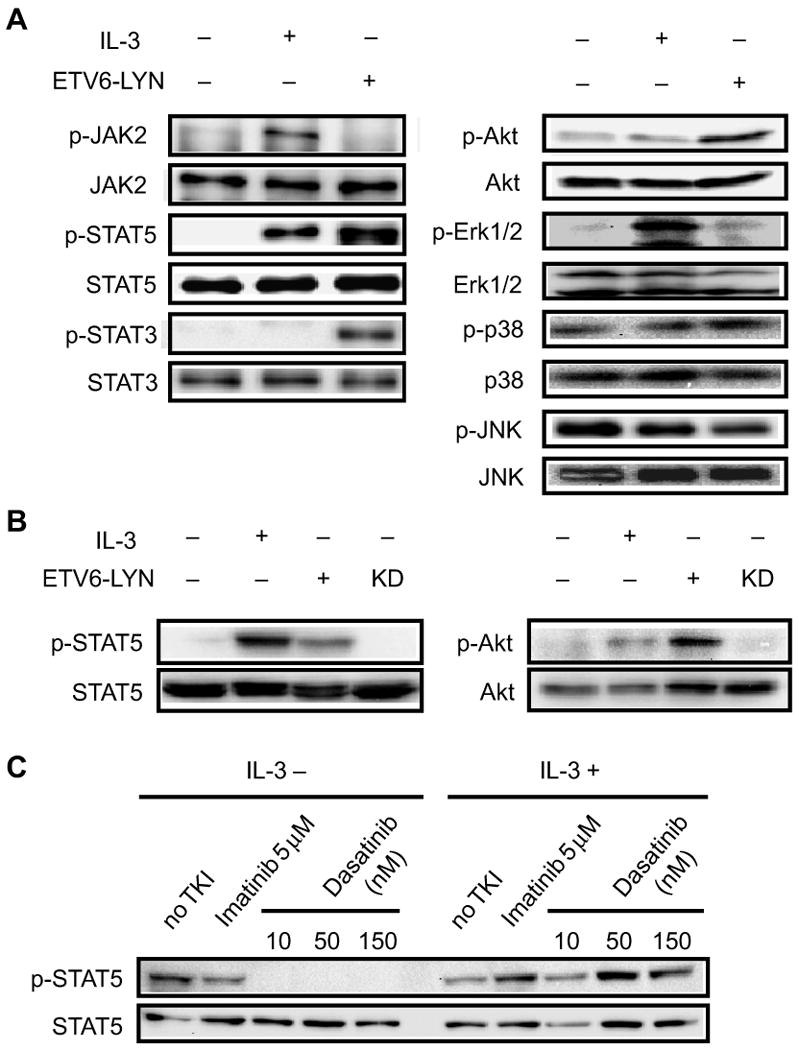
(A) Signalling pathways activated by ETV6-LYN in Ba/F3 cells. Parental Ba/F3 were deprived of IL-3 for 12 h and then further incubated with or without IL-3 for 15 min. BaF3/ETV6-LYN cells were deprived of IL-3 for 12 h. The cells were lysed and the phosphorylation status of JAK2, STAT5, STAT3, Akt, Erk1/2, p38, and JNK were examined by Western blotting. To detect Erk1/2, p38, and Akt, equal amount of whole cell lysate were used and probed with anti-phospho-Erk1/2, anti-phospho-p38, or anti-phospho-Akt antibody (upper panels), and reprobed with anti-Erk1/2, anti-p38, or anti-Akt antibody (lower panels). To detect JNK, JAK2, STAT3 and STAT5 activation, JNK, JAK2, STAT3 or STAT5 proteins were immunoprecipitated and probed with anti-phospho-JNK, anti-phospho-JAK2, or anti-phosphotyrosine antibody, and reprobed with anti-JNK, anti-JAK2, anti-STAT3, or anti-STAT5 antibody. (B) Kinase activity of ETV6-LYN KD in Ba/F3 cells. Parental Ba/F3 cells were deprived of IL-3 for 12 h and then further incubated with or without IL-3 for 15 min. BaF3/ETV6-LYN and BaF3/ETV6-LYN KD cells were deprived of IL-3 for 12 h. The cells were lysed and the phosphorylation status of STAT5 and Akt was examined by Western blotting as in (A). (C) STAT5 activation by ETV6-LYN vs. canonical cytokine signalling pathway. BaF3/ETV6-LYN cells were treated with dasatinib or imatinib with or without IL-3. The phosphorylation status of STAT5 was examined as in (A).
ETV6-LYN interacts with STAT5
The JAK2/STAT5 pathway has been demonstrated to be essential for induction of MPN by ETV6-JAK2 and ETV6-PDGFRB in mice (Schwaller et al, 2000; Cain et al, 2007). Furthermore, we and others have reported that mice that received transplants of cells expressing a constitutively active mutant of STAT5A but not STAT3 developed a lethal MPN (Schwaller et al, 2000; Kato et al, 2005). Based on these findings, we hypothesized that ETV6-LYN directly activates STAT5. We first performed subcellular fractionation experiments to identify the subcellular distribution of ETV6-LYN. ETV6-LYN was detected only in the cytoplasmic fraction of BaF3/ETV6-LYN cells (Fig 3A). We then examined whether ETV6-LYN and STAT5 could physically interact. Cytoplasmic proteins were recovered from 293T cells transfected with Flag-ETV6-LYN and endogenous STAT5B was immunoprecipitated using an anti-STAT5B antibody. Of note, ETV6-LYN was co-immunoprecipitated with STAT5B (Fig 3B). These results indicate that ETV6-LYN physically interacts with STAT5.
Fig 3. Subcellular distribution of ETV6-LYN and STAT5.
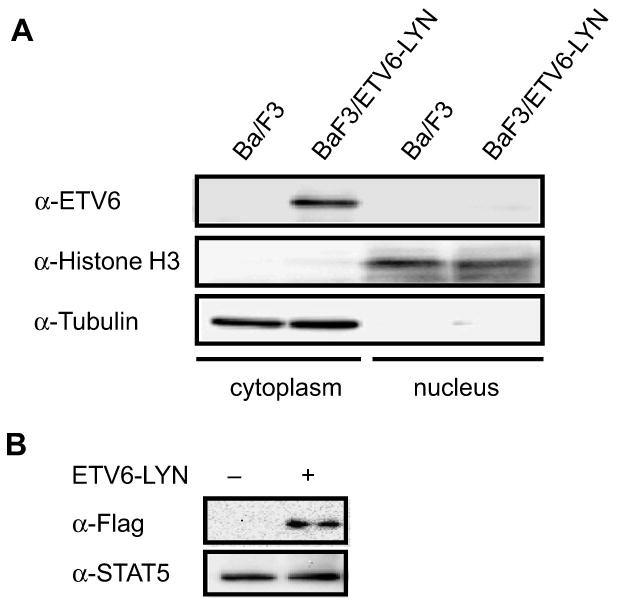
(A) Detection of ETV6-LYN by subcellular fractionation. The cytoplasmic and nuclear fractions were prepared from BaF3/ETV6-LYN cells and ETV6-LYN was detected by Western blotting using an anti-ETV6 antibody. (B) Physical interaction between ETV6-LYN and STAT5. Cytoplasmic proteins were prepared from 293T cells transiently transfected with Flag-ETV6-LYN, and endogenous STAT5 was immunoprecipitated using an anti-STAT5B antibody. The immunoprecipitates were probed with anti-Flag and anti-STAT5B antibodies.
ETV6-Lyn directly phosphorylates STAT5
We next examined whether ETV6-LYN directly phosphorylated STAT5 or not in vitro. ETV6-LYN and ETV6-LYN KD were immunoprecipitated from BaF3/ETV6-LYN cultured without IL-3 and BaF3/ETV6-LYN KD cells cultured with IL-3, respectively using an anti-ETV6 antibody. STAT5 was immunoprecipitated from Ba/F3 cells deprived of IL-3 for 12 h using an anti-STAT5B antibody. STAT5 was phosphorylated when incubated with ETV6-LYN at 30°C in the presence of ATP, but not when incubated with ETV6-LYN KD, at 4°C, or in the absence of ATP (Fig 4A).
Fig 4. ETV6-LYN directly phosphorylates STAT5.
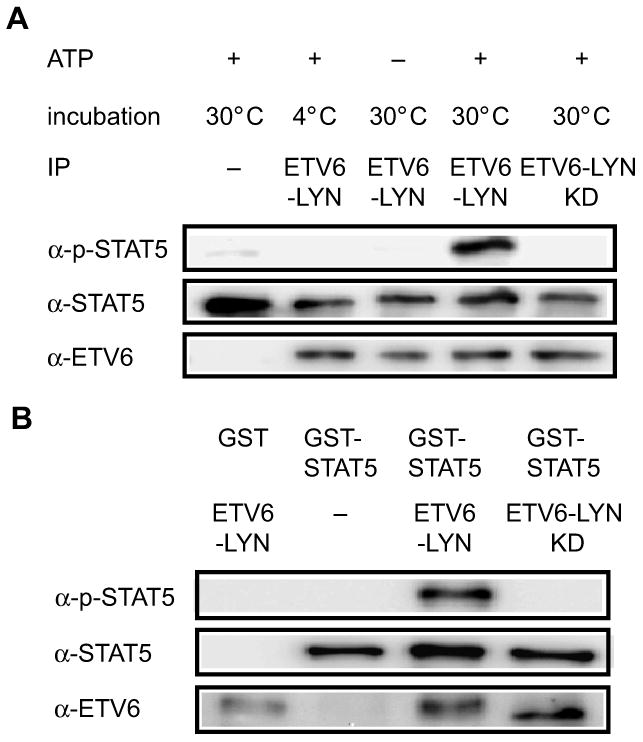
(A) In vitro kinase assays using ETV6-LYN and STAT5 immunoprecipitates. ETV6-LYN and ETV6-LYN KD were immunoprecipitated from BaF3/ETV6-LYN cultured without IL-3 and BaF3/ETV6-LYN KD cells cultured with IL-3, respectively, using an anti-ETV6 antibody. STAT5 was immunoprecipitated from Ba/F3 cells deprived of IL-3 for 12 h using an anti-STAT5B antibody. The in vitro kinase assay was performed under the indicated conditions. The proteins were then detected by Western blot analysis using anti-phosphotyrosine and anti-ETV6 antibodies. (B) In vitro kinase assays using ETV6-LYN and GST-STAT5A. GST-STAT5A and in vitro-translated ETV6-LYN and ETV6-LYN KD proteins were subjected to in vitro kinase assay under the indicated conditions. The proteins were then detected by Western blot analysis using anti-phosphotyrosine and anti-ETV6 antibodies.
To rule out the possibility that the other kinases co-immunprecipitated with LYN or STAT5 were responsible for STAT5 phosphorylation, we prepared GST-STAT5A and ETV6-LYN and ETV6-LYN KD proteins synthesized by in vitro transcription/translation using reticulocyte lysate. ETV6-LYN but not ETV6-LYN KD phosphorylated GST-STAT5A in vitro (Fig 4B), further supporting direct action of ETV6-LYN on STAT5.
ETV6-LYN does not promote proliferation of Stat5-/- HSCs in vitro
To determine the biological significance of STAT5 activation by ETV6-LYN in primary haematopoietic cells, we tested the effect of ETV6-LYN on HSCs in a Stat5-null background. Stat5-/- mice show perinatal lethality due to severe anaemia combined with other physiological defects (Cui et al, 2004). Therefore, we purified CD150+KSL haematopoietic stem/progenitor cells from E14.5 fetal livers of Stat5-/- embryos, which lack both the Stat5a and Stat5b genes (Cui et al, 2004), and transduced them with ETV6-LYN. As we reported previously (Tanaka et al, 2010), ETV6-LYN enhanced the colony-forming activity of wild-type HSCs in the presence of cytokines. Notably, ETV6-LYN promoted the formation of high proliferative potential (HPP) colonies larger than 1 mm in diameter (Fig 5A). ETV6-LYN also supported the formation of colonies even under cytokine-free conditions. In contrast, Stat5-/- haematopoietic stem/progenitor cells showed profoundly impaired colony-forming activity as previously reported (Li et al, 2007), and mostly gave rise to low proliferative potential (LPP) colonies smaller than 1 mm in diameter. ETV6-LYN did not promote the formation of colonies by Stat5-/- HSCs in the presence of cytokines and failed to support the colony-forming process under cytokine-free conditions. Morphological analyses of the colonies revealed that ETV6-LYN supported the differentiation of wild-type CD150+KSL cells into all myeloid lineages, including neutrophils, macrophages, erythroblasts, and megakaryocytes, even in the absence of cytokines, but had no effect on the differentiation of Stat5-/- cells which exclusively differentiated into neutrophils and macrophages in vitro (Fig 5B).
Fig 5. ETV6-LYN does not induce fatal MPN in the absence of STAT5.

(A) Colony-forming capacity of Stat5-/- HSCs expressing ETV6-LYN. CD150+KSL haematopoietic stem/progenitor cells from E14.5 fetal livers of wild-type and Stat5-/- embryos were transduced with either control vector or the ETV6-LYN retrovirus. Colonies were formed in the presence and absence of cytokines (20 ng/ml murine SCF, 20 ng/ml murine IL-3, 2 units/ml human erythropoietin, and 50 ng/ml human thrombopoietin). Colony numbers with the indicated size per 150 CD150+KSL cells are depicted as the mean ± SD for triplicate cultures. C and EL denote control and ETV6-LYN, respectively. (B) Proportion of colony types. The colonies were recovered and examined by microscopy to determine their composition (n; neutrophil, m; macrophage, E; erythroblast, M; megakaryocyte). C and EL denote control and ETV6-LYN, respectively. (C) Survival of recipient mice infused with wild-type and Stat5-/- cells transduced with ETV6-LYN. Wild-type and Stat5-/- c-Kit+ cells from E14.5 fetal livers were transduced with ETV6-LYN and transplanted into lethally irradiated B6-Ly5.1 mice. One out of the 5 recipient mice transplanted with Stat5-/- cells expressing ETV6-LYN died following a non-haematological accident (*).
STAT5 plays a crucial role in ETV6-LYN-induced MPN
To examine the involvement of STAT5 in the transformation properties of ETV6-LYN in vivo, we transduced purified c-Kit+ haematopoietic stem/progenitor cells from E14.5 wild-type and Stat5-/- fetal livers with ETV6-LYN and transplanted them into lethally irradiated B6-Ly5.1 mice. All of the 11 mice transplanted with wild-type cells expressing ETV6-LYN died within 3 months after transplantation (Fig 5C). The mice showed leucocytosis in peripheral blood (Table 1) with increasing numbers of Gr-1+Mac-1+ myeloid cells (data not shown). As we reported previously (Tanaka et al, 2010), they also had hypercellular BM and huge splenomegaly with destroyed white pulp caused by extramedullary haematopoiesis. Massive fibrosis was observed in both BM and spleen in their terminal stage of the disease while abnormal megakaryocytopoiesis was evident rather than fibrosis in their early stage of the disease (data not shown). In contrast, 5 mice transplanted with Stat5-/- cells expressing ETV6-LYN remained healthy over 6 months except for one mouse which died of a non-haematological accident (Fig 5C). These findings were confirmed by a repeated experiment and all recipients (n=5) that received ETV6-LYN-expressing Stat5-/- cells survived over 6 months (data not shown). The contribution of ETV6-LYN-expressing cells in peripheral blood was very low and the mice showed a normal range of white blood cell counts in peripheral blood (Table 1).
Table1. Peripheral blood data of recipient mice.
| Day 30 | Day 60 | Day 180 | |
|---|---|---|---|
| WT | |||
| WBC (× 109/l) | 27.94±44.331 | 28.338±33.882 | ** |
| (3.9 - 148.4) | (5.4 - 103.3) | ||
| n=10 | n=8 | ||
| Chimerism (%)* | 71.1±16.5 | 80.2±13.7 | |
| (60.0±13.2) | (72.8±19.4) | ||
| Stat5-/- | |||
| WBC (× 109/l) | 2.02±0.42 | 6.4±1.487 | 7.525±4.152 |
| (1.4 - 2.5) | (4.9 - 8.5) | (3.4 - 11.1) | |
| n=5 | n=5 | n=4 | |
| Chimerism (%)* | 12.9±6.94 | 5.00±3.0 | 1.90±1.75% |
| (4.69±1.86) | (1.04±0.56) | (1.01±0.96) |
Percentage of chimerism in total CD45.2 donor cells. Percentage of chimerism in CD45.2+GFP+ cells expressing ETV6-LYN is indicated in parentheses.
All mice were dead.
Because Stat5-/- HSCs have a severe repopulation defect (Yao et al, 2006; Dai et al, 2007; Li et al, 2007), the attenuation in disease development in the absence of Stat5 could be largely attributed to the defective repopulation capacity of Stat5-/- HSCs. We therefore analysed BM and spleen of all surviving mice at 6 months post-transplant. The capacity of ETV6-LYN to induce MPN with myelofibrosis was profoundly attenuated in the absence of Stat5 (Supplementary Figure S1F). Nonetheless, compared to the histology of the mice transplanted with GFP-transduced wild-type control cells, their BM was hypercellular and had an increased number of megakaryocytes (Supplementary Figure S1C), which is one of the characteristics of ETV6-LYN-induced disease observed in the early stage, but not in the terminal fibrotic stage. Extramedullary haematopoiesis was also evident in spleen (Supplementary Figure S1I) and the splenic white pulp was destroyed in some mice (Supplementary Figure S1J). Furthermore, the capacity of Stat5-/- fetal liver cells to home to BM was demonstrated to be intact (Supplementary Figure S1K). These findings indicate that ETV6-LYN-expressing Stat5-/- fetal liver cells could repopulate in recipient mice, but cannot induce severe MPN with myelofibrosis in the absence of Stat5.
Discussion
ETV6-LYN is a new member of the ETV6-PTK family. In ETV6-PTK proteins, the PNT domain of ETV6 serves as an oligomerization module and induces the constitutive activation of the PTK domain through autophosphorylation (Bohlander 2005). In this study, we confirmed that ETV6-LYN functions as an active kinase through oligomerization and activates STAT3 and STAT5 in a JAK2-independent manner. The kinase activity was totally dependent on the portion of LYN as the ETV6-LYN kinase-dead mutant did not have any kinase activity at all. Our findings correspond well to those of Abe et al. (2004) who identified ETV6-LYN fusion genes in a screening of new partners of ETV6 using ETV6-cDNA libraries. These authors cloned 25 independent artificial fusion proteins that supported cytokine-independent growth of 32D cells. Among them, 12 clones were ETV6-LYN fusion genes. Because the deletion of the PNT domain of this ETV6-LYN fusion protein resulted in a loss of transforming activity, the transformation capacity of the ETV6-LYN fusion protein was thus considered to depend on the PNT domain of ETV6. Altogether, this evidence indicates that the ETV6-LYN fusion protein is constitutively active through oligomerization and activates downstream signalling pathways responsible for the pathogenesis of MPN.
We clearly showed that ETV6-LYN directly activates STAT5 and the capacity of ETV6-LYN to induce MPN with myelofibrosis was profoundly attenuated in the absence of STAT5. The JAK2-STAT5 pathway is the major pathway affected by genomic mutations or activated by causative chimeric fusions in MPN. Several fusions have been reported to activate STAT5, including ETV6-JAK2, and ETV6-PDGFRB, and all of them largely depend on STAT5 for their tumorigenic activity (Schwaller et al, 2000; Cain et al, 2007). Essentially, expression of constitutively active STAT5A is enough to induce MPN-like disease (Schwaller et al, 2000; Kato et al, 2005). Interestingly, however, we have previously demonstrated that expression of constitutively active STAT5A in CD34-KSL HSCs but not in CD34+KSL multipotent progenitor cells (MPPs) induced fatal MPN in mice (Kato et al, 2005). These findings indicate that constitutive activation of STAT5A promotes self-renewal of HSCs but cannot confer a self-renewal capacity to MPPs, although it strongly enhances the proliferative capacity of MPPs. Thus, augmentation of the self-renewal capacity of HSCs by STAT5A is crucial for induction of MPN, a group of HSC disorders. In this regard, our findings provide the ETV6-LYN-STAT5 axis as a novel pathway to transmit aberrant self-renewal signals in MPN.
The SRC family kinases have also been implicated in the activation of STAT pathways (Silva 2004). Activation of STAT3 by v-Src was originally identified (Silva 2004) but a series of in vitro studies in insect cells showed that SRC, HCK, LYN, FYN, and FGR are all capable of tyrosine phosphorylating STAT3 (Schreiner et al, 2002). Among SRC family kinases, LYN has been characterized to play a major role in B cells (Xu et al, 2005), but recent studies revealed that LYN is also involved in cytokine receptor signalling such as erythropoietin receptor signalling (Tilbrook et al, 1997). LYN physically interacts with the erythropoietin receptor and is proposed to play a role in activation of the STAT5 pathway (Chin et al, 1998). Our findings of ETV6-LYN in this study support the role of the SRC family kinases in the regulation of STAT pathways and implicate active LYN in the alternative pathway for STAT activation in pathological cytokine signalling.
Of interest, SRC family kinases LYN, HCK, and FGR are reportedly dispensable for ETV6-PDGFRB-mediated MPN in mice, whereas STAT5, which is directly activated by ETV6-PDGFRB, is indispensable (Cain et al, 2007). By contrast, a requirement of SRC kinases LYN, HCK, and FGR is reported for BCR-ABL1-induced B-lymphoblastic leukaemia but not CML (Hu et al, 2004). SRC kinases are generally regarded to be important but not crucial downstream targets of BCR-ABL1 kinase in chronic-phase CML. However, other reports have shown that overexpression and activation of LYN are hallmarks of cells from patients with imatinib-resistant, BCR-ABL1 mutation-negative CML, although the regulatory mechanism of LYN activity remained obscure (Donato et al, 2003; Ptasznik et al, 2004). Notably, these patients with BCR-ABL1-independent resistance responded to dasatinib, an inhibitor of BCR-ABL1 and LYN, as well as other SRC family members (Donato et al, 2003). These results suggest that therapies targeting both BCR-ABL1 and LYN may prove beneficial in certain circumstances for imatinib-resistant CML. Furthermore, SRC family kinases appeared constitutively activated in most AML cases. Among the SRC family kinases, LYN is highly expressed and constitutively phosphorylated at a high level in AML cells. LYN knockdown by small interfering RNA strongly inhibited the proliferation of primary AML cells (Dos Santos et al, 2008). Aberrantly activated LYN in AML and imatinib-resistant CML might augment their leukaemic activity by constitutively activating STAT5 as in the case of ETV6-LYN. Furthermore, STAT5 has recently been implicated in leukaemia stem cell self-renewal in AML patients (Heuser et al, 2009). In these reports, however, the expression and kinase activity of LYN were not evaluated in relation to those of STAT5. Thus, it would be of importance to evaluate the contribution of LYN to the activation of STAT5 in those patients.
MPN is often accompanied by myelofibrosis. In this study, ETV6-LYN also induced massive myelofibrosis in a short time. Myelofibrosis is thought to be the consequence of an excessive release/leakage of growth factors within the BM by cells from pathological haematopoietic clones, especially by necrotic megakaryocytes. Transforming growth factor-β1 (TGF-β1) is speculated to be one of the major causative growth factors that activate mesenchymal cells (Martyré et al, 1994). The capacity of ETV6-LYN to induce myelofibrosis was well correlated with its capacity to induce MPN, indicating again the major role of STAT5 signalling in myelofibrosis. Correspondingly, expression of the constitutively active STAT5 alone is sufficient to induce myelofibrosis associated with MPN in mice (Schwaller et al, 2000). So far, we have identified the ETV6-LYN fusion only in one case of IMF. In this regard, it would be intriguing to test whether there are any activating mutations of LYN in IMF. Finally, as evident in Figure 2C, dasatinib but not imatinib is effective in inhibiting catalytic activity of ETV6-LYN. Thus, dasatinib could be an effective therapeutic agent to treat patients with ETV6-LYN.
Supplementary Material
Acknowledgments
We thank Dr. T. Kitamura and W. Pear for providing the pMXs-puro vector and MigR1 vector, respectively. This work was supported in part by Grants-in-Aid for scientific research (#20591110) and the Global COE Program (Global Centre for Education and Research in Immune System Regulation and Treatment), MEXT, Japan, a Grant-in-aid for Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation (JST), and a grant from the Takeda Science Foundation. LH was supported by the international program of the National Institutes of Health (NIDDK).
Footnotes
Authorship Contributions: Y.T. designed and performed experiments, interpreted the data, and wrote the paper; H.T., M.T., M.Y., A.S., S.M., C.W., S.T., C.O., and E.S. performed experiments; N.Y. designed in vitro phosphorylation experiments and provided materials; K.Y. designed the experiments; L.H. provided Stat5+/- mice and instructed the experiments using Stat5+/- mice; and C.N. and A.I. designed experiments and were responsible for execution of the research, interpretation of the data, and writing the paper.
Conflict of interest: The authors declare no conflict of interest.
References
- Abe A, Emi N, Kanie T, Imagama S, Kuno Y, Takahashi M, Saito H, Naoe T. Expression cloning of oligomerization-activated genes with cell-proliferating potency by pseudotype retrovirus vector. Biochemical Biophysical Research Communications. 2004;320:920–926. doi: 10.1016/j.bbrc.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- Bohlander SK. ETV6: a versatile player in leukemogenesis. Seminars in Cancer Biology. 2005;15:162–174. doi: 10.1016/j.semcancer.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Cain JA, Xiang Z, O'Neal J, Kreisel F, Colson A, Luo H, Hennighausen L, Tomasson MH. Myeloproliferative disease induced by TEL-PDGFRB displays dynamic range sensitivity to Stat5 gene dosage. Blood. 2007;109:3906–3914. doi: 10.1182/blood-2006-07-036335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood. 1998;91:3734–3745. [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Molecular and Cellular Biology. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Chen Y, Di L, Podd A, Li G, Bunting KD, Hennighausen L, Wen R, Wang D. Stat5 is essential for early B cell development but not for B cell maturation and function. Journal of Immunology. 2007;179:1068–1079. doi: 10.4049/jimmunol.179.2.1068. [DOI] [PubMed] [Google Scholar]
- Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–698. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- Dos Santos C, Demur C, Bardet V, Prade-Houdellier N, Payrastre B, Recher C. A critical role for Lyn in acute myeloid leukemia. Blood. 2008;111:2269–2279. doi: 10.1182/blood-2007-04-082099. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes and Development. 2008;22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser M, Sly LM, Argiropoulos B, Kuchenbauer F, Lai C, Weng A, Leung M, Lin G, Brookes C, Fung S, Valk PJ, Delwel R, Löwenberg B, Krystal G, Humphries RK. Modeling the functional heterogeneity of leukemia stem cells: role of STAT5 in leukemia stem cell self-renewal. Blood. 2009;114:3983–3993. doi: 10.1182/blood-2009-06-227603. [DOI] [PubMed] [Google Scholar]
- Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nature Genetics. 36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, van Lohuizen M, Nakauchi H. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–851. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Nakayama Y, Ikeda K, Fukushima Y, Matsuda D, Horimoto S, Yamaguchi N. Trafficking of Lyn through the Golgi caveolin involves the charged residues on αE and αI helices in the kinase domain. Journal of Cell Biology. 2004;165:641–652. doi: 10.1083/jcb.200403011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Iwama A, Tadokoro Y, Shimoda K, Minoguchi M, Akira S, Tanaka M, Miyajima A, Kitamura T, Nakauchi H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. Journal of Experimental Medicine. 2005;202:169–179. doi: 10.1084/jem.20042541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Koshino Y, Shibata F, Okia T, Nakajima H, Nosaka T, Kumagai H. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Experimental Hematology. 2003;31:1007–1014. [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. New England Journal of Medicine. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112:2190–2198. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Wang Z, Zhang Y, Kang Z, Haviernikova E, Cui Y, Hennighausen L, Moriggl R, Wang D, Tse W, Bunting KD. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Experimental Hematology. 2007;35:1684–1694. doi: 10.1016/j.exphem.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyré MC, Romquin N, Le Bousse-Kerdiles MC, Chevillard S, Benyahia B, Dupriez B, Demory JL, Bauters F. Transforming growth factor-beta and megakaryocytes in the pathogenesis of idiopathic myelofibrosis. British Journal of Haematology. 1994;88:9–16. doi: 10.1111/j.1365-2141.1994.tb04970.x. [DOI] [PubMed] [Google Scholar]
- Palacios R, Steinmetz M. Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell. 1985;41:727–734. doi: 10.1016/s0092-8674(85)80053-2. [DOI] [PubMed] [Google Scholar]
- Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, DeAngelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nature Medicine. 2004;10:1187–1189. doi: 10.1038/nm1127. [DOI] [PubMed] [Google Scholar]
- Schreiner SJ, Schiavone AP, Smithgall TE. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. Journal of Biological Chemistry. 2002;277:45680–45687. doi: 10.1074/jbc.M204255200. [DOI] [PubMed] [Google Scholar]
- Schwaller J, Parganas E, Wang D, Cain D, Aster JC, Williams IR, Lee CK, Gerthner R, Kitamura T, Frantsve J, Anastasiadou E, Loh ML, Levy DE, Ihle JN, Gilliland DG. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Molecular Cell. 2000;6:693–704. doi: 10.1016/s1097-2765(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Silva C. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigensis. Oncogene. 2004;23:8017–8123. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Nakaseko C, Miyagi S, Takeda Y, Ozawa S, Ohwada C, Cho R, Nishimura M, Saito Y, Iwama A. Clonal expansion of non-leukemic cells expressing two novel MLL-ELL variants differing in transforming activity. Leukemia. 2008;22:861–864. doi: 10.1038/sj.leu.2404954. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Takeuchi M, Takeda Y, Sakai S, Abe D, Ohwada C, Sakaida E, Shimizu N, Saito Y, Miyagi S, Iwama A, Nakaseko C. Identification of a novel TEL-Lyn fusion gene in primary myelofibrosis. Leukemia. 2010;24:197–200. doi: 10.1038/leu.2009.167. [DOI] [PubMed] [Google Scholar]
- Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA, Barosi G, Verstovsek S, Birgegard G, Mesa R, Reilly JT, Gisslinger H, Vannucchi AM, Cervantes F, Finazzi G, Hoffman R, Gilliland DG, Bloomfield CD, Vardiman JW. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110:1092–1097. doi: 10.1182/blood-2007-04-083501. [DOI] [PubMed] [Google Scholar]
- Tilbrook PA, Ingley E, Williams JH, Hibbs ML, Klinken SP. Lyn tyrosine kinase is essential for erythropoietin-induced differentiation of J2E erythroid cells. EMBO Journal. 1997;16:1610–1619. doi: 10.1093/emboj/16.7.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22:9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, Hennighausen L, O'Shea JJ. Stat5a/b are essential for normal lymphoid development and differentiation. Proceedings of the National academy of Sciences of the United States of America. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S, Van Etten RA. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.