

Abstract
Programmed cell death of cardiomyocytes following myocardial ischemia increases biomechanical stress on the remaining myocardium, leading to myocardial dysfunction that may result in congestive heart failure or sudden death. Nogo-A is well-characterized as a potent inhibitor of axonal regeneration and plasticity in the central nervous system, however, the role of Nogo-A in non-nervous tissues is essentially unknown. In this study, Nogo-A expression was shown to be significantly increased in cardiac tissue from patients with dilated cardiomyopathy and from patients who have experienced an ischemic event. Nogo-A expression was clearly associated with cardiomyocytes in culture and was localized predominantly in the endoplasmic reticulum. In agreement with the findings from human tissue, Nogo-A expression was significantly increased in cultured neonatal rat cardiomyocytes subjected to hypoxia/reoxygenation. Knockdown of Nogo-A in cardiomyocytes markedly attenuated hypoxia/reoxygenation-induced apoptosis, as indicated by the significant reduction of DNA fragmentation, phosphatidylserine translocation, and caspase-3 cleavage, by a mechanism involving the preservation of mitochondrial membrane potential, the inhibition of ROS accumulation, and the improvement of intracellular calcium regulation. Together, these data demonstrate that knockdown of Nogo-A may serve as a novel therapeutic strategy to prevent the loss of cardiomyocytes following ischemic/hypoxic injury.
Keywords: myocardial infarction, ischemic cardiomyopathy, cell death, reticulon-4A
Introduction
Cardiomyocyte loss following myocardial ischemia results in increased biomechanical stress on the remaining myocardium, leading to contractile dysfunction, left ventricular remodeling, and congestive heart failure [1-3]. While initial cardiomyocyte loss is primarily due to necrosis, apoptosis plays a key role in the damage of tissue bordering and distant from the infarcted myocardium subsequent to reperfusion [4-7]. Apoptosis is a highly regulated program of cell death that contributes significantly to the gradual decline of left ventricular function following ischemia/reperfusion (I/R) injury [8, 9]. Cardiomyocyte apoptosis following I/R can be mediated by activation of the death receptor pathway (extrinsic pathway), the mitochondria-dependent pathway (intrinsic pathway), or endoplasmic reticulum (ER) stress [10-15]. While death receptor-mediated apoptosis and ER stress are known to play a role in I/R injury, the mitochondria-dependent pathway serves as the critical integration site for a number of pro-apoptotic signals in the cardiomyocyte [16]. Following the sudden availability of oxygen during reperfusion, there is a burst of reactive oxygen species (ROS) production, increased mitochondrial permeability transition, decreased mitochondrial membrane potential (ΔΨm), and the release of pro-apoptotic factors, such as cytochrome c, ultimately leading to the activation of caspase-3 and apoptosis [17-20]. Protection of mitochondrial function is therefore a promising therapeutic strategy for the treatment of myocardial infarction.
Members of the reticulon (RTN) family of proteins contain a C-terminal reticulon-homology domain and are highly enriched in the membranes of the endoplasmic reticulum [21]. The largest of the RTN proteins, Nogo-A (RTN4-A), contains well-characterized regions essential for inhibiting neurite outgrowth and plasticity in the central nervous system (CNS) [22-26]. Following CNS injury, axonal regeneration, neuronal plasticity, and functional recovery are promoted using antibodies that neutralize Nogo-A function [27-29].
In addition to being highly expressed in the brain and spinal cord, Nogo-A is clearly detectable in other tissues, including the heart, where its function remains largely unclear despite evidence that it may play a role in normal cardiac development [22, 30-32]. Nogo-A expression in the CNS has been shown to be significantly up-regulated in several models of injury, including hypoxia/ischemia [33-38]. Interestingly, Nogo-A has been proposed as a potential indicator of heart failure due to its elevated expression in genetic models of dilated cardiomyopathy (DCM) and its increased mRNA expression in end-stage heart failure in humans [39, 40].
To better determine the relationship between Nogo-A up-regulation and heart failure, we evaluated Nogo-A protein expression in human left ventricular tissue from DCM and ischemic hearts and determined, in vitro, which cell type is primarily responsible for its expression. Nogo-A expression was found to be significantly increased in left ventricular tissue from DCM and ischemic hearts, as well as in cultured cardiomyocytes subjected to H-R injury. The pathophysiological significance of Nogo-A up-regulation in H-R-induced cardiomyocyte apoptosis was determined by knocking down its expression. Knockdown of Nogo-A in cardiomyocytes was found to significantly inhibit H-R-induced activation of the intrinsic pathway of apoptosis. Based on these findings, we propose that Nogo-A may serve as novel therapeutic target in the treatment of ischemic/hypoxic-related cardiovascular injury.
Materials and Methods
Human Tissue
Left ventricular (LV) tissue from patients with dilated, ischemic, or non-failing hearts was obtained from the Cardiovascular Institute Tissue Bank at Loyola University Medical Center. Acquired LV tissue was frozen in liquid nitrogen and immediately stored at −80°C. All surgical procedures and tissue harvesting were approved by the Loyola University Medical Center Institutional Review Board and were conducted in accordance with NIH guidelines.
Cell Culture
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health, using protocols approved by the Institutional Animal Care and Use Committee. Neonatal rat ventricular myocytes (NRVMs) were isolated from 1-2 day old Sprague-Dawley rats as previously described [41], plated on gelatin-coated 60 mm2 dishes (2.5×106 - 3.0×106 cells/60 mm2) in serum-free PC-1 medium (Lonza, Walkersville, MD), and allowed to attach for 14-18 h. Cells were subsequently maintained in (4:1) DMEM/medium 199 (HyClone Laboratories Inc., Logan, UT) containing (100 U/mL) penicillin/(100 μg/mL) streptomycin (Invitrogen, Carisbad, CA). Adult rabbit cardiomyocytes were prepared as previously described [42]. Neonatal rat fibroblasts were isolated via a pre-plating procedure during the preparation of NRVMs. Adherent cells, highly enriched in fibroblasts, were maintained in DMEM containing (100 U/mL) penicillin/(100 μg/mL) streptomycin and 10% FBS (HyClone Laboratories Inc., Logan, UT) and used after 4 passages.
Adenovirus Preparation and Infection
Nogo-A siRNA, targeting nucleotides 856-874 of the rat Nogo-A cDNA sequence, was designed as previously described [43]. Nogo-A sense and anti-sense oligonucleotides (Integrated DNA Technologies, Inc., Coralville, IA, USA), flanked by BamHI and HindIII restriction endonuclease sites, were annealed, subcloned into GenScript pRNA-H1.1/Adeno, and sequenced. The shNogo-A adenovirus, empty vector adenovirus control, and Nogo-A adenovirus containing the sequence for full length human Nogo-A were constructed using AdEasy Adenoviral Vector System (Stratagene, La Jolla, CA). Nogo-A cDNA containing plasmid was generously provided by Dr. Martin Schwab. Adenovirus expressing scrambled shRNA sequence was obtained from Vector Biolabs (Philadelphia, PA). Adenoviruses were amplified, purified, and titered as previously described [41]. NRVMs were infected after 24 h of culture at a multiplicity of viral infection (MOI) of 50 (Ad-shScr and Ad-shN) or 10 (Ad-Con and Ad-Nogo-A) and allowed 48 h to express the transgene prior to hypoxia/reoxygenation (H-R).
Hypoxia/Reoxygenation Injury
NRVMs were subjected to hypoxia using GasPak EZ Anaerobe Pouch System (Becton, Dickinson, and Company, Sparks, MD). Culture dishes were placed inside pouches containing gas generating sachets and incubated at 37°C for 24 h, unless noted otherwise. Following hypoxia, the media was changed with (4:1) DMEM/medium 199 and culture dishes were reoxygenated for 2 h. Normoxic controls were subjected to 24 h normoxia followed by a change of media and 2 h of additional normoxia. Cells subjected to ischemia were treated in the same manner, except that culture media was changed to standard ischemic medium [1.13 mM CaCl2, 0.5 mM KCl, 0.3 mM KH2PO4, 0.5 mM MgCl2, 0.4mM MgSO4, 128mM NaCl, 4mM NaHCO3, and 10mM HEPES] during hypoxia and control medium [ischemic medium supplemented with 0.3 mM Na2PO4 and 10mM d-glucose] during reoxygenation.
Immunofluorescence Staining
NRVMs were cultured on 4-well Lab-Tek Chamber Slides (Nalge Nunc International Corp., Naperville, IL) at a density of 3 × 105 cells/well. Cells were fixed with 4% paraformaldehyde (4°C) for 3 min, followed by 100% methanol (−20°C) for 1 min, and rinsed briefly with phosphate buffered saline (PBS). Slides were incubated at room temperature (rt) in PBS/ 0.5% Triton X-100 for 15 min, washed twice with PBS/0.1% Triton X-100 for 10 min, then blocked for 1 h with blocking buffer (PBS/0.1% Triton X-100/1% normal goat serum). Slides were incubated with 11μg/mL mouse anti-Nogo-A antibody (11c7), 1/200 rabbit anti-SERCA2a (Badrilla Ltd., Leeds, UK), or 1/1000 rabbit anti-VDAC (BioVision, Mountain View, CA) in blocking buffer overnight at 4°C. Cells were washed three times with PBS/0.1% Triton X-100, then incubated with 1/200 anti-mouse IgG-Alexa Fluor 488, 1/200 anti-rabbit IgG-Texas Red (Invitrogen, Carisbad, CA), and 50 ng/mL Hoechst 33342 (Sigma Aldrich, St. Louis, MO) in blocking buffer for 1 h at rt. Cells were washed with PBS/0.1% Triton X-100, mounted with Vectasheild (Vector Laboratories, Inc., Burlingame, CA), and imaged with a Zeiss Axiovert 100 microscope (Carl Zeiss Microimaging, LLC, Thornwood, NY)
Immunoblots
Cells were harvested in ice cold lysis buffer [44] with protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO), sonicated, and centrifuged at 16,000g for 5 min at 4°C to remove cellular debris. Extracted proteins were separated on 10% or 12% SDS-polyacrylamide gels, electrophoretically transferred onto nitrocellulose membrane, and incubated overnight with 3μg/mL anti-Nogo-A (11c7), 1/1000 anti-Nogo-A/B (Imgenex, San Diego, CA), 1/1000 anti-Grp94, 1/000 anti-Grp78, 1/1000 anti-PDI (Assay Designs, Ann Arbor, MI), 1/1000 anti-CHOP, 1/500 anti-cleaved Caspase-3, 1/1000 anti-Bcl-2, 1/1000 anti-Bcl-xL (Cell Signaling Technology, Beverly, MA), or 1/5000 anti-GAPDH (Research Diagnostics, Inc., Flanders, NJ) in 3% non-fat milk/tris-buffered saline/0.1%Tween-20. Membranes were then incubated for 30 min with 1/5000 anti-mouse IgG, 1/5000 anti-rabbit IgG (Cell Signaling Technology, Beverly, MA), or 1/5000 anti-rat IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) and detected with enhanced chemiluminescence (Pierce, Rockford, IL).
TUNEL Assay
NRVMs were cultured on 10 mm2 glass cover slips, infected with adenovirus, and subjected to H-R or normoxia as above. Following H-R, cells were fixed with 4% paraformaldehyde for 15 min at rt, permeabilized with ice cold 0.1% Triton-X 100/0.1% sodium citrate for 2 min, incubated with fluorescein conjugated TUNEL reaction mixture (Roche Applied Science, Quebec, Canada) for 1 h at 37°C, and incubated with 100 μg/mL Hoechst 33342 for 5 min at rt. Cover slips were mounted on glass slides with Vectasheild (Vector Laboratories Inc., Burlingame, CA) and TUNEL positive cells were quantified from ≥ 200 cells from ≥ 5 randomly chosen fields per cover slip.
Annexin-V Binding Assay
NRVMs were cultured (2 × 105 cells/cover slip), infected with adenovirus, and subjected to H-R or normoxia. Following H-R, cells were washed briefly with PBS and binding buffer (10 mM HEPES, 140 mMNaCl, and 2.5 mM CaCl2, pH 7.4.), then incubated with 1/3 Annexin V-Alexa 488 (Molecular Probes, Eugene, OR) in binding buffer for 15 min at rt. Necrotic cells were stained with 1μg/mL propidium iodide in Tyrode solution [135 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM d-glucose, 10 mM HEPES] for 15 min at rt. Cells were washed with binding buffer, mounted on glass slides with Vectashield, and phosphatidylserine was detected using fluorescence microscopy. Fluorescence intensity was quantified from ≥ 5 randomly chosen fields per cover slip using ImageJ software.
DNA Laddering
NRVMs, previously infected with adenovirus and subjected to H-R or normoxia, were harvested from 60 mm2 dishes with ice cold PBS and centrifuged at 700g for 10 min at 4°C. Pellets were re-suspended in tris/EDTA (TE)/0.2% Triton X-100, incubated on ice for 10 min, and centrifuged at 13,000g for 15 min at 4°C. Supernatants were collected and incubated for 1 h at 37°C with 60μg/mL RNase A. SDS (0.5%) and proteinase K (20μg/mL) were added and samples were incubated for 1 h at 50°C. To precipitate nucleosomal DNA, 0.1 volume 5M NaCl and 1 volume isopropanol (−20°C) were added, samples were incubated on ice for 10 min, and centrifuged for 15 min at 4°C. Pellets were re-suspended in TE buffer and DNA was separated on 2% agarose gel with a 1kb ladder.
Subcellular Fractionation
NRVMs were cultured on 100 mm2 dishes, infected with shScramble or shNogo-A adenovirus, and subjected to H-R or normoxia as above. Cells were harvested with a cell scraper in ice cold PBS and centrifuged at 700g for 10 min at 4°C to obtain a crude nuclear fraction. Mitochondrial and crude cytosolic fractions were prepared from 8.0 × 106 cells per experimental condition using a mitochondrial isolation kit (Pierce, Rockford, IL) according to the manufacturer’s instructions. Crude cytosolic fractions were further centrifuged at 100,000g for 90 min to obtain microsomal pellets and cytosolic fractions. All pellets were solubilized in lysis buffer.
Mitochondrial Membrane Potential (ΔΨm)
NRVMs were cultured (2.5 × 105 cells/cover slip), infected with adenovirus, and subjected to normoxia or 24 h hypoxia. Following H-R, cells were loaded with 50 nM tetramethylrhodamine ethyl ester (TMRE) in Tyrode solution for 20 min at rt. Differences in ΔΨm were determined by measuring TMRE fluorescence intensity at excitation and emission wavelengths of 543 and 575-640 nm respectively with confocal microscopy using a Zeiss LSM 410. The TMREfluorescence intensity of each cell (F) was obtained by subtracting the average fluorescence of the extracellular area (background) from the average fluorescence within the entire cell excluding the nucleus. Cells were perfused with1 μM of the mitochondrial uncoupler, p-trifluoromethoxy carbonyl cyanide phenyl hydrazone (FCCP), in Tyrode solution. Data was reported as F/F1 , where F1 is equal to the minimum fluorescence signal obtained following the administration of FCCP, expressed as a percentage of Ad-shScr infected cells at normoxia prior to the addition of FCCP. Quantification of mitochondrial fluorescence intensity was determined using ImageJ software.
Reactive Oxygen Species (ROS) Generation
NRVMs were cultured (2.5 × 105 cells/cover slip), infected with adenovirus, and subjected to normoxia or 24 h hypoxia followed by 4 h reoxygenation. Following H-R, cells were loaded with 20μM 2′,7′-Dichlorofluorescein diacetate (DCFDA) in Krebs buffer/0.5% Pluronic F-127 (Sigma Aldrich, St. Louis, MO) for 40 min at rt. Differences in ROS generation were determined by measuring DCF fluorescence intensity at excitation and emission wavelengths of 543 and 575-640 nm respectively. To confirm that the fluorescence signals detected reflected ROS generation, cells were perfused with 1M H2O2. Quantification of fluorescence intensity was determined by ImageJ software.
Ca2+ Transients
NRVMs were cultured (2.5 × 105 cells/cover slip), infected with adenovirus, and subjected to normoxia or 24 h hypoxia followed by 4 h reoxygenation. Cells were loaded with 2μM Indo-1 AM containing 0.04% Pluronic F-127 for 20 min at rt, perfused with Tyrode solution, and stimulated at 1Hz with 40V using a 6 ms pulse duration. Ca2+ transient recordings were obtained by measuring fluorescence intensity at excitation and emission wavelengths of 340 and 405/485 nm respectively and analyzed using IonOptix software (IonOptix, LLC, Milton, MA).
Statistical Analysis
Data are expressed as the mean ± SEM of n observations unless otherwise noted. Statistical significance between groups was determined by Student’s t-test. Significance between multiple groups was determined by one-way ANOVA with Tukey’s post hoc analysis. In each case, p < 0.05 was considered statistically significant.
Results
Nogo-A expression is increased in human DCM and ischemic hearts
Nogo-A expression was evaluated in left ventricular tissue from patients with DCM, and from patients who suffered an ischemic event. Patients with DCM exhibited 2.8 ± 0.2 fold higher Nogo-A expression compared to patients with non-failing hearts (Fig. 1A, C). Similarly, tissue from patients who had experienced an ischemic event exhibited 2.6 ± 0.3 fold higher Nogo-A expression compared to patients with non-failing hearts (Fig. 1B, C). These findings are consistent with previous studies that show increased Nogo-A expression in rodent models of DCM [39, 40] and increased Nogo-A mRNA in human end-stage heart failure [39], and raise questions as to the role of Nogo-A in I/R injury, since myocardial ischemia can often lead to DCM and congestive heart failure [45].
Figure 1.
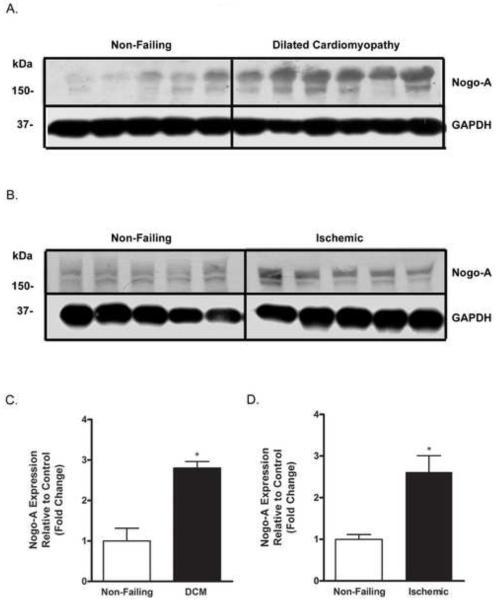
Nogo-A expression is increased in human DCM and ischemic hearts. Immunoblot analysis of Nogo-A expression in left ventricular tissue from A) non-failing hearts and hearts with DCM; and B) non-failing hearts and hearts that had experienced an ischemic event. Quantitative comparison of Nogo-A expression normalized to GAPDH between C) non-failing (open bar) and DCM hearts (closed bar), and D) non-failing (open bar) and ischemic hearts (closed bar). Data shown are the means ± SEM. *p < 0.01 (n≥5); Student’s t test.
Nogo-A is expressed in cardiomyocytes
While Nogo-A expression in the heart has been previously demonstrated, it is unclear which cell types are responsible for its expression. Neonatal and adult cardiomyocytes were immunostained for the presence of Nogo-A and the cardiac isoform of sarcoplasmic/endoplasmic reticulum calcium ATPase (Serca2a). Nogo-A expression was observed in both neonatal and adult cardiomyocytes and was co-localized to a large degree with Serca2a, consistent with the known ER expression pattern of RTN family proteins (Fig. 2A). Comparison between myocytes and fibroblasts obtained from the same tissue source revealed that fibroblasts (NRVFs) contribute relatively little to the expression of Nogo-A, while myocytes (NRVMs) exhibit robust expression (Fig. 2B,C).
Figure 2.

Nogo-A is expressed in neonatal and adult cardiomyocytes. A) Immunofluorescent detection of Nogo-A (green) and Serca2a (red) showing co-localization in neonatal rat ventricular myocytes (NRVM) and rabbit adult cardiomyocytes (ACM). Scale bar: 10μM. B) Immunoblot comparing Nogo-A expression between NRVMs and neonatal rat ventricular fibroblasts (NRVF) originating from the same hearts. C) Quantitative comparison of Nogo-A expression normalized to GAPDH between NRVMs (open bar) and NRVFs (closed bar). Data shown are the means ± SEM. *p < 0.01 (n≥4); Student’s t test.
Nogo-A expression is increased in cardiomyocytes following H-R
Considering the increase in Nogo-A expression observed in ischemic human ventricular tissue, we evaluated the expression of Nogo-A in a well established culture model of H-R injury. Following H-R, NRVMs exhibited a 2.4 ± 0.2 fold increase in Nogo-A expression compared to NRVMs subjected to normoxia alone (Fig. 3A, B). These findings are consistent with the degree of increased Nogo-A expression seen in human tissue (Fig. 1) and are in agreement with previous studies showing increased Nogo-A expression in neurons following hypoxia-ischemia [34, 38, 46].
Figure 3.

Nogo-A expression is increased in cardiomyocytes following H-R. A) Immunoblot analysis of Nogo-A expression in NRVMs subjected to either normoxia or H-R. B) Quantitative comparison of Nogo-A expression normalized to GAPDH between NRVMs subjected to normoxia (open bar) or H-R (closed bar). C) Immunoblot demonstrating knockdown of Nogo-A expression following infection with Ad-shN compared to Ad-shScr in NRVMs subjected to normoxia or hypoxia-reoxygenation (H-R). D) Quantitative comparison of Nogo-A expression normalized to GAPDH between NRVMs infected with Ad-shScr (open bars) or Ad-shN (closed bars) and subjected to normoxia or H-R. E) Immunoblot demonstrating over-expression of Nogo-A following infection with Ad-Nogo-A compared to Ad-Con in NRVMs subjected to normoxia or H-R. F) Quantitative comparison of Nogo-A expression normalized to GAPDH between NRVMs infected with Ad-Con (open bars) or Ad-Nogo-A (closed bars) and subjected to normoxia or H-R. Data shown are the means ± SEM. *p < 0.01 (n=5); One-way ANOVA with Tukey’s post hoc analysis.
To determine the effects of cardiomyocyte Nogo-A expression following H-R, shScramble, shNogo-A, empty vector, and Nogo-A over-expression adenoviral constructs (Ad-shScr ,Ad-shN, Ad-Con, and Ad-Nogo-A) were generated. NRVMs were infected with either Ad-shScr, Ad-shN, Ad-Con, or Ad-Nogo-A and subsequently subjected to normoxia or H-R. Infection with Ad-shN resulted in approximately 60% knockdown of Nogo-A expression, after either normoxia or H-R, compared to Ad-shScr infected controls (Fig. 3C,D). Knockdown of Nogo was specific for the Nogo-A isoform, as Nogo-B expression was unaffected by Ad-shN infection (Fig. 3C). Infection with Ad-Nogo-A resulted in an approximate 4 fold increase in Nogo-A expression at normoxia, and a 6 fold increase after H-R, compared to Ad-Con infected controls (Fig. 3E,F).
Knockdown of Nogo-A inhibited H-R-induced DNA fragmentation
It is well established that subjecting cultured cardiomyocytes to H-R induces cell death via apoptosis [11, 47], the hallmark of which being the appearance of DNA fragmentation [48]. To determine if Nogo-A plays a role in H-R-induced apoptosis, NRVMs were infected with Ad-shScr or Ad-shN, subjected to normoxia or H-R, and DNA fragmentation was measured by TUNEL staining and DNA laddering. Following H-R, NRVMs infected with Ad-shScr exhibited a significant increase in TUNEL-positive staining compared to normoxia (43.0 ± 2.3 vs. 18.2 ± 2.7%) and demonstrated extensive internucleosomal DNA degradation indicated by the appearance of DNA laddering (Fig. 4A,B,C). In contrast, NRVMs infected with Ad-shN exhibited no significant change in TUNEL-positive staining between normoxia and H-R (17.0 ± 2.9 vs. 18.6 ± 2.6%) and did not demonstrate internucleosomal DNA degradation (Fig. 4A,B,C). Conversely, NRVMs infected with Ad-Nogo-A exhibited a modest increase in TUNEL-positive staining at normoxia (19.58 ± 1.6 vs. 14.54 ± 1.8) and a significant increase following H-R (35.16 ± 2.9 vs. 24.89 ± 0.6; p< 0.01) compared to Ad-Con infected cells.
Figure 4.

Knockdown of Nogo-A protects cardiomyocytes from H-R induced DNA fragmentation. NRVMs were infected with either shScr or shN adenovirus, and subjected to normoxia or H-R. A) DNA fragmentation was analyzed using TUNEL staining. Shown are representative images of nuclei stained with Hoechst 33342 and TUNEL reaction mixture. Scale bar: 10μM. B) Quantitative comparison of TUNEL positive cells between Ad-shScr or Ad-shN infected NRVMs subjected to normoxia (open bars) or H-R (closed bars). Data shown are the means ± SEM. *p < 0.01 (n=5); One-way ANOVA with Tukey’s post hoc analysis. C) Internucleosomal DNA was isolated from NRVMs and analyzed by agarose gel electrophoresis. Lane 1, 1 kb molecular weight markers. Lane 2, DNA from NRVMs infected with shScr adenovirus and subjected to normoxia. Lane 3, DNA from NRVMs infected with shN adenovirus and subjected to normoxia. Lane 4, DNA from NRVM infected with shScr adenovirus and subjected to H-R. Lane 5, DNA from NRVMs infected with shN adenovirus and subjected to H-R.
Knockdown of Nogo-A inhibited H-R-induced phosphatidylserine translocation
In cells undergoing early apoptosis, phosphatidylserine (PS) is translocated from the cytoplasmic surface of the plasma membrane to the extracellular surface [49]. To determine if knockdown of Nogo-A affected H-R-induced PS translocation, NRVMs were stained with Annexin-V, while propidium iodide (PI) was used as a counterstain to distinguish between apoptotic and necrotic cells. Following H-R, NRVMs infected with Ad-shScr exhibited a 2.8 ± 0.1 fold increase in Annexin-V binding to PS compared to normoxia, while NRVMs infected with Ad-shN exhibited no significant change in Annexin-V binding between normoxia and H-R (0.7 ± 0.1 vs. 0.9 ± 0.2) (Fig. 5A,B). Conversely, following H-R, NRVMs infected with Ad-Con exhibited a significant 2.7 ± 0.4 fold increase (p< 0.05) in Annexin-V binding compared to normoxia, while NRVMs infected with Ad-Nogo-A exhibited a significantly higher 6.6 ± 0.5 fold increase (p< 0.001) in Annexin-V binding following H-R compared to normoxia. Cells that were positively stained with Annexin-V showed an absence of PI staining, indicating that cell death was not due to necrosis.
Figure 5.
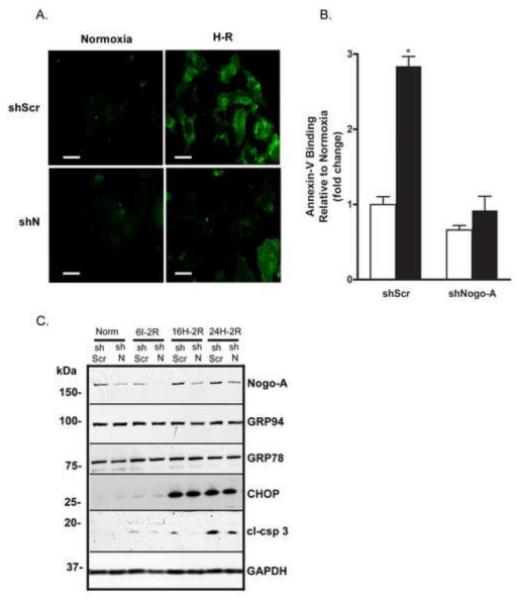
Knockdown of Nogo-A inhibited H-R-induced phosphatidylserine translocation and caspase-3 cleavage without affecting ER stress. A) Representative immunofluorescent images showing Alexa 488-conjugated Annexin V binding on Ad-shScr or Ad-shN infected NRVMs subjected to normoxia or H-R. Scale bar: 10μM. B) Quantitative comparison of Annexin-V-Alexa 488 fluorescence between Ad-shScr or Ad-shN infected NRVMs subjected to normoxia (open bars) or H-R (closed bars). Data shown are the means ± SEM. *p < 0.01 (n=5); One-way ANOVA with Tukey’s post hoc analysis. C) Immunoblot analysis of Nogo-A, GRP 94, GRP 78, CHOP, cleaved caspase-3 (cl-csp 3), and GAPDH expression in NRVMs infected with shScr or shN adenovirus and subjected to normoxia, 6 h ischemia with 2h reoxygenation (6I-2R), 16 h of hypoxia with 2 h reoxygenation (16H-2R) or 24 h hypoxia with 2 h reoxygenation (24H-2R). Images shown are representative of ≥ 3 independent experiments.
Knockdown of Nogo-A inhibited H-R-induced caspase-3 cleavage without affecting ER stress
ER stress has been shown to occur in myocardial infarction, as well as in cultured cardiomyocytes subjected to H-R, and is known to play a role in H-R-induced apoptosis [15, 50, 51]. As Nogo-A is highly expressed in the ER, we determined whether knockdown of Nogo-A modulated the induction of the H-R-induced ER stress response by measuring expression of common ER stress markers: glucose-regulated protein 94 (GRP 94), GRP 78 (also known as immunoglobulin heavy chain-binding protein (BiP)), and C/BEP homologous protein (CHOP) (also known as growth-arrest- and DNA-damage inducible gene 153 (GADD153)) [51, 52]. NRVMs were infected with Ad-shScr or Ad-shN and subjected to either 6 h ischemia, 16 h hypoxia, or 24 h hypoxia, followed by 2 h reoxygenation. Following ischemia/hypoxia, NRVMs infected with Ad-shScr expressed active, cleaved caspase-3 (cl-csp 3) proportionate to the duration of injury and exhibited induction of ER stress markers, most noticeably CHOP (Fig. 5C). By comparison, NRVMs infected with Ad-shN consistently expressed lower levels of cl-csp 3 following each injury regimen. However, there were no differences in the expression of GRP 94, GRP 78, or CHOP between NRVMs infected with Ad-shScr or Ad-shN following any of the injury regimens, indicating that Nogo-A knockdown did not inhibit H-R-induced apoptosis by modulating ER stress (Fig. 5C).
Knockdown of Nogo-A prevented H-R-induced ΔΨm reduction
Previous studies have demonstrated that dissipation of the mitochondrial membrane potential ΔΨm is an early event in the apoptotic cell death process that results in mitochondrial membrane permeabilization, production of ROS, and the release of pro-apoptotic factors, such as cytochrome c, into the cytosol [16]. ΔΨm in NRVMs subjected to H-R was determined by measuring the TMRE fluorescence intensity normalized to the residual fluorescence signal obtained with FCCP. Following H-R, NRVMs infected with Ad-shScr exhibited a 50.7 ± 5.1% decrease in TMRE fluorescence compared to Ad-shScr infected NRVMs at normoxia (Fig. 6A, B). In contrast, the normalized TMRE fluorescence intensity in Ad-shN-infected cells was not significantly different from Ad-shScr normoxic controls at normoxia (97.9 ± 6.9% of control) or following H-R (89.5 ± 6.0% of control) (Fig. 6A, B).
Figure 6.

Knockdown of Nogo-A prevented H-R-induced reduction of ΔΨm. A) Representative confocal images of TMRE fluorescence prior to and following administration of FCCP in NRVMs infected with shScr or shN adenovirus and subjected to normoxia or H-R. Scale bar: 10μM. B) Time course of ΔTMRE fluorescence prior to and following administration of FCCP for Ad-shScr infected NRVMs at normoxia (open circles), Ad-shScr infected NRVMs following 24 h hypoxia (closed circles), Ad-shN infected NRVMs at normoxia (open triangles) and Ad-shN infected NRVMs following 24 h hypoxia (closed triangles). Data are the normalized fluorescence (F/F1) expressed as a percentage of Ad-shScr normoxic controls prior to FCCP. Data shown are the means ± SEM. *p < 0.001 for 25≥ cells per field; One-way ANOVA with Tukey’s post hoc analysis.
Knockdown of Nogo-A prevented H-R-induced ROS production and cytochrome c release
It is widely known that ROS are generated by mitochondria during reperfusion following ischemia/hypoxia and play an important role in the induction of apoptotic cell death in cardiomyocytes [53, 54]. The generation of ROS in NRVMs subjected to H-R was determined by measuring DCF fluorescence intensity. Following H-R, NRVMs infected with Ad-shScr exhibited increased DCF fluorescence compared to normoxia (77.0 ± 3.5 vs. 41.0 ± 1.5 a.u.), indicative of reperfusion-induced ROS generation (Fig. 7A, B). In contrast, following H-R, DCF fluorescence intensity in Ad-shN-infected cells (41.3 ± 1.9 a.u.) was not significantly different from Ad-shScr-infected cells at normoxia (Fig. 7A, B). In addition, DCF fluorescence in Ad-shN-infected cells at normoxia (59.0 ± 2.7 a.u.) was modestly increased compared to Ad-shScr-infected cells at normoxia or Ad-shN-infected cells after H-R (Fig. 7A, B).
Figure 7.

Knockdown of Nogo-A prevented H-R-induced ROS generation and release of cytochrome c. A) Representative images of DCF fluorescence in NRVMs infected with shScr or shN adenovirus and subjected to normoxia or H-R. Scale bar: 10μM. B) Quantitative comparison of DCF fluorescence between NRVMs infected with Ad-shScr (open bars) or Ad-shN (closed bars) and subjected to normoxia or H-R. Data shown are the means ± SEM. *p < 0.01; **p < 0.001 (n=5); One-way ANOVA with Tukey’s post hoc analysis. C) Immunoblot analysis of cytosolic cytochrome c expression in NRVMs infected with shScr or shN adenovirus and subjected to normoxia or H-R.
As expected, cytosolic cytochrome c expression was readily detectable in Ad-shScr infected cells following H-R, suggesting mitochondrial membrane permeability was increased. Ad-shN-infected cells, however, exhibited greatly reduced cytosolic cytochrome c expression following H-R compared to Ad-shScr control. Consistent with previous data demonstrating a modest ROS increase in Ad-shN-infected cells at normoxia, cytosolic cytochrome c was also detected in NRVMs infected with Ad-shN at normoxia.
Knockdown of Nogo-A prevented H-R-induced abnormal Ca2+ cycling
During hypoxia/ischemia, depletion of energy reserves results in the progressive elevation of intracellular Ca2+. Persistently high concentrations of intracellular Ca2+ eventually lead to cell death by necrosis or apoptosis [55]. Since Nogo-A is primarily located in the ER, we investigated the possibility that knockdown of Nogo-A modulates Ca2+ cycling following H-R. Following H-R, NRVMs infected with Ad-shN exhibited significant decreases in diastolic Ca2+ (0.61 ± 0.03 vs. 2.26 ± 0.17 F405/485) and transient decay (tau) (0.138 ± 0.012 vs. 0.307 ± 0.020 s) compared to NRVMs infected with Ad-shScr (Fig. 8C,D,E), without affecting transient amplitudes or time to peak (Fig. 8A,B). Interestingly, diastolic Ca2+ in Ad-shN-infected cells at normoxia (1.81 ± 0.11 F405/485) was modestly increased compared to Ad-shScr-infected cells at normoxia or Ad-shN-infected cells after H-R.
Figure 8.

Knockdown of Nogo-A prevented H-R-induced abnormal Ca2+ cycling. Quantitative comparison of Ca2+ transient amplitudes A), time to peak B), diastolic calcium C), and transient decay (tau) D) between NRVMs infected with Ad-shScr (open bars) or Ad-shN (closed bars) and subjected to normoxia or H-R. Data shown are the means ± SEM. *p < 0.05; **p<0.001; One-way ANOVA with Tukey’s post hoc analysis. E) Representative averaged calcium transients in NRVMs infected with shScr (solid line) or shN (dotted line) following H-R.
In agreement with the data presented above, Nogo-A knockdown also prevented H-R-induced necrosis as determined by PI staining. Following H-R, NRVMs infected with Ad-shScr exhibited significant necrosis compared to normoxia (39.7 ± 1.3 vs. 12.5 ± 0.5 %). By comparison, NRVMs infected with Ad-shN showed no significant increase in necrosis following H-R compared to normoxia (15.8 ± 1.1 vs. 12.4 ± 1.6 %).
Discussion
We demonstrate here the expression of the RTN protein, Nogo-A, in isolated cardiomyocytes, and the significant up-regulation of Nogo-A in human DCM and following I/R injury. The major finding of this study is that knockdown of Nogo-A protects cardiomyocytes against H-R-induced apoptosis, which was confirmed using a series of approaches to measure the hallmarks of apoptotic cell death. Nogo-A knockdown significantly inhibited H-R-induced DNA fragmentation, non-necrotic PS translocation, caspase-3 cleavage and cytochrome c release. We postulate that reduction of Nogo-A expression may prove useful in the management of ischemic cardiomyopathy.
In addition to Nogo-A, the B isoform of Nogo was abundantly expressed in isolated cardiomyocytes. Nogo-B is thought to be a regulator of vascular remodeling and homeostasis and has been shown to interact with both Bcl-xL and Bcl-2, thereby reducing their anti-apoptotic activity by modulating their mitochondrial localization [56, 57]. We rule out the possibility that Nogo-B could be responsible for the inhibition of apoptosis observed following H-R, since knockdown of Nogo-A was observed in the absence of any change in Nogo-B expression.
Conditions that lead to the accumulation of unfolded proteins in the lumen of the ER, such as H-R, activate a signaling program known as the unfolded protein response (UPR) [58]. The UPR is an adaptive attempt to relieve ER stress by increasing ER-associated degradation (ERAD) of unfolded proteins, decreasing protein synthesis, and inducing the synthesis of ER chaperones that facilitate protein folding and prevent unfolded protein accumulation [59]. Three transmembrane sensors in the ER, ATF6 (activating transcription factor 6), IRE1 (inositol-requiring enzyme 1), and PERK (double-stranded RNA-dependent protein kinase-like ER kinase), are responsible for the initiation of signaling cascades in the UPR, and induce transcription of CHOP, a potent pro-apoptotic factor that represses expression of Bcl-2 [14]. We found the expression of ER stress markers to be unaffected by Nogo-A knockdown following different durations of injury. Consistent with these findings, the expression of Bcl-2, Bcl-xL, and Bax following H-R were unaffected by Nogo-A knockdown in whole cell lysates, and mitochondrial fractions showed no change in Bcl-2 or Bcl-xL localization. We also observed no protein-protein interaction between Nogo-A and Bcl-2, Bcl-xL, or Bax (data not shown). These findings suggest that the inhibition of H-R-induced apoptosis by Nogo-A knockdown was not due to a modulation of Bcl-2 family protein expression via CHOP, nor through modulation of Bcl-2 or Bcl-xL localization.
Disrupting the interaction of Nogo-A with its cognate receptor using neutralizing anti-Nogo-A antibodies has been shown to significantly increase axonal sprouting and functional recovery in animal models of spinal cord injury and stroke [29, 60-62]. However, interfering with the interaction between Nogo-A and its receptor prior to stroke has been shown to increase p38/MAPK, and decrease Akt and ERK 1/2 activity, leading to a decrease in neuronal survival [63]. Although we cannot rule out the possibility that the inhibition of H-R-induced apoptosis by Nogo-A knockdown occurs through the modulation of an interaction between Nogo-A and its receptor(s), we suggest this is an unlikely explanation as we observed no change in p38/MAPK, Akt, or ERK 1/2 activity in Ad-shN infected cells following H-R (data not shown).
The mitochondrial permeability transition pore (MPTP) is a large, non-selective channel spanning the inner mitochondrial membrane that is a critical mediator of cell death. MPTP opening is favored by high concentrations of intracellular calcium, Pi, and ROS, all of which are elevated as a consequence of H-R injury. H+ diffusion through the open MPTP results in the dissipation of ΔΨm, leading to the loss of ATP production, the generation of ROS, mitochondrial swelling and rupture, and the release of several pro-apoptotic factors [64, 65]. Knockdown of Nogo-A was shown to protect cardiomyocytes from H-R-induced apoptosis by preventing an elevation in intracellular Ca2+, preserving ΔΨm, inhibiting ROS accumulation, and inhibiting cytochrome c release. It has yet to be determined, however, whether Nogo-A knockdown protects against H-R-induced apoptosis by affecting MPTP pore formation or opening.
The observations that Ad-shN-infected NRVMs exhibited modest elevations of ROS generation, cytochrome c release, and intracellular Ca2+ at normoxia are consistent with the idea that knockdown of Nogo-A may result in a sub-lethal, partial uncoupling of mitochondrial respiration. Mitochondrial uncouplers are known to generate ROS, and raise intracellular Ca2+ [66, 67], and agents that partially uncouple mitochondrial respiration have been shown to protect cardiomyocytes from ischemic/hypoxic injury [67-72]. Considering that we have been unable to demonstrate Nogo-A expression in mitochondria by either fractionation or immunocytochemistry (Suppl. Fig. 1), the potential mitochondrial uncoupling associated with Nogo-A knockdown is likely to occur through indirect means.
The precise mechanism in which the knockdown of expression of a residential ER protein preserves mitochondrial integrity following H-R remains to be determined. Of particular future interest is the mechanism(s) by which Nogo-A knockdown improves Ca2+ cycling following H-R. In addition, subsarcolemmal and interfibrillar mitochondria differ in structure, function, and the ability to mediate cardioprotection by ischemic pre-conditioning [73, 74]. It will be important to determine which mitochondria population is responsible for mediating the protective effects of Nogo-A knockdown or whether there are any differential effects of Nogo-A knockdown on the two populations.
In summary, we show that knockdown of Nogo-A protects cardiomyocytes from apoptosis by significantly inhibiting hypoxia/reoxygenation-induced mitochondrial membrane permeabilization, indicated by the preservation of ΔΨm , the inhibition of ROS accumulation, and the inhibition of cytochrome c release. Although the significance of these findings remains to be evaluated in vivo, we postulate that Nogo-A may serve as novel therapeutic target in the treatment of I/R injury.
Supplementary Material
Acknowledgements
The authors wish to thank Dr. Martin Schwab for providing invaluable help and insight, Dr. Kyle Henderson and the Loyola Cardiovascular Institute Tissue Repository for assistance procuring human left ventricular tissue, Rekha Iyengar for the isolation of neonatal cardiomyocytes, and the Falk Foundation for support. This study was supported by grants from the NIH/NINDS (1R01NS40960), the AHA (SDG 02354112), the NHLBS (HL62426), the Department of Veteran Affairs, and the Loyola Neuroscience Institute. Dr. Sarkey is supported by the NIH (PAF 2009-03479).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None Declared
References
- [1].Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic Res Cardiol. 1998;93(Suppl 3):8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- [2].Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM, et al. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2001;280:H2726–31. doi: 10.1152/ajpheart.2001.280.6.H2726. [DOI] [PubMed] [Google Scholar]
- [3].Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–72. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- [4].Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol. 1996;28:2005–16. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- [5].Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–8. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hofstra L, Liem IH, Dumont EA, Boersma HH, van Heerde WL, Doevendans PA, et al. Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet. 2000;356:209–12. doi: 10.1016/s0140-6736(00)02482-x. [DOI] [PubMed] [Google Scholar]
- [7].Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–3. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- [8].Regula KM, Kirshenbaum LA. Apoptosis of ventricular myocytes: a means to an end. J Mol Cell Cardiol. 2005;38:3–13. doi: 10.1016/j.yjmcc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- [9].Webster KA. Programmed death as a therapeutic target to reduce myocardial infarction. Trends Pharmacol Sci. 2007;28:492–9. doi: 10.1016/j.tips.2007.07.004. [DOI] [PubMed] [Google Scholar]
- [10].Lee Y, Gustafsson AB. Role of apoptosis in cardiovascular disease. Apoptosis. 2009;14:536–48. doi: 10.1007/s10495-008-0302-x. [DOI] [PubMed] [Google Scholar]
- [11].Tanaka M, Ito H, Adachi S, Akimoto H, Nishikawa T, Kasajima T, et al. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ Res. 1994;75:426–33. doi: 10.1161/01.res.75.3.426. [DOI] [PubMed] [Google Scholar]
- [12].Lee P, Sata M, Lefer DJ, Factor SM, Walsh K, Kitsis RN. Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol. 2003;284:H456–63. doi: 10.1152/ajpheart.00777.2002. [DOI] [PubMed] [Google Scholar]
- [13].Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol. 2006;291:H1411–20. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Szegezdi E, Duffy A, O’Mahoney ME, Logue SE, Mylotte LA, O’Brien T, et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349:1406–11. doi: 10.1016/j.bbrc.2006.09.009. [DOI] [PubMed] [Google Scholar]
- [15].Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–82. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- [16].Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- [17].Ferrari R, Ceconi C, Curello S, Cargnoni A, De Giuli F, Visioli O. Occurrence of oxidative stress during myocardial reperfusion. Mol Cell Biochem. 1992;111:61–9. doi: 10.1007/BF00229575. [DOI] [PubMed] [Google Scholar]
- [18].Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, et al. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996;384:53–7. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- [19].Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- [20].Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta. 2006;1757:1297–300. doi: 10.1016/j.bbabio.2006.03.017. [DOI] [PubMed] [Google Scholar]
- [21].Oertle T, Schwab ME. Nogo and its paRTNers. Trends Cell Biol. 2003;13:187–94. doi: 10.1016/s0962-8924(03)00035-7. [DOI] [PubMed] [Google Scholar]
- [22].Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002;22:3553–67. doi: 10.1523/JNEUROSCI.22-09-03553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, et al. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci. 2003;23:5393–406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–44. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- [25].Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, et al. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383–4. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- [26].Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–9. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- [27].Brosamle C, Huber AB, Fiedler M, Skerra A, Schwab ME. Regeneration of lesioned corticospinal tract fibers in the adult rat induced by a recombinant, humanized IN-1 antibody fragment. J Neurosci. 2000;20:8061–8. doi: 10.1523/JNEUROSCI.20-21-08061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Merkler D, Metz GA, Raineteau O, Dietz V, Schwab ME, Fouad K. Locomotor recovery in spinal cord-injured rats treated with an antibody neutralizing the myelin-associated neurite growth inhibitor Nogo-A. J Neurosci. 2001;21:3665–73. doi: 10.1523/JNEUROSCI.21-10-03665.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Markus TM, Tsai SY, Bollnow MR, Farrer RG, O’Brien TE, Kindler-Baumann DR, et al. Recovery and brain reorganization after stroke in adult and aged rats. Ann Neurol. 2005;58:950–3. doi: 10.1002/ana.20676. [DOI] [PubMed] [Google Scholar]
- [30].Hu WH, Hausmann ON, Yan MS, Walters WM, Wong PK, Bethea JR. Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP) J Neurochem. 2002;81:36–45. doi: 10.1046/j.1471-4159.2002.00788.x. [DOI] [PubMed] [Google Scholar]
- [31].Nath AK, Krauthammer M, Li P, Davidov E, Butler LC, Copel J, et al. Proteomic-based detection of a protein cluster dysregulated during cardiovascular development identifies biomarkers of congenital heart defects. PLoS One. 2009;4:e4221. doi: 10.1371/journal.pone.0004221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].O’Neill P, Whalley K, Ferretti P. Nogo and Nogo-66 receptor in human and chick: implications for development and regeneration. Dev Dyn. 2004;231:109–21. doi: 10.1002/dvdy.20116. [DOI] [PubMed] [Google Scholar]
- [33].Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–15. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang H, Yao Y, Jiang X, Chen D, Xiong Y, Mu D. Expression of Nogo-A and NgR in the developing rat brain after hypoxia-ischemia. Brain Res. 2006;1114:212–20. doi: 10.1016/j.brainres.2006.07.056. [DOI] [PubMed] [Google Scholar]
- [35].Hunt D, Coffin RS, Prinjha RK, Campbell G, Anderson PN. Nogo-A expression in the intact and injured nervous system. Mol Cell Neurosci. 2003;24:1083–102. doi: 10.1016/j.mcn.2003.09.002. [DOI] [PubMed] [Google Scholar]
- [36].Meier S, Brauer AU, Heimrich B, Schwab ME, Nitsch R, Savaskan NE. Molecular analysis of Nogo expression in the hippocampus during development and following lesion and seizure. Faseb J. 2003;17:1153–5. doi: 10.1096/fj.02-0453fje. [DOI] [PubMed] [Google Scholar]
- [37].Eslamboli A, Grundy RI, Irving EA. Time-dependent increase in Nogo-A expression after focal cerebral ischemia in marmoset monkeys. Neurosci Lett. 2006;408:89–93. doi: 10.1016/j.neulet.2006.08.056. [DOI] [PubMed] [Google Scholar]
- [38].Cheatwood JL, Emerick AJ, Schwab ME, Kartje GL. Nogo-A expression after focal ischemic stroke in the adult rat. Stroke. 2008;39:2091–8. doi: 10.1161/STROKEAHA.107.507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bullard TA, Protack TL, Aguilar F, Bagwe S, Massey HT, Blaxall BC. Identification of Nogo as a novel indicator of heart failure. Physiol Genomics. 2008;32:182–9. doi: 10.1152/physiolgenomics.00200.2007. [DOI] [PubMed] [Google Scholar]
- [40].Gramolini AO, Kislinger T, Alikhani-Koopaei R, Fong V, Thompson NJ, Isserlin R, et al. Comparative proteomics profiling of a phospholamban mutant mouse model of dilated cardiomyopathy reveals progressive intracellular stress responses. Mol Cell Proteomics. 2008;7:519–33. doi: 10.1074/mcp.M700245-MCP200. [DOI] [PubMed] [Google Scholar]
- [41].Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation. 1997;96:4343–8. doi: 10.1161/01.cir.96.12.4343. [DOI] [PubMed] [Google Scholar]
- [42].Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pradhan AD, Case AM, Farrer RG, Tsai SY, Cheatwood JL, Martin JL, et al. Dendritic Spine Alterations in Neocortical Pyramidal Neurons following Postnatal Neuronal Nogo-A Knockdown. Dev Neurosci. 2010;32:313–20. doi: 10.1159/000309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–33. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Heusch G, Schulz R, Rahimtoola SH. Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol. 2005;288:H984–99. doi: 10.1152/ajpheart.01109.2004. [DOI] [PubMed] [Google Scholar]
- [46].Li S, Carmichael ST. Growth-associated gene and protein expression in the region of axonal sprouting in the aged brain after stroke. Neurobiol Dis. 2006;23:362–73. doi: 10.1016/j.nbd.2006.03.011. [DOI] [PubMed] [Google Scholar]
- [47].Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O’Neill L, et al. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99:2635–43. doi: 10.1172/JCI119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gorczyca W, Bigman K, Mittelman A, Ahmed T, Gong J, Melamed MR, et al. Induction of DNA strand breaks associated with apoptosis during treatment of leukemias. Leukemia. 1993;7:659–70. [PubMed] [Google Scholar]
- [49].van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- [50].Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am J Physiol Heart Circ Physiol. 2009;297:H1711–9. doi: 10.1152/ajpheart.00553.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–75. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1999;99:2934–41. doi: 10.1161/01.cir.99.22.2934. [DOI] [PubMed] [Google Scholar]
- [54].Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987;84:1404–7. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Piper HM, Siegmund B, Ladilov Yu V, Schluter KD. Calcium and sodium control in hypoxic-reoxygenated cardiomyocytes. Basic Res Cardiol. 1993;88:471–82. doi: 10.1007/BF00795413. [DOI] [PubMed] [Google Scholar]
- [56].Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, et al. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–8. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- [57].Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. A novel protein, RTN-XS, interacts with both Bcl-XL and Bcl-2 on endoplasmic reticulum and reduces their anti-apoptotic activity. Oncogene. 2000;19:5736–46. doi: 10.1038/sj.onc.1203948. [DOI] [PubMed] [Google Scholar]
- [58].Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- [59].Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- [60].Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–72. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- [61].Papadopoulos CM, Tsai SY, Alsbiei T, O’Brien TE, Schwab ME, Kartje GL. Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann Neurol. 2002;51:433–41. doi: 10.1002/ana.10144. [DOI] [PubMed] [Google Scholar]
- [62].Seymour AB, Andrews EM, Tsai SY, Markus TM, Bollnow MR, Brenneman MM, et al. Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. J Cereb Blood Flow Metab. 2005;25:1366–75. doi: 10.1038/sj.jcbfm.9600134. [DOI] [PubMed] [Google Scholar]
- [63].Kilic E, ElAli A, Kilic U, Guo Z, Ugur M, Uslu U, et al. Role of Nogo-A in neuronal survival in the reperfused ischemic brain. J Cereb Blood Flow Metab. 2010;30:969–84. doi: 10.1038/jcbfm.2009.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol. 2009;104:181–8. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–4. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
- [66].Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998;513( Pt 3):819–33. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–21. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- [68].Brennan JP, Berry RG, Baghai M, Duchen MR, Shattock MJ. FCCP is cardioprotective at concentrations that cause mitochondrial oxidation without detectable depolarisation. Cardiovasc Res. 2006;72:322–30. doi: 10.1016/j.cardiores.2006.08.006. [DOI] [PubMed] [Google Scholar]
- [69].Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–7. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- [70].An J, Rhodes SS, Jiang MT, Bosnjak ZJ, Tian M, Stowe DF. Anesthetic preconditioning enhances Ca2+ handling and mechanical and metabolic function elicited by Na+-Ca2+ exchange inhibition in isolated hearts. Anesthesiology. 2006;105:541–9. doi: 10.1097/00000542-200609000-00018. [DOI] [PubMed] [Google Scholar]
- [71].Sedlic F, Pravdic D, Ljubkovic M, Marinovic J, Stadnicka A, Bosnjak ZJ. Differences in production of reactive oxygen species and mitochondrial uncoupling as events in the preconditioning signaling cascade between desflurane and sevoflurane. Anesth Analg. 2009;109:405–11. doi: 10.1213/ane.0b013e3181a93ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, et al. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–90. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- [73].Riva A, Tandler B, Loffredo F, Vazquez E, Hoppel C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am J Physiol Heart Circ Physiol. 2005;289:H868–72. doi: 10.1152/ajpheart.00866.2004. [DOI] [PubMed] [Google Scholar]
- [74].Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, et al. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol. 2009;104:141–7. doi: 10.1007/s00395-009-0007-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.