

Abstract
Introduction
Accelerated cardiovascular disease in patients with type I diabetes (TID) is a well-described condition and serious clinical obstacle. At present, the notion that early atherogenesis is largely dependent on sustained hyperglycemia remains in question. We hypothesize that an alteration in T lymphocyte homeostasis may result in early vascular inflammation which might amplify subsequent blood vessel injury in euglycemia.
Methods
A murine of carotid arterial ligation was employed to induce neointimal hyperplasia (NIH) in wild type and non-obese diabetic (NOD) mice. Additionally, adoptive transfer of NOD splenocytes into immunodeficient NOD mice (NOD.scid) was undertaken to evaluate the influence of restored autoimmunity on NIH development.
Results
Interestingly, compared to wild type mice, the NOD demonstrate a significant increase in neointimal area. Conversely, the NOD.scid reveal almost no evidence of vascular injury. While evidence of early vascular inflammation can be detected in the injured NOD vasculature, uninjured contralateral vessels and those of the NOD.scid have minimal T cell infiltration. Following reconstitution of autoimmune responses via NOD splenocyte adoptive transfer, accelerated vascular pathology is restored.
Conclusions
These observations suggest that autoimmunity, in the setting of impending hyperglycemia, may contribute to accelerated vascular inflammation and subsequent pathology.
Keywords: Neointimal hyperplasia, inflammation, autoimmunity, adoptive transfer
Introduction
Pathogenesis of accelerated cardiovascular disease in patients with type I diabetes (T1D) is under intense investigation. At present, there appears to be a clear correlation between sustained hyperglycemia and atherogenesis (1, 2). However, soluble markers of inflammation remain after intensive hyperglycemic therapy, suggesting a vascular pathology initiated prior to, or concurrent with, the development of T1D (3). Autoimmune destruction of pancreatic β-cells is facilitated by T lymphocyte infiltration and robust production of reactive oxygen species. While destruction of these β-cells results in T1D, it is unclear whether the vasculature is an early target of this immune dysregulation.
The non-obese diabetic (NOD) mouse is a well-established model of spontaneous T1D and has been used extensively by investigators to dissect the immune components associated with disease pathogenesis. Not surprisingly, immunodeficient NOD mice, or mice that have mutations in costimulatory pathways, fail to develop T1D (4). While there has been little application of this model to the investigation of diabetic vascular complications, we have previously identified a distinct vasculopathy in the NOD during the pre-diabetic phase (5). We hypothesize that a breakdown in T cell homeostasis triggers early inflammation and endothelial dysfunction that may amplify vascular injury regardless of glycemic status.
In this study, we employ a low shear-stress model of carotid arterial injury to characterize the natural history of luminal pathology prior to the onset of spontaneous T1D in the autoimmune model. Secondly, we sought to determine the influence of a competent immune response in this process.
Methods
Animals
All experimental protocols were approved by the University of Colorado Animal Review Committee. Age and weight-matched animals of the following strains were used in all experiments: C57/Bl6, NOD and NOD.scid (severe combined immunodeficient). NOD and NOD.scid breeding mice were initially acquired from The Jackson Laboratory or the Barbara Davis Center for Childhood Diabetes (Denver, CO) Experimental animals were monitored for diabetes by checking urine glucose levels (Diastix, Bayer) and hyperglycemia was confirmed using a One Touch Ultra glucometer (Life Scan, Milpitas, CA). In an effort to remove the potential pathologic influence of sustained hyperglycemia on endothelial injury, all mice underwent carotid ligation and subsequent histological evaluation prior to the development of hyperglycemia. Glucose levels were closely monitored both pre- and post-procedure for the duration of the experiment. Mice that developed T1D (blood glucose levels > 15mM) prior to histologic evaluation, were excluded from the studies.
Murine model of low shear-stress injury
Cessation of carotid arterial flow, as a vascular model of endothelial shear-stress injury, was carried out as previously described (6). Briefly, general anesthesia was achieved by intraperitoneal injection of Avertin [250 mg/kg body weight, supplemental dose 75 mg/kg]. A midline incision was made in the neck with the subcutaneous tissue retracted cephalad. The carotid artery is gently dissected free from the nerve and jugular vein and ligated at the level of the bifurcation with 6–0 prolene suture. The skin is then closed with 5–0 prolene suture in running fashion.
Splenocyte adoptive transfer
While under anesthesia with Avertin, NOD.scid mice received 2 × 107 NOD spleen cells injected intravenously into the retro orbital sinus (ROS). This was done concurrently with the carotid ligation. The spleen cells were from NOD donors that were not diabetic.
NIH and morphometric analysis
At 28 days following carotid ligation the animals were euthanized in accordance with the guidelines set forth by the American Veterinary Medical Association Panel on Euthanasia. For morphometric analysis, animals were euthanized with subsequent intracardiac injection of 4% paraformaldehyde. Both the right and left carotid arteries were harvested, embedded in paraffin, and sectioned for hematoxylin and eosin staining. Serial cross sections were taken along the length of the vessel at 100μm intervals. Qualitative visual inspection of these specimens revealed the beginning of luminal stenosis. At this point, 30–40 sections were taken at 5μm intervals. Multiple samples (6–8) were subject to quantitative morphometic analysis. Plain images were taken on the confocal microscope, and the following structures were identified: lumen, internal elastic lamina (IEL), external elastic lamina, and neointima. Intimal areas (lumen to IEL) and medial areas (IEL to external elastic lamina) were measured using Slidebook 4.2 software (Cross-platform digital imaging, processing and analysis software for advanced microscopy applications).
Immunohistochemistry
Murine carotid arteries and spleen were embedded in OCT medium (frozen blocks) and cut into 5μm sections. Several sections from each mouse were stained as follows: incubation with 10% goat blocking serum, then a 1:50 dilution of rat anti-mouse CD4 (BD Pharmingen #550280). Detection for CD4 was with goat anti-rat Alexa Fluor 594 secondary (Molecular Probes #A11007) at a 1:200 dilution. The sections were then incubated in 5% rat serum and 5% rabbit serum. Excess serum was moved by tapping the slide and a rat anti-mouse CD8α antibody conjugated to FITC (Molecular Probes #MCD0801) was added at a 1:50 dilution. For detection and amplification of the signal an Alexa Fluor 488 Signal Amplification Kit for FITC (Molecular Probes #A-11053) was used. This kit includes Rabbit anti-FITC antibody (1:70 dilution) and Goat anti-rabbit antibody conjugated to Alexa Fluor 488 (1:125 dilution). All antibodies were diluted in 1X Tris Buffered Saline (TBS) pH7.6 and all washes between antibody incubations were with 1X TBS pH7.6. Some sections were also stained with rabbit anti-human/mouse CD3ε (Dako #A0452), in which case the antibody was added to the same tube with CD4 at a 1:100 dilution. The secondary for CD3 was goat anti-rabbit conjugated to Cy5 (Zymed #81-6116) and was added at a 1:200 dilution. All sections were mounted with VectaShield Mounting Media for Fluorescence with DAPI (Vector Labs #H-1200). Negative controls were included for each stain. Images were obtained with a Leica widefield microscope with DAPI, FITC, TRITC, and Cy5 fluorescent filters and 10X, 20X and 40X objectives. A Leica DFC350FX monochrome digital camera was used as well as Leica software. One spleen section was imaged using a Zeiss widefield microscope with an Apotome attachment.
Statistical analysis
Values are given as means ± SE. Analysis of variance with Bonferroni-Dunn post hoc analysis was used to imate whether differences between experimental groups were statistically significant. Statistical significance was accepted within 95% confidence limits.
Results
T lymphocytes localize to the carotid artery early post-injury in the NOD vasculature
At 7 days post-carotid arterial ligation, the NOD animals, show minimal luminal injury proximal (common carotid artery) to the prolene suture at the level of the bifurcation (Figure 1A). However, numerous CD3+ (yellow), CD4+ (red) and CD8+ (green) cells can be identified in the adventitia (Figure 1C, D) as well as the perivascular tissue (1B). Importantly, the internal and external elastic laminae were completely preserved, as well as the endothelial monolayer. Conversely, a minimal number of T cells (CD3, CD4, CD8) are observed in the contralateral uninjured vessel at the same time point (Figure 2A–C). Again, the internal and external elastic laminae are intact.
Figure 1. Localization of CD3+ T cells to the NOD vasculature.

Early following carotid ligation the NOD vessels show no evidence of structural injury (A). However, using immunofluorescence numerous adventitial/perivascular T lymphocytes can be identified (B–D).
Figure 2. Minimal T cell infiltration in the contralateral NOD vessels.

The contralateral uninjured arteries demonstrate no evidence of structural injury at 7 days (A [10X]). Similarly, the adventitia show minimal presence of T cells (B [10X], C [40X]) in the vessel wall.
To better characterize the contribution of a competent immune response to vascular pathology we performed carotid arterial ligation in immunodeficient mice (NOD.scid). These animals demonstrate minimal evidence of injury (n=8), at either the early (7 days, Figure 3A, B) or late (28 days, Figure 3C) time periods, suggesting a competent immune system is required for the whole spectrum of post-injury vascular remodeling.
Figure 3. Lack of significant inflammation in the injured NOD.scid arteries.
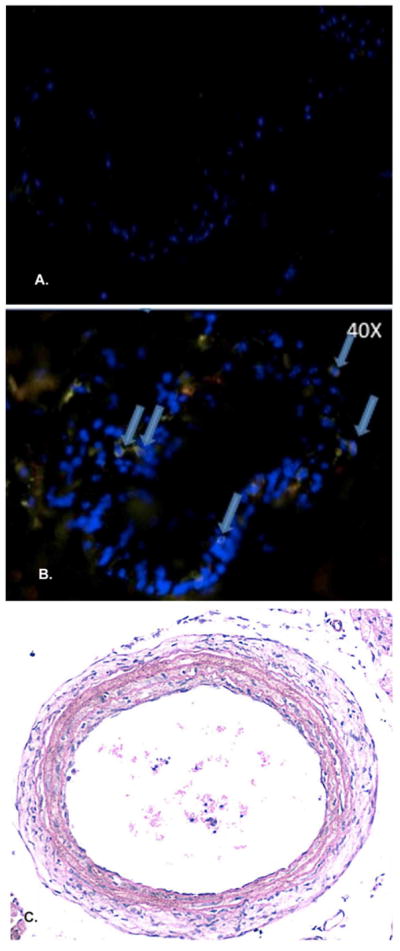
In the setting of immunodeficiency, the NOD.scid shows the presence of few T cells (arrows) in the injured arterial wall at 7 days post-ligation (A [10X], B [40X]). Importantly, the NOD.scid demonstrates no appreciable neoinitimal formation at the 28 day time point (C).
Confirmation of CD4+/CD8+ cells in the NOD spleen
To confirm the presence of CD4/CD8 T cells prior to splenoctye adoptive transfer, splenic tissue was evaluated via IHC for both CD4 and CD8 positivity. Not surprisingly, 5μm tissue sections reveal robust positivity for both CD4+ (red) and CD8+ (green) in the germinal center (Figure 4A [10X], and 4B [40X]). A similar degree of positivity can be identified at the periphery (Figure 4C [40X]). Using the Zeiss Apotome to flatten Z stacks into one image, with addition of DAPI nuclear stain (blue), CD4/CD8 staining is greatly enhanced.
Figure 4. Identification of CD4+/CD8+ T cells in the NOD spleen.
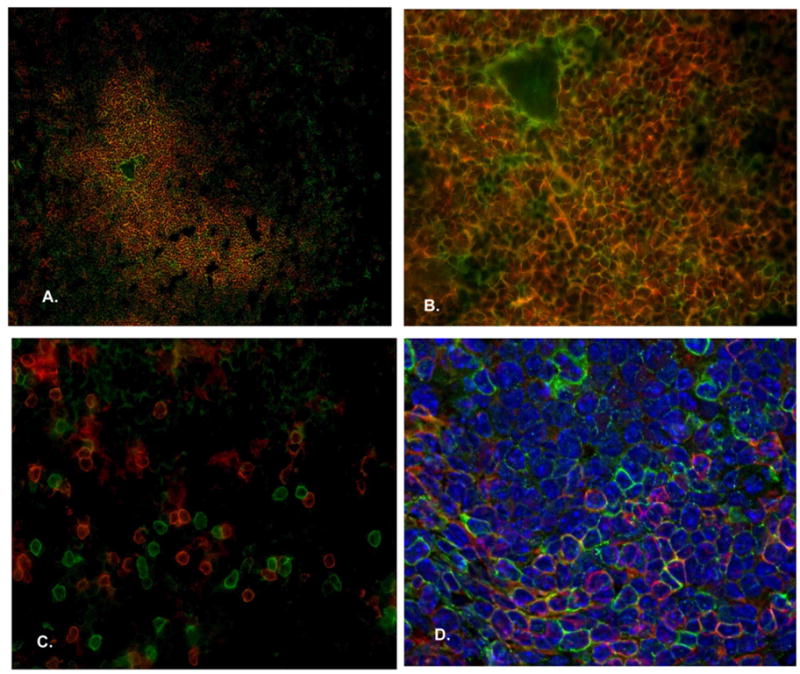
IHC confirms the presence of both CD4+ (red) and CD8+ (green) T cells in the lymph nodes of NOD donor mice (A [10X], B [20X], C/D [40X]) prior to adoptive tranfer into immunodeficient recipients.
Reconstitution of NIH following splenocyte adoptive transfer
The effects of complete carotid occlusion were evalutated in both C57/Bl6 and NOD mice. At 28 days post-ligation, NOD mice develop a significant vascular injury with profound neointimal hyperplasia and complete luminal occlusion and poorly defined internal and external elastic lamina (Figure 5A). In contrast, non-autoimmune C57/Bl6 mice develop hyperplasia and subsequent stenosis but the lumina remain patent. Quantitatively, the total intimal area in NOD carotid (n=8) was significantly larger than in the C57/Bl6 carotids (n=8) (53,310±2,151 vs. 25,424±2151 μm2, p<0.05). Interestingly, adoptive transfer of splenocytes from the autoimmune NOD into the NOD.scid (n=8), to generate chimeric mice, recapitulates the severity and magnitude of the obliterative lesions observed in the NOD (Figure 6A). Furthermore, these hyperplastic lesions look histologically similar. Quantitatively, there is no difference in the total intimal area between groups (Figure 6B) (62,694±3,996 vs. 53,310±2,151 μm2, ns).
Figure 5. Accelerated injury in the NOD vasculature.

At 28 days post-injury the C57/Bl6 mice develop significant intimal occlusion. While these vessels demonstrate significant stenosis, the lumen remains patent (A). The injured NOD vasculature accelerates to complete occlusion in the same time course. Quantitatively, the neointimal areas are significantly different (B, *p < 0.05).
Figure 6. Reconstitution of the accelerated lesion following splenocyte transfer.
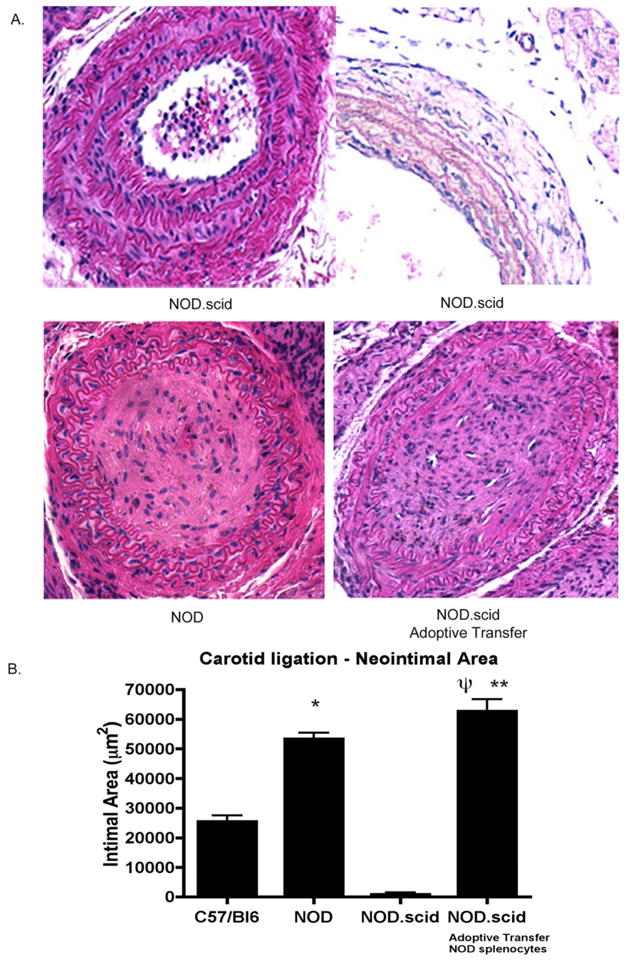
Following NOD splenocyte transfer, and concomitant ligation, the immunodeficient mice develop a robust pathologic response. As such, the accelerated injury is reconstituted (A). Quantitativley, the intimal area of the chimeric mice is significantly greater than either the NOD.scid (Ψp< 0.05) or the C57/Bl6 animals (**p < 0.05).
Discussion
Severe vascular disease in the form of arteriosclerosis, extracellular matrix remodeling, and alterations in gene expression have been previously identified in T1D (7–9). Specifically, the vascular endothelium is a sensitive target of the inflammatory response in several autoimmune diseases including systemic lupus erythematosus and rheumatoid arthritis (10–12). While the endothelium is a dynamic structure involved in tissue homeostasis, luminal inflammation and subsequent dysfunction result in clinically significant end-organ injury. Clearly, these early proinflammatory events result in vascular smooth muscle cell proliferation/migration, and ultimately neointimal formation and lipid deposition (13, 14). However, a clear link between autoimmunity, endothelial injury, and accelerated cardiovascular disease in T1D has not been established.
The identification of diabetogenic T cells, anti-islet antibodies, and marked histologic infiltrates strongly suggest a pathologic correlate between inflammation and sustained hyperglycemia. In general, pancreatic β-cell destruction is the result of a T cell-mediated autoimmune process directed against a specific antigen (or set of antigens) which have yet to be identified. Several recent findings suggest a similar vascular pathology in the setting of autoimmunity. In an experimental model of T1D, autoantibodies against endothelial antigens have been observed (15). Similar observations have been reported in diabetic patients (16). Interestingly, diabetes is transferable to immunodeficient NOD mice via a specific set of diabetogenic T cells which are CD40+ (17). Conversely, immunodeficient animals fail to develop T1D (18). While a competent immune response is pathologically required for the development of T1D, the concept of early vascular involvement continues to evolve (19).
Several pre-clinical observations in the NOD model document early vascular endothelial inflammation and dysfunction in euglycemia. Interestingly, indices of inflammation and oxidative stress are consistently elevated in the NOD vasculature. Recently, our laboratory addressed the pathobiological concept that vascular injury in T1D-prone animals might occur prior to sustained elevations in serum glucose levels. Using the NOD model, we observed that 10-week old animals demonstrate significant vascular endothelial dysfunction which is causally related to oxidant stress (5). Furthermore, treatment with a superoxide dismutase mimetic, or native antioxidant enzymes, inhibited tissue oxidant stress and restored normal endothelium-dependent relaxation. As impaired endothelial-dependent vasodilation, and paradoxical vasoconstriction, can be dissociated from elevated blood glucose, these observations strengthen the argument that vascular injury occurs prior to symptomatic T1D.
In this preliminary study, we carry the previous ex-vivo findings one step further. Not surprisingly, T lymphocytes (both CD4+ and CD8+) can be localized to the vasculature early in the post-ligation phase, with minimal infiltrate in the contralateral uninjured vessels. Few T cells are noted in immunodeficient animals. Following carotid arterial ligation, wild type mice clearly develop a distinct luminal stenosis. This injury is nearly undetectable in the NOD.scid. Importantly, the NOD demonstrates a more robust vascular injury at the same time interval. Following reconstitution of a competent immune response from the autoimmune phenotype, this accelerated response is restored.
While these observations are encouraging with regard to establishing a link between autoimmunity and accelerated euglycemic vascular injury in T1D, they must be considered with several caveats. Primarily, the NOD splenocyte preparation infused into the immunodeficient animals contains all splenic contents. As such, we are unable to specifically identify which immune compartment is responsible for this early vascular pathology. Clearly, the next phase of this initiative will be devoted to this line of investigation. Secondly, using a wild type splenocyte preparation in the NOD.scid would theoretically enhance the current series of observations attempting to differentiate the influence of the autoimmune phenotype versus not. However, the existing immunologic barriers make a graft versus host reaction a real concern with regard to post-operative mortality as well as a possible confounding effect on vascular injury.
Cumulatively, these data suggest that the same autoimmune-mediated tissue injury which results in pancreatic β-cell injury and subsequent T1D, may be at least partly involved in early cardiovascular injury. However, exactly which component of the immune response is predominantly involved in endothelial pathology is not clear. Future directions include identifying these cell types at various time points as well as examining several epigenetic changes which may be coincident with immune-mediated dysfunction.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Forrest KY, Becker DJ, Kuller LH, Wolfson SK, Orchard TJ. Are predictors of coronary heart disease and lower-extremity arterial disease in type 1 diabetes the same? A prospective study. Atherosclerosis. 2000;148:159–169. doi: 10.1016/s0021-9150(99)00217-8. [DOI] [PubMed] [Google Scholar]
- 2.Koivisto VA, Stevens LK, Mattock M, Ebeling P, Muggeo M, Stephenson J, et al. Cardiovascular disease and its risk factors in IDDM in Europe. EURODIAB IDDM Complications Study Group. Diabetes care. 1996;19:689–697. doi: 10.2337/diacare.19.7.689. [DOI] [PubMed] [Google Scholar]
- 3.Schaumberg DA, Glynn RJ, Jenkins AJ, Lyons TJ, Rifai N, Manson JE, et al. Effect of intensive glycemic control on levels of markers of inflammation in type 1 diabetes mellitus in the diabetes control and complications trial. Circulation. 2005;111:2446–2453. doi: 10.1161/01.CIR.0000165064.31505.3B. [DOI] [PubMed] [Google Scholar]
- 4.Tonkin DR, He J, Barbour G, Haskins K. Regulatory T cells prevent transfer of type 1 diabetes in NOD mice only when their antigen is present in vivo. J Immunol. 2008;181:4516–4522. doi: 10.4049/jimmunol.181.7.4516. [DOI] [PubMed] [Google Scholar]
- 5.Ling X, Cota-Gomez A, Flores NC, Hernandez-Saavedra D, McCord JM, Marecki JC, et al. Alterations in redox homeostasis and prostaglandins impair endothelial-dependent vasodilation in euglycemic autoimmune nonobese diabetic mice. Free Rad Biol Med. 2005;39:1089–1098. doi: 10.1016/j.freeradbiomed.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscl Throm Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 7.Andreassen TT, Oxlund H. Changes in collagen and elastin of the rat aorta induced by experimental diabetes and food restriction. Acta endocrinologica. 1987;115:338–344. doi: 10.1530/acta.0.1150338. [DOI] [PubMed] [Google Scholar]
- 8.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 9.Giugliano D, Ceriello A, Paolisso G. Diabetes mellitus, hypertension, and cardiovascular disease: which role for oxidative stress? Metabolism: clinical and experimental. 1995;44:363–368. doi: 10.1016/0026-0495(95)90167-1. [DOI] [PubMed] [Google Scholar]
- 10.Ciarla MV, Bocciarelli A, Di Gregorio S, Tordi A, Cotroneo P, Marra G, et al. Autoantibodies and endothelial dysfunction in well-controlled, uncomplicated insulin-dependent diabetes mellitus patients. Atherosclerosis. 2001;158:241–246. doi: 10.1016/s0021-9150(01)00440-3. [DOI] [PubMed] [Google Scholar]
- 11.Petty RG, Pottinger BE, Greenwood RM, Pearson JD, Mahler RF. Diabetes is associated with a high incidence of endothelial-binding antibodies which do not correlate with retinopathy, von Willebrand factor, angiotensin-converting enzyme or C-reactive protein. Diabetes Res. 1991;17:115–123. [PubMed] [Google Scholar]
- 12.Wangel AG, Temonen M, Brummer-Korvenkontio M, Vaheri A. Anti-endothelial cell antibodies in nephropathia epidemica and other viral diseases. Clin Exp Immunol. 1992;90:13–17. doi: 10.1111/j.1365-2249.1992.tb05824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmerman MA, Reznikov LL, Raeburn CD, Selzman CH. Interleukin-10 attenuates the response to vascular injury. J Surg Res. 2004;121:206–213. doi: 10.1016/j.jss.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 14.Zimmerman MA, Reznikov LL, Sorensen AC, Selzman CH. Relative contribution of the TNF-alpha receptors to murine intimal hyperplasia. American J Physiol. 2003;284:R1213–1218. doi: 10.1152/ajpregu.00434.2002. [DOI] [PubMed] [Google Scholar]
- 15.Quintana FJ, Cohen IR. Autoantibody patterns in diabetes-prone NOD mice and in standard C57BL/6 mice. J Autoimmun. 2001;17:191–197. doi: 10.1006/jaut.2001.0544. [DOI] [PubMed] [Google Scholar]
- 16.Jones DB, Wallace R, Frier BM. Vascular endothelial cell antibodies in diabetic patients. Association with diabetic retinopathy. Diabetes care. 1992;15:552–555. doi: 10.2337/diacare.15.4.552. [DOI] [PubMed] [Google Scholar]
- 17.Balasa B, Krahl T, Patstone G, Lee J, Tisch R, McDevitt HO, et al. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J Immunol. 1997;159:4620–4627. [PubMed] [Google Scholar]
- 18.Yoon JW, Jun HS, Santamaria P. Cellular and molecular mechanisms for the initiation and progression of beta cell destruction resulting from the collaboration between macrophages and T cells. Autoimmunity. 1998;27:109–122. doi: 10.3109/08916939809008041. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerman MA, Flores SC. Autoimmune-mediated oxidative stress and endothelial dysfunction: implications of accelerated vascular injury in type I diabetes. J Surg Res. 2009;155:173–178. doi: 10.1016/j.jss.2008.04.026. [DOI] [PubMed] [Google Scholar]