

Abstract
Angiocidin, a tumor-associated peptide, has been previously shown to inhibit tumor progression by blocking angiogenesis. We now show that angiocidin has a direct inhibitory effect on tumor cell proliferation. MDA-MB-231 breast cancer cells were inhibited from proliferating in the presence of epidermal growth factor (EGF) and angiocidin. Angiocidin transfected breast cancer cells also displayed growth inhibition in vitro and failed to develop significant tumors in mice as compared to vector controls. The anti-proliferative effect of angiocidin was reversed by treating the cells with the epidermal growth factor receptor (EGFR) inhibitor 4557W, a potent tyrosine kinase inhibitor. Consistent with these results, we found that treatment of breast cancer cells with angiocidin induced a 2.3 fold increase in EGFR tyrosine 845 phosphorylation while no change in phosphorylation was observed in the remaining 16 phosphorylation sites of EGFR and those of its family members as measured by a human EGFR phosphorylation array. Treatment of breast cancer cells with angiocidin also resulted in the activation of nuclear factor κB (Nf-κB) and the de novo up-regulation of many down-stream genes transcribed by Nf-κB, including cytokines, inflammatory mediators and the cell cycle inhibitor p21waf1. Therefore, angiocidin is a peptide that not only inhibits tumor angiogenesis but directly induces inhibition of tumor growth progression through the activation of EGFR and down-stream genes transcribed by Nf-κB.
Keywords: Angiocidin, breast cancer, proliferation, Nf-κB pathway, cytokines, EGFR
Introduction
Angiocidin is a 41 kilodalton protein first isolated from lung carcinoma by affinity chromatography using the immobilized peptide CSVTCG present in the type 1 repeat region of thrombospondin-1, an important matrix protein that mediates tumor progression (Tuszynski et al., 1993). Angiocidin has a high degree of homology with two other proteins, anti-secretory factor (ASF) and S5a, with the exception of three additional amino acids at positions 769–771 on angiocidin (Zhou et al., 2004). ASF is a plasma-bound protein known to inhibit cholera toxin-induced intestinal fluid secretion in rats (Johansson et al., 1997). S5a is the polyubiquitin-binding subunit of the 26S proteasome, which is responsible for degradation of polyubiquitinated proteins (Walters et al., 2002). Like its homologue S5a, angiocidin also contains polyubiquitin-binding motifs, which have been shown to bind polyubiquitinated proteins on the surface of endothelial cells with high affinity (Dimitrov et al., 2005).
High levels of angiocidin have been found in the extracellular matrix of many types of cancers, including breast cancer, which suggests that it plays a role in tumor progression. Additionally, several studies have shown that recombinant angiocidin or its matrix binding domain has considerable anti-tumor activity (Sabherwal et al., 2006; Zhou et al., 2004). For example, in a mouse model of lung cancer iv injection of angiocidin reduced tumor growth by more than 90% (Zhou et al., 2004).
The precise mechanism of the anti-tumor activity of angiocidin has yet to be determined. Our previous studies suggest that angiocidin blocks tumor angiogenesis through its ability to bind extracellular matrix and block adhesion (Sabherwal et al., 2006) or block proteasome activity by competing with endogenous ubiquitin recognition subunits (Dimitrov et al., 2005). Recently, we made the unexpected discovery that when hematologic tumor cells such as leukemia cells were treated with angiocidin, they stop proliferating and undergo differentiation (Gaurnier-Hausser et al., 2008). This activity was due to activation of the Nf-κB pathway and the subsequent release of a cocktail of growth inhibitory and differentiation inducing cytokines that act on the cell in an autocrine manner (Gaurnier-Hausser and Tuszynski, 2009). Based on these data, we postulated that the same kind of mechanism might also explain in part the anti-tumor activity of angiocidin in solid tumors.
In this study we show that breast cancer cells treated with angiocidin undergo growth arrest and activation of EGFR and the Nf-κB pathway. We show that the consequence of this is inhibition of tumor growth in vivo. Remarkably, angiocidin just like in the leukemia system up-regulates Nf-κB responsive genes that inhibit proliferation directly through proteins such as the cell cycle inhibitor p21waf1 or indirectly through secreted cytokines by an autocrine mechanism. These studies further define the anti-tumor activity of angiocidin.
Materials and methods
Materials
MDA-MB-231 cells were originally obtained from American Type Culture Collection (Manassas, VA). High glucose Dulbecco’s modified Eagle Medium (DMEM) was obtained from Mediatech (Herndon, VA) and GIBCO, through Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS), bovine serum albumin (BSA), and L-glutamine were purchased from HyClone (Logan, UT). Rabbit monoclonal Anti-p65, anti-IκBα, and anti-phospho-IκBα antibodies were obtained from Cell Signaling Technologies (Boston, MA). Rabbit monoclonal anti-phospho-p50 (Ser 337) was purchased from Santa Cruz (Santa Cruz, CA). Rabbit polyclonal anti-TLR2 antibody was purchased from Abcam (Cambridge, MA). Mouse monoclonal anti-p21 antibody was purchased from BioLegend (San Diego, CA). Mouse monoclonal anti-p53 antibody was obtained from CalBioChem (Gibbstown, NJ). Mouse monoclonal anti-β-actin antibody was obtained from Sigma-Aldrich (St. Louis, MO). Goat anti-rabbit IgG/horseradish peroxidase (HRP) and rabbit anti-mouse IgG/HRP conjugates were purchased from BioRad (Hercules, CA). Nuclear Extraction Kit was obtained from Panomics (Fremont, CA). Human CCL2/MCP-1 ELISA kit was obtained from eBioscience (San Diego, CA). RayBio Human Cytokine Antibody Array (Array 3) was purchased from RayBiotech, Inc. (Norcross, GA). RT² qPCR-Grade RNA Isolation Kit, RT² SYBR Green Master Mix, RT² First Strand Kit, and qRT-PCR array were purchased from SA Biosciences (Frederick, MD). For nuclear isolation and lysis, the Panomics Nuclear Extraction Kit was used (Affymetrix, Inc. Santa Clara, CA). Cells were lysed according to the manufacturer’s protocol and stored at − 80° C.
Anchorage-independent Growth
Soft agar assay was performed in 6 well plates where a base 0.8% Noble Agar (Difco Laboratories, MI) mixed with 10% FBS containing DMEM was coated. An aliquot of 5,000 cells per well was mixed in 0.4% DMEM-agar and overlaid on the base agar. The plates were incubated at 37° C in 5% CO2 for 10–14 days. Colony formation was checked under the Olympus IMT-2 microscope with a 4X objective and digital images were obtained with a Kodak DC120 (Eastman Kodak, NY) camera device and the Photoenhanser software from PictureWorks Technologies.
Animal Studies
Athymic mice were obtained from Charles River Laboratories and housed in the University’s Animal Facility. The animals were nude/nude genetype and they lacked a functioning immune system so rejection of human cells did not occur. 6 female and aged matched animals per test group were injected in the mammary fat pad with 107 transfected cells as was previously reported (Zhou et al., 2004). 6 animals per group were injected on the right flank with 107 transfected cells. Animals were examined every other day for tumor size as assessed with a caliper. The tumor volume was estimated using the formula length X width2/2. Six weeks later the animals were euthanized with CO2 asphyxiation and the tumors were fixed, paraffin embedded, sectioned and examined by immunohistochemical staining for the expression of angiocidin in the tumors.
Cytokine and EGFR Phosphorylation Antibody Array Analysis
MB-231 cells were seeded in a 6-well plate and left until 75% confluent. Cells were then placed in 2% FBS DMEM for 2 hours prior to a six-hour treatment or 24-hour treatment period with 10 μg/ml angiocidin for the cytokine array analysis and a 6 hour treatment with 10 μg/ml angiocidin for the EGFR array analysis. Untreated cells were maintained in 2% FBS without angiocidin for the same amount of time. Following treatment, media was collected from both treated and untreated samples and kept on ice until applying it to the antibody array membranes. Prior to application of media onto membranes, membranes were placed in 2 ml of blocking buffer for thirty minutes at room temperature. Media was then incubated with membrane overnight at 4° C. Membranes were washed with provided wash buffers. Following the washes, membranes were incubated with biotin-conjugated anti-cytokine antibodies or biotin-conjugated anti-phospho EGFR antibodies in blocking buffer overnight at 4° C. The next day, membranes were washed and incubated overnight at 4° C with 2 ml of 1,000-fold diluted HRP-conjugated streptavidin in blocking buffer. Following this overnight incubation, membranes were washed and detection buffers applied to the membranes. The membranes were then detected using autoradiography film.
PCR Array Analysis
In order to perform qRT-PCR of the toll-like receptor (TLR) pathway genes, cells were either treated with 10 μg/ml angiocidin for 24 hours or left untreated in 2% FBS DMEM. Cells were then lysed and RNA isolated using the protocol from the manufacturer (SABiosciences). RNA was isolated in a sterile, RNase-free environment. RNA concentration and purity were then determined using NanoDrop machinery and the quality of RNA was confirmed with an RNA gel. The RNA was then reverse-transcribed using the RT2 First Strand Kit according to the manufacturer’s protocol. One microgram of each sample was used for the first strand kit and was combined with a genomic DNA elimination mixture before undergoing reverse transcription polymerase chain reaction (PCR). The cDNA templates were then combined with SYBR green in the amount of 1.4 ml per sample of RNA. The cDNA/SYBR green mixture was applied in equal amounts (25 μl per well) to the 96-well plate, which contained primers for various TLR pathway genes. The plates were spun down at 600 rpm to rid the samples of bubbles that may have occurred due to pipetting. The plates were then kept on ice until running RT-PCR for 2.5 hours. Following RT-PCR, data was analyzed using SABiosciences analysis program on the SABiosciences website.
Proliferation Assay
Cell proliferation was measured using the Alamar blue assay as previously described (Zhou et al., 2004).
Sample Preparation and Protein Assay
To perform cell lysis and protein collection, cells were washed twice with sterile phosphate-buffered saline (PBS). Cell Stripper was applied for a period of 20 minutes in 37° C, 5% CO2. Detached cells were spun at 700 rpm to obtain a cell pellet. Cells were lysed using either a 2% sodium dodecyl sulfate (SDS) lysis buffer or a 1% nonyl phenoxylpolyethoxylethanol–40 (NP-40) lysis buffer containing protease and phosphatase inhibitors and kept on ice for the duration of lysis. Lysates were then sonicated and spun at 15,000 rpm to obtain a pellet of debris and supernatant containing cellular proteins. The supernatant was saved and the total protein concentration determined using Pierce’s micro bicinchoninic acid (BCA) protein assay according to the manufacturer’s protocol. Cell lysates were aliquoted at 20 μg/tube and preserved at − 20° C for short-term storage or at − 80° C for long-term storage.
Immunohistochemical Staining
Cells were grown to 80% confluency in a 6 well chamber slide. Cells were then fixed in 3% paraformaldehyde for 3 min. After washing slides in PBS, samples were incubated with 5% normal horse serum in PBS for 1 hour at room temperature and then they were incubated with a monoclonal anti-angiocidin IgG (developed in our laboratory) at a concentration of 2 μg/ml for 1 hour at room temperature. As a negative control mouse IgG was used in every experiment. Slides were developed as previously described (Zhou et al., 2004).
Preparation of Angiocidin Transfected Cells
The angiocidin gene was cloned into the BamHI and EcoRI sites of the pcDNA3.1/zeo vector (OpenBiossystems, Huntsville, AL). The angiocidin insert was generated as a PCR product that had the Kozak sequence, at the beginning and the stop codon, TAG, at the end of the insert. Sequencing of the vectors using the T7 primer confirmed the sequence of the insert. MDA-MB-231cells were transfected with purified DNA isolated by the Wizard Plus kit (Promega, Madison, WI). The DNA is incorporated into the cells using the Superfect transfection reagent (Qiagen, Valencia, CA). Cells were plated in 6 well plates and when the cells reached 80% confluency transfection was performed. 12.5 l of the reagent was used as well as 2.5 g of the DNA, with a minimal concentration of 0.1 g/l. Superfect-DNA complex formation was performed in a serum- free and antibiotic- free medium at room temperature for 10 min. Cells were then incubated with the superfect-DNA complex at 37°C for 3–4 hours. Then media was changed and 48 hours post transfection cells were selected with zeocin (400 g/ml) antibiotic. Several clones (12 each) of the vector control, and sense breast cancer cells were isolated using cloning rings. Out of the 24 total clones isolated there were 3 clones (1 control, and 2 sense) that were kept for further analysis. Clones were assessed for their angiocidin protein level expression by western blot analysis.
Purification of Recombinant Angiocidin
Recombinant his-tagged angiocidin was expressed in E. coli and purified on a nickel- Sepharose column as previously described (Zhou et al., 2004).
Western Blot Analysis
Cell lysates were combined with a 1% SDS/bromophenol blue running buffer. 5% B-mercaptoethanol was added fresh just before heating the lysates in boiling water for five minutes. Following heating, 20 ug of lysates were run on a 6–12% polyacrylamide gel under reducing conditions. Proteins were transferred onto a PVDF membrane overnight at 4° C. The polyacrylamide gel was stained with BioSafe stain and the PVDF membrane was blocked for 1 hour in 5% milk/TBST or in 5% BSA/Tris-buffered saline with 0.1% Tween-20 (TBST). Following blocking, membranes were incubated overnight at 4° C with the manufacturer’s recommended dilution of antibody (typically 1:1000 in 5% BSA/TBST). Following incubation with primary antibody, membrane was washed three times in TBST and incubated with a 1:10,000 dilution of secondary antibody in TBST for one hour. The secondary antibody was either goat anti-rabbit IgG/HRP or rabbit anti/mouse IgG/HRP. An Enhanced Chemiluminescence (ECL) Plus kit was used to detect protein levels via chemiluminescence. Following detection, the membrane was maintained in Tris-buffered saline (TBS) in 4° C. To strip the membrane of the antibodies, a 0.2N solution of NaOH was applied to the membrane for anywhere from 5–10 minutes depending on intensity of bands. The membrane was then washed in water once and in PBS three times and re-blocked in 5% milk/TBST.
Results
Angiocidin Inhibits Breast Cancer Proliferation
We evaluated the effect of angiocidin on the proliferation of MB-231 breast cancer under various experimental conditions. After a 16 hour culture of MB-231 breast cancer cells in serum-free media containing 10 μg/ml of angiocidin, cells rounded up and detached (Figure 1). The proliferation rate of the rounded cells was inhibited by more than 80% (Figure 1) and if the cells were cultured for more than 24 hours under these conditions they underwent apoptosis (data not shown). To further evaluate the effect of angiocidin on the proliferation of breast cancer cells, cells were transfected with angiocidin-containing vector or with the control vector. These cells expressed about 2 fold more angiocidin as assessed by immunohistochemical staining and Western blot analysis (Fig 2A and B). The angiocidin-transfected breast cancer cells failed to develop significant colonies in soft agar and or tumors in aythmic mice as compared to vector controls (Fig. 2C and D). This effect was not due to clonal variation since two separate clones (sense 1 and sense 2) developed tumors that were at least 6 fold smaller than controls (Fig. 2D).
Fig.1.
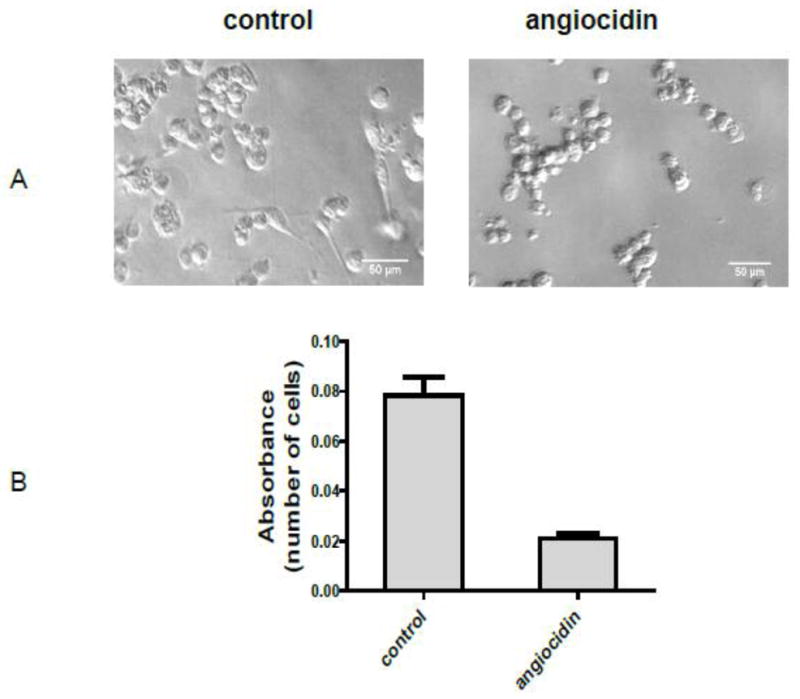
Morphological appearance and growth of angiocidin-treated MB-231 cells. MDA-MB-231 cells were seeded in 0.1% BSA DMEM without recombinant angiocidin and incubated at 37° C for sixteen hours. After sixteen hours the cells were photographed. The number of adherent, spread cells resembling the morphology of normal MB-231 cells in four representative fields was counted. The percentage of adherent, spindle-shaped cells was determined to be 36 out of 225, or 16% (panel A). Cell proliferation was determined in a 96 well plate using the Alamar blue proliferation assay under the same culture conditions (Panel B). Experiments were repeated three times and the results of a representative experiment are shown in the figure.
Fig. 2.
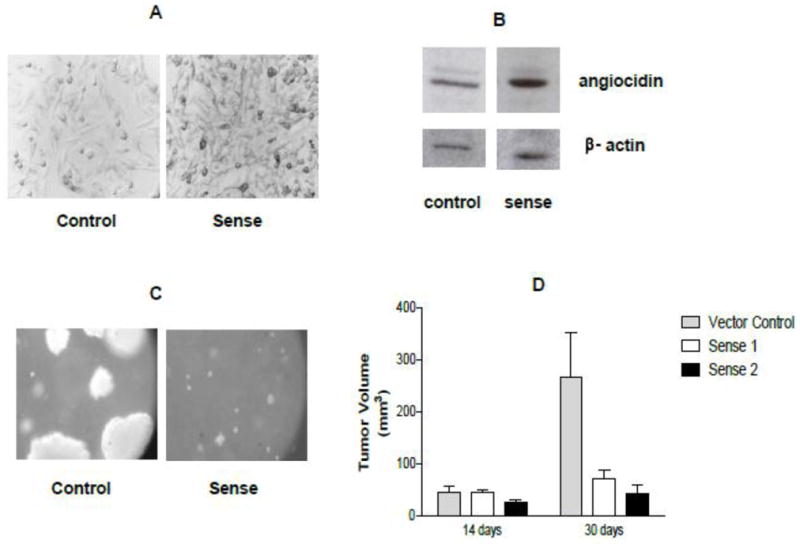
Angiocidin-transfected MDA-MB-231 tumor cells develop smaller tumors in mice and smaller colonies in soft agar. MDA-MB-231 breast cancer cells were transfected with angiocidin using the pcDNA3.1 transfection system as described by Invitrogen. At least two angiocidin over-expressing clones were selected sense 1 and 2 as well as the vector control. The panels marked sense show the sense 1 clone. Similar results were obtained with sense 2 clones. Panel A show paraformaldehyde-fixed cells grown in culture and stained with anti-angiocidin antibody and developed with DAB (100×). Panel B shows Western blot of cells grown in panel A. Panel C shows colonies grown in soft agar as seen by phase contrast (50x mag). Panel D shows tumor volumes of cells grown in mice (n=6 per group ± std). In vitro experiments were repeated three times and the results of a representative experiment are shown in the figure.
Angiocidin Activates EGFR
To begin to investigate the mechanism of angiocidin-induced inhibition of breast cancer proliferation, we postulated that angiocidin may be a ligand for a growth factor receptor and signal growth arrest. We chose to investigate EGFR since this receptor is activated by a number of ligands (Yarden, 2001) and that some extracellular ligands such as decorin can induce growth inhibitory signals in breast cancer (Reed et al., 2005). We found that angiocidin inhibited breast cancer proliferation in serum containing media supplemented with EGF (Fig. 3A). This growth inhibiting activity of angiocidin was completely blocked with 4557W, a potent tyrosine kinase inhibitor specific for EGFR. Angiocidin induced a dose-dependent up-regulation of p21waf1, a major cyclin-dependent kinase inhibitor that regulates the G1 phase of the cell cycle (Fig. 3B, C). When angiocidin-treated breast cancer cells were analyzed for phosphorylation changes of the EGFR by a phosphorylation antibody array that detects all the major phosphorylated tyrosine and serine sites of EGFR and its closely related family members ErbB2, 3 and 4, we found that tyrosine 845 (Tyr845) of EGFR was phosphorylated 2 fold more as compared to media-treated cells (Fig 3D, top and bottom panel). Since Tyr845 of EGFR has been shown to signal up-regulation of p21waf1 by decorin in A431 carcinoma cells (Iozzo et al., 1999), our results are consistent with a mechanism of angiocidin-mediated growth inhibition involving activation of EGFR with the downstream up-regulation of p21waf1.
Fig. 3.
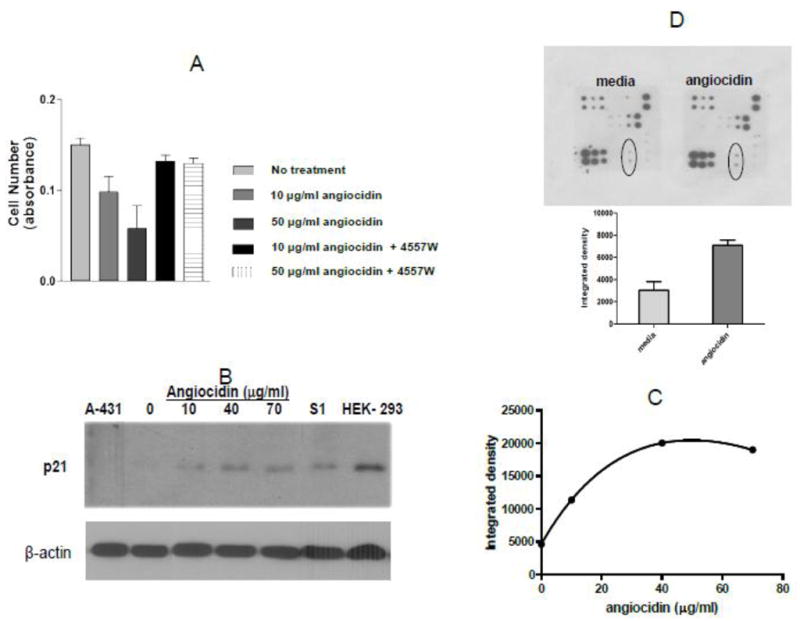
Angiocidin inhibits proliferation of MB-231 breast cancer cells through activation of EGFR and up-regulation of p21waf1. Panel A- Cells were grown in DMEM media containing 10% serum and 10 ng/ml EGF either alone or treated with 10 μg/ml or 50 μg/ml angiocidin in the presence or absence of 10 μg/ml EGFR inhibitor 4557W and proliferation was measured after 24 hours using the Alamar blue proliferation assay. Panel B- Cells were grown in DMEM media containing 2% serum for 24 hours in six well plates and then treated with various concentrations of angiocidin for an additional 24 hours, harvested and blotted for p21waf1. Cell lysate from A-431 was used as the negative control and cell lysate from HEK-293 cells was the positive control. Panel C- p21waf1 positive bands in Panel B quantitated by densitometry using Image J software. Panel D- EGFR phosphoarray results (top-stained array, bottom- spots encircled in the stained array were quantitated by densitometric analysis of the spots using Image J software ). Experiments were repeated two times and the results of a representative experiment are shown in the figure.
Angiocidin activates genes transcribed by NFkB
Since activation of EGFR in breast cancer has been shown to activate the NFkB signaling pathway in breast cancer (Biswas and Iglehart, 2006) and we previously showed that angiocidin activates the NFkB pathway in leukemia cells (Gaurnier-Hausser et al., 2008), we investigated if angiocidin could activate NFkB and stimulate transcription of NFkB responsive genes. We found that angiocidin promoted the time-dependent nuclear localization of NFkB protein (p65) with the concomitant loss of cytoplasmic p65 expression (Fig.4B). Loading controls for the nuclear fractions (p53) and actin for the cytoplasmic fractions were constant over time indicating that the observed changes were not due to differential loading (Fig. 4B). We also found that angiocidin promoted the phosphorylation of IkBα, the major cytoplasmic inhibitor of NFkB, and decreased total IkB, further providing evidence that angiocidin activates the NFkB pathway (Fig. 4B). The presence of higher molecular forms of phospho-NFkB seen in cell extracts from cells treated with 20 μg/ml angiocidin could be ubiquitinated forms of the protein since phosphorylation of the protein has been shown to signal its degradation by the proteasome (Walters et al., 2002).
Fig. 4.
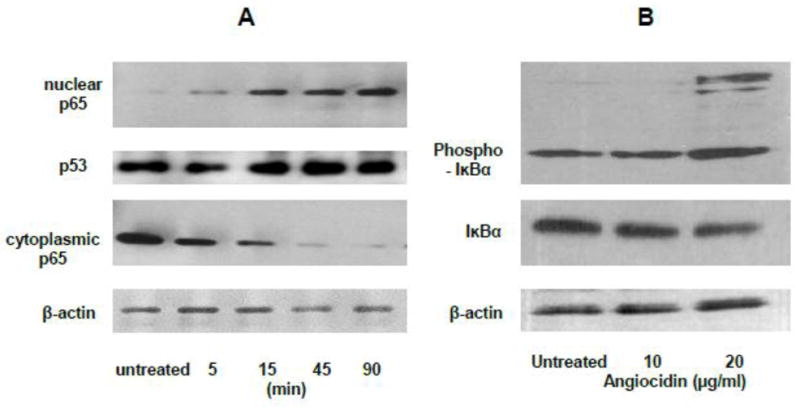
Angiocidin promotes the activation of NFκB and phosphorylation of IκBα. Panel A-NFκB p65 Western Blot of Nuclear and cytoplasmic extracts of MB-231 cells treated with 10 μg/ml of angiocidin in 2% FBS DMEM for a period of time ranging from 5 minutes to 90 minutes. Panel B-Phospho-IKBα Western Blot of MB-231 cells treated for twenty-four hours with 0 μg/ml, 10 μg/ml, or 20 μg/ml of angiocidin in DMEM containing 0.1% BSA. Experiments were repeated three times and the results of a representative experiment are shown in the figure.
Activation of the NFkB pathway results in the transcription of many important genes that mediate proliferation, cell survival and cell death (Karin et al., 2002). One of the NFkB responsive genes that we discovered to be up-regulated was p21waf1, a key regulator of tumor growth (Fig. 3B). Additionally, important mediators of cell growth, immunity and cell differentiation are also known to be transcribed by NFkB (Karin et al., 2002). Therefore, we investigated whether angiocidin up-regulated important cytokines and chemokines as well as inflammatory mediators that could impact cell growth. We found several genes up-regulated by angiocidin using a PCR toll-like receptor array, which measures many genes transcribed by NFkB (Figure 5A, table 1). Notable among these were interleukins, interferons, map kinase genes and toll-like receptor genes that play an important role in growth and immunity. Similarly, angiocidin induced secretion of MCP-1, Il-8, angiogenin, and GM-CSF, important cellular mediators of growth and differentiation (Figure 5B, Table 2). This cocktail of factors can act on tumor cells in a paracrine manner and impacting cell proliferation directly or indirectly by stimulating innate immunity. For example, the secreted cocktail of cytokines can be potent stimulators of the innate immune system, while others such as the interferons can be potent inhibitors of breast cancer proliferation (Gooch et al., 2000).
Fig. 5.
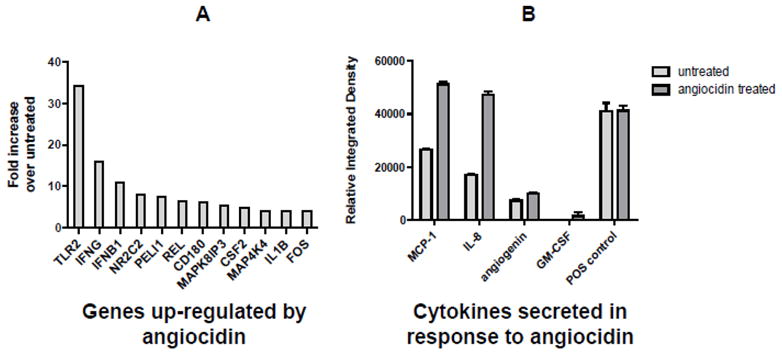
Angiocidin up-regulates cytokines, immune and inflammatory mediators in MB-231 cells. Panel A-Genes up-regulated 4 fold or greater in angiocidin-treated cells as detected with a PCR Toll- like receptor pathway array. Panel B- Cytokines detected in conditioned media from untreated and angiocidin-treated MB-231 using a cytokine array. Details of the array analysis are described in Material and Methods.
Table 1.
Genes up-regulated more than 4 fold in angiocidin-treated MB-231 cells as detected by PCR Array
| Gene | Function | Fold Increase | Reference |
|---|---|---|---|
| TLR2 | Immune cell receptor, promotes cytokine production through NFkB pathway, improve chemotherapy | 34.3 | (Garay et al., 2007) |
| IFNG | Immune cytokine, inhibits breast cancer proliferation | 16 | (Iacopino et al., 1997) |
| IFNB1 | Immune cytokine, inhibits breast cancer proliferation | 10.6 | (Sica et al., 1987) |
| NR2C2/TR4 | Orphan nuclear receptor | 8 | (Shyr et al., 2002) |
| PELI1 | E3 ubiquitin ligase, promotes cytokine synthesis | 7.5 | (Chang et al., 2009) |
| REL | a member of the NFκB family of transcription factors. | 6.5 | (Biswas et al., 2004) |
| CD180 | a member of the Toll-like receptors | 6.1 | (Aderem and Ulevitch, 2000) |
| MAPK8IP3 | protein kinase, regulates the activity of numerous protein kinases of the JNK signaling pathway | 5.3 | (Davis, 2000) |
| CSF2/GM- CSF | Anti-tumorigenic cytokine in breast cancer | 4.9 | (Eubank et al., 2009) |
| MAP4K4 | Protein kinase, upregulated p53 and drives c- Jun NH2-terminal kinase (JNK) Mediated apoptosis | 4.0 | (Miled et al., 2005) |
| IL1B | Inflammatory cytokine, promotes leukocyte differentiation, inhibits breast proliferation | 4.0 | (Roy et al., 2006) |
| c-FOS | Proto-oncogene important in breast cancer proliferation | 4.0 | (van der Burg et al., 1989) |
Table 2.
Cytokines up-regulated in angiocidin-treated MB-231 cells as detected by Cytokine Array
| Gene | Function | Fold Increase | Reference |
|---|---|---|---|
| MCP-1 | Inflammatory cytokine, promotes innate immunity | 1.92 | (Fujimoto et al., 2009) |
| IL-8 | Inflammatory cytokine, promotes innate immunity, stimulates breast cancer proliferation | 2.7 | (Nicolini et al., 2006) |
| CSF2/GM- CSF | Anti-tumorigenic cytokine in breast cancer | N/A | (Eubank et al., 2009) |
| Angiogenin | ribonuclease, promotes angiogenesis | 1.3 | (Nilsson et al., 2010) |
Discussion
Angiocidin is a unique tumor suppressive peptide that targets both endothelial cells and tumor cells. We have previously shown that angiocidin can induce apoptosis in endothelial cells by binding to tumor extracellular matrix (Sabherwal et al., 2006), transglutaminase (L’Heureux D et al., 2009) and cell surface integrins (Sabherwal et al., 2006). These interactions initiate a signaling cascade that results in the suppression of tumor progression of solid tumors in vivo by inhibiting tumor angiogenesis.
Recently, we asked the question if angiocidin could directly act on tumor cells to make them less tumorigenic. To answer this question we first evaluated the effect of angiocidin on hematologic tumors (Gaurnier-Hausser et al., 2008) since our previous studies showed that angiocidin could activate T cells and monocytes (Kremlev et al., 2008). Using a well-studied leukemic cell line (THP-1), we showed that angiocidin inhibited its proliferation (Gaurnier-Hausser and Tuszynski, 2009). However, more remarkably, treatment of THP-1 cells with angiocidin induced them to differentiate into a normal-like monocytic phenotype (Gaurnier-Hausser et al., 2008). In the presence of angiocidin THP-1 cells regained the ability to phagocitize, expressed monocyte markers, antigen presenting molecules and secreted a cocktail of cell differentiating cytokines. These phenotypic changes were initiated by activation of the NFKB pathway.
In the present study we decided to extend our observations obtained in the leukemia system to a solid tumor system. We chose breast cancer because in previous studies we could show that angiocidin bound breast cancer cells through α2β1 integrin (Sabherwal et al., 2006) and that the integrin and matrix binding domain was critical for the anti-tumor activity of the full-length molecule (Zhou et al., 2004).
To evaluate the role of angiocidin in breast cancer progression we pursued two approaches. The first was to treat breast cancer cells in culture and evaluate the effect of exogenously added angiocidin on breast cancer proliferation and the second was to transfect breast cancer cells with angiocidin and evaluate the tumorigenicity of the resulting stable transfectants. When MB-231 breast cancer cells were treated with angiocidin in serum-free media for 16 hours they rounded-up and showed a marked inhibition of growth (Fig. 1). After prolonged growth under these conditions the cells became apoptotic (data not shown). These results were similar to those obtained for endothelial cells (Zhou et al., 2004). In the second approach, we isolated two stable MB-231 angiocidin transfectants that expressed 2–3 fold more angiocidin than vector controls. The angiocidin-transfected cells failed to develop significant colonies in soft agar as compared to vector controls (Fig. 2). Even more importantly, both angiocidin-transfected clones developed tumors in aythmic mice that were more than 90% smaller than vector controls (Fig. 2). These results directly show that angiocidin inhibits tumor proliferation and progression both in vitro and in vivo.
We next investigated possible mechanisms of angiocidin growth inhibition. We postulated that angiocidin may signal growth inhibition through a growth factor receptor that associates with integrins on the cell surface which we have shown previously bind angiocidin (Sabherwal et al., 2006). We decided to investigate whether angiocidin could signal through EGFR since EGFR interacts with integrins and many different ligands are known to signal through EGFR including matrix proteins. For example, decorin interacts with α2β1 integrins and EGFR to signal growth inhibition of many different cancer cells (Grant et al., 2002). Our first indication that EGFR might be involved in angiocidin-mediated growth inhibition of MB-231 cells was the observation that angiocidin inhibited growth of MB-231 cells in serum-containing media supplemented with EGF (Fig. 3A). In Figure 1 we showed that angiocidin inhibited growth of MB-231 cells in serum-free media but that this effect could be rescued with serum (data not shown). However, in serum-containing media supplemented with EGF, angiocidin inhibited growth in a dose-dependent manner (Fig. 3A). However, when the specific EGFR tyrosine kinase inhibitor 4557W was added the inhibitory effect of angiocidin was completely abrogated (Fig 3A). Western blots of cells treated under these conditions showed that angiocidin up-regulated p21waf1, a potent cell cycle inhibitor. (Fig. 3B and C). When we examined the tyrosine and serine phosphorylation state of EGFR in the angiocidin-treated cells, we found that only tyrosine 845 was phosphorylated by more than 2 fold as compared to untreated cells (Fig. 3D). Consistent with our results, previous studies have shown that tyrosine 845 mediates the growth inhibitory signals in A-431 carcinoma cells (Sato et al., 1995).
Activation of NFkB occurs in response to extracellular chemical stresses, various cytokines and growth stimuli, resulting in the direct induction of hundreds of genes whose cellular influences extend well beyond those of the immune system, where its essential role was first appreciated (Karin et al., 2002). In this study, we demonstrated that angiocidin rapidly activates NFκB in MB-231 cells. Within 15 minutes of angiocidin treatment, NFκB translocated to the nucleus and the major inhibitor of NFκB, IκBα was phosphorylated (Fig. 4A and B). These results were similar to those obtained with THP-1 leukemia cells in our previously published studies (Gaurnier-Hausser et al., 2008). We found that as with THP-1 cells, angiocidin up-regulated a number of cytokines and genes associated with inflammation and immunity (Fig. 5A and B and Table 1). The functional significance of all these genes is unknown at this time. Clearly, some of the up-regulated cytokines such as the interferons are growth inhibitory while others such as IL-8 are growth promoting. While these factors may on balance be difficult to determine if they are positive or negative regulators of tumor progression, it is clear that the majority play a role in innate immunity. It is possible that angiocidin could render tumors more susceptible to immune surveillance and thereby inhibit tumor progression. Therefore, it seems likely that angiocidin may play an important role in tumor immunology (Gaurnier-Hausser and Tuszynski, 2009).
In summary, we have shown that angiocidin directly inhibits breast cancer progression through its effect on breast cancer cell proliferation. This inhibitory activity is mediated by EGFR and downstream genes transcribed by NFκB. Our studies further underscore the remarkable anti-tumor activity of the peptide angiocidin and strongly provide evidence for the development of this peptide for the treatment of breast cancer.
Acknowledgments
This study was supported in part by a grant from the National Institute of Health R01 88931 to GPT and the Pennsylvania State Tobacco Settlement funds.
Abbreviations
- Rel
C-Rel proto-oncogene protein
- MAPK8IP3
C-jun-amino-terminal kinase-interacting protein 3
- EGFR
epidermal growth factor receptor
- CSF2/GM-CSF
Granulocyte-macrophage colony-stimulating factor
- IκBα
Inhibitor of Nf-κB
- IFNG
Interferon gamma
- IFNB1
Interferon beta 1
- IL1B
Interleukin 1 beta
- IL-8
Interleukin 8
- MAP4K4
Mitogen-activated protein kinase kinase kinase kinase 4
- MCP-1
monocyte chemotactic protein-1
- Nf-κB
nuclear factor κB
- Peli1
Protein pellino homolog 1
- SDS-PAGE
sodium dodecyl sulfate-polyacylamide gel electrophoresis
- NR2C2/TR4
Testicular receptor 4
- TLR2
Toll-like receptor 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- Biswas DK, Iglehart JD. Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J Cell Physiol. 2006;209:645–52. doi: 10.1002/jcp.20785. [DOI] [PubMed] [Google Scholar]
- Biswas DK, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, et al. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol. 2009;10:1089–95. doi: 10.1038/ni.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Dimitrov S, et al. The endothelial apoptotic activity of angiocidin is dependent on its polyubiquitin binding activity. Br J Cancer. 2005;93:662–669. doi: 10.1038/sj.bjc.6602773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubank TD, et al. Granulocyte macrophage colony-stimulating factor inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor-educated macrophages. Cancer Res. 2009;69:2133–40. doi: 10.1158/0008-5472.CAN-08-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto H, et al. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int J Cancer. 2009;125:1276–84. doi: 10.1002/ijc.24378. [DOI] [PubMed] [Google Scholar]
- Garay RP, et al. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur J Pharmacol. 2007;563:1–17. doi: 10.1016/j.ejphar.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Gaurnier-Hausser A, et al. The novel angiogenic inhibitor, angiocidin, induces differentiation of monocytes to macrophages. Cancer Res. 2008;68:5905–14. doi: 10.1158/0008-5472.CAN-07-6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaurnier-Hausser A, Tuszynski GP. The immunomodulatory role of angiocidin, a novel angiogenesis inhibitor. Curr Pharm Des. 2009;15:1937–48. doi: 10.2174/138161209788453149. [DOI] [PubMed] [Google Scholar]
- Gooch JL, et al. The role of p21 in interferon gamma-mediated growth inhibition of human breast cancer cells. Cell Growth Differ. 2000;11:335–42. [PubMed] [Google Scholar]
- Grant DS, et al. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene. 2002;21:4765–77. doi: 10.1038/sj.onc.1205595. [DOI] [PubMed] [Google Scholar]
- Iacopino F, et al. Natural interferon-alpha activity in hormone-sensitive, hormone-resistant and autonomous human breast-cancer cell lines. Int J Cancer. 1997;71:1103–8. doi: 10.1002/(sici)1097-0215(19970611)71:6<1103::aid-ijc29>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Iozzo RV, et al. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem. 1999;274:4489–92. doi: 10.1074/jbc.274.8.4489. [DOI] [PubMed] [Google Scholar]
- Johansson E, et al. Antisecretory factor suppresses intestinal inflammation and hypersecretion. Gut. 1997;41:642–5. doi: 10.1136/gut.41.5.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, et al. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kremlev SG, et al. Angiocidin promotes pro-inflammatory cytokine production and antigen presentation in multiple sclerosis. J Neuroimmunol. 2008;194:132–42. doi: 10.1016/j.jneuroim.2007.11.003. [DOI] [PubMed] [Google Scholar]
- L’Heureux DZ, et al. The interaction of angiocidin with tissue transglutaminase. Exp Mol Pathol. 2009 doi: 10.1016/j.yexmp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miled C, et al. A genomic map of p53 binding sites identifies novel p53 targets involved in an apoptotic network. Cancer Res. 2005;65:5096–104. doi: 10.1158/0008-5472.CAN-04-4232. [DOI] [PubMed] [Google Scholar]
- Nicolini A, et al. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006;17:325–37. doi: 10.1016/j.cytogfr.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Nilsson UW, et al. Angiogenin regulation by estradiol in breast tissue: tamoxifen inhibits angiogenin nuclear translocation and antiangiogenin therapy reduces breast cancer growth in vivo. Clin Cancer Res. 2010;16:3659–69. doi: 10.1158/1078-0432.CCR-10-0501. [DOI] [PubMed] [Google Scholar]
- Reed CC, et al. Decorin prevents metastatic spreading of breast cancer. Oncogene. 2005;24:1104–10. doi: 10.1038/sj.onc.1208329. [DOI] [PubMed] [Google Scholar]
- Roy D, et al. Levels of IL-1 beta control stimulatory/inhibitory growth of cancer cells. Front Biosci. 2006;11:889–98. doi: 10.2741/1845. [DOI] [PubMed] [Google Scholar]
- Sabherwal Y, et al. Integrin alpha2beta1 mediates the anti-angiogenic and anti-tumor activities of angiocidin, a novel tumor-associated protein. Exp Cell Res. 2006;312:2443–53. doi: 10.1016/j.yexcr.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Sato K, et al. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem Biophys Res Commun. 1995;215:1078–87. doi: 10.1006/bbrc.1995.2574. [DOI] [PubMed] [Google Scholar]
- Shyr CR, et al. Modulation of estrogen receptor-mediated transactivation by orphan receptor TR4 in MCF-7 cells. J Biol Chem. 2002;277:14622–8. doi: 10.1074/jbc.M110051200. [DOI] [PubMed] [Google Scholar]
- Sica G, et al. Effect of natural beta-interferon on cell proliferation and steroid receptor level in human breast cancer cells. Cancer. 1987;60:2419–23. doi: 10.1002/1097-0142(19871115)60:10<2419::aid-cncr2820601011>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Tuszynski GP, et al. Identification and characterization of a tumor cell receptor for CSVTCG, a thrombospondin adhesive domain. J Cell Biol. 1993;120:513–21. doi: 10.1083/jcb.120.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg B, et al. Direct effects of estrogen on c-fos and c-myc protooncogene expression and cellular proliferation in human breast cancer cells. Mol Cell Endocrinol. 1989;64:223–8. doi: 10.1016/0303-7207(89)90149-4. [DOI] [PubMed] [Google Scholar]
- Walters KJ, et al. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry. 2002;41:1767–77. doi: 10.1021/bi011892y. [DOI] [PubMed] [Google Scholar]
- Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- Zhou J, et al. Cloning and characterization of angiocidin, a tumor cell binding protein for thrombospondin-1. J Cell Biochem. 2004;92:125–46. doi: 10.1002/jcb.20076. [DOI] [PubMed] [Google Scholar]