

Abstract
The in vitro treatment of vascular smooth muscle cells (VSMC) with angiotensin II (Ang II) causes Janus kinase 2 (Jak2) to interact with the Ang II type 1 receptor (AT1–R) resulting in enhanced cell growth. However, the role that Jak2 plays in AT1-R-mediated vascular cell growth and remodeling in vivo is less clear. We hypothesized that in vivo, Jak2 plays a rate limiting role in Ang II-mediated neointima formation following vascular injury. Using the Cre-loxP system, we conditionally ablated Jak2 from the VSMC of mice. We found that these mice are protected from Ang II-mediated neointima formation following iron chloride-induced vascular injury. In addition, the VSMC Jak2 null mice were protected from injury-induced vascular fibrosis and the pathological loss of the contractile marker, smooth muscle α-actin. Finally, when compared to controls, the VSMC Jak2 null mice exhibited significantly less Ang II-induced VSMC proliferation and migration in vitro and in vivo and more apoptosis. These results suggest that Jak2 plays a central role in the causation of Ang II-induced neointima formation following vascular injury and may provide a novel target for the prevention of neointima formation.
Keywords: Jak2, Neointima, Angiotensin II, Vascular Injury
1. Introduction
Damage to the vascular endothelial cell (EC) layer often caused by stenting, angioplasty, or bypass surgery is one of the initial key events in the pathogenesis of various vascular diseases including neointima formation, atherosclerosis, restenosis, and hypertension. It causes phenotypic switching of vascular smooth muscle cells (VSMC) from a contractile to a synthetic phenotype characterized by increased cell proliferation, migration, and production of extracellular matrix [1, 2]. Neointima formation resulting in vascular EC injury is also characterized by a general loss of critical VSMC contractile markers including smooth muscle α-actin (SMA), myosin heavy chain (SM-MHC), SM22α and calponin (CNN) [2, 3]. However, the mechanistic processes that trigger these phenotypic alterations are still not fully understood.
Janus kinase 2 (Jak2) is well known for its role in hematopoiesis and cytokine signaling. Mice that are completely Jak2 null die at embryonic day 12.5 due to impaired hematopoiesis and profound anemia [4, 5]. Conversely, mutations that lead to hyper-kinetic Jak2 kinase activity result in various hematological diseases characterized increased cell proliferation and hematopoiesis [6]. As a consequence, numerous Jak2 inhibitors are currently under pre-clinical and clinical investigation for their potential in the treatment of Jak2 mediated hematological diseases, but to date, none have been approved.
It is well accepted that the Jak2 signaling pathway couples to the angiotensin II type 1 receptor (AT1-R) [7]. However, the physiological and/or pathological importance of this coupling is poorly understood in the in vivo setting. Angiotensin II (Ang II) has been shown to exhibit growth promoting effects and migration in cultured VSMCs [8, 9]. In addition, chronic infusion of Ang II induces VSMC proliferation in normal and injured vessels in vivo [10, 11]. Unfortunately, there have been conflicting reports as to which kinase signaling pathway(s) mediate(s) Ang II-induced cell proliferation and migration. For instance, some studies have shown that Ang II mediates its growth promoting effects via the Mitogen-Activated Protein (MAP) kinase pathway in vitro [12]. On the other hand, studies have reported a correlative involvement of the Jak/STAT signaling pathway in the growth factor-like signaling properties of Ang II and subsequent vascular neointima formation [13, 14]. Previous attempts to determine the specific kinase pathway involved in Ang II-induced neointima formation have been limited by the lack of specific kinase inhibitors and the lack of conditional knockout animal models. For example, AG490 has been used in these types of studies [17], but in addition to inhibiting Jak2, it also inhibits Jak3 and MAP kinase [15]. Thus, it is still unclear which kinase signaling pathway acts downstream of the AT1-R to mediate VSMC proliferation, migration, and neointima formation.
Previous studies have long shown that Ang II binding causes physical interaction of Jak2 with the AT1-R resulting in the subsequent activation of the Signal Transducers and Activators of Transcription (STAT) proteins [16]. Activated STATs in turn form homo- and hetero-dimers which translocate into the nucleus and bind cis-inducible elements resulting in the activation of growth promoting genes [17, 18]. However, there is no evidence indicating that the pathological AT1-R-mediated growth effects occur exclusively through Jak2. The current study was aimed at determining whether Jak2 plays a central role in the pathogenesis of Ang II-mediated neointima formation following injury. Using an approach whereby we conditionally deleted Jak2 from the VSMC of mice, our data indicate for the first time that Jak2 plays a rate liming role in the causation of Ang II-induced neointima formation following vascular injury in vivo.
2. Materials and Methods
2.1. Animals
Male mice generated on an FVB background strain were used in these experiments. All procedures using laboratory animals were approved by the Institutional Animal Care and Use Committee at the University of Florida. Animals were maintained in accordance with NIH standards established in the Guidelines for the Care and Use of Experimental Animals.
2.2. Generation of knockout mice
VSMC deletion of Jak2 was achieved by crossing mice carrying loxP sites around the first coding exon of the Jak2 gene [19] with mice expressing Cre recombinase under the control of the SM22α promoter [20]. Specifically, male mice of genotype SM22αCreJak2fl/+ were crossed with Jak2fl/fl females, which resulted in mice whose VSMCs are devoid of Jak2 (SM22αCre(+)Jak2fl/fl). Genotyping was done by PCR using primers 5′-GCTAAACATGCTTCATCGTCGGTC and 5′-CAGATTACGTATATCCTGGCAGCG in the Cre coding region, 5′-ATTCTGAGATTCAGGTCTGAGC and 5′-CTCACAACCATCTGTATCTCAC in the Jak2 coding region, and 5′-GTCTATACACCACCACTCCTG and 5′-GAGCTGGAAAGATAGGTCAGC to identify the null Jak2 allele. Expression of VSMC SM22αCre was confirmed by crossing mice expressing SM22αCre(+)Jak2fl/fl with Rosa26 β-galactosidase reporter mice, and X-Gal (5-bromo-4-chloro-3-indolyl β-D-galactoside) staining was analyzed as previously described [21].
2.3. Vascular injury model
Iron chloride-induced vascular injury was carried out as previously described with a few modifications [22]. Briefly, mice at 3 months of age were anaesthetized using isoflurane. An incision was made directly on top of the right common carotid artery and vascular injury was induced by applying a sterile Q-tip saturated with 10% ferric chloride (Sigma, St. Louis, MO, USA) for 3 minutes. The left common carotid artery was exposed by blunt dissection, but not injured and thus served as a contralateral control. At the same time, the animals received an Alzet Model 1004 osmotic minipump (Alzet Corp) for subcutaneous infusion of 1,000 ng/kg/min of Ang II [23]. The incision was closed and the animals were allowed to recover. To prevent thrombosis, animals were injected subcutaneously with 20 units of heparin prior to surgery. Animals were euthanized at 7 days (n=6 for each genotype) or 14 days (n=6 for each genotype) following vascular injury. The carotid arteries were fixed using 10% formalin and processed for histological determination of neointima formation.
2.4. Histology
Tissue samples were prepared for histology as previously described [24]. Briefly, tissues were fixed overnight at 4°C in 10% buffered formalin (Fisher Scientific, Pittsburgh, PA). The tissues were subsequently dehydrated through a graded ethanol series, paraffin embedded and sectioned. Five micrometer sections were stained with hematoxylin and eosin (H&E) for morphological analysis. Transverse sections of the carotid arteries were subjected to morphometry for assessing the intima/media ratio (I/M ratio). Other tissues were stained with Masson’s trichrome using a kit (87019 Richard-Allan Scientific) for analysis of fibrosis.
2.5. Immunohistochemistry
5 micron sections mounted on gelatin-coated slides were dewaxed in ethanol, rehydrated, then blocked in 3% H2O2 followed by 5% normal goat serum. Sections were exposed to the primary antibody overnight at 4°C, washed, and then treated with the biotinylated secondary antibody. After secondary antibody incubation, the samples were washed, exposed to the avidin-peroxidase reagent (Vectastain Elite, Vector Laboratories, Burlingame, CA), and reacted with diaminobenzidine to produce a brown reaction product. The sections were dehydrated in ethanol, mounted with Permount, and observed by light microscopy. Immunohistological detection of anti-smooth muscle α-actin (CM001B) was carried out using the Rat on Mouse AP-Polymer Kit (Biocare Medical) according to the manufacturer’s instructions. Anti-Jak2 (ab39636 Abcam), anti-phospho-Jak2 (Ab32101 Abcam), anti-phospho-STAT5 (Ab32364 Abcam), and Ki-67 (M7249 DAKO) immunohistochemical detection was performed as previously described [24]. Apoptosis detection was carried out using a TUNEL staining kit (S7100 Chemicon) according to the manufacturer’s instructions.
2.6. Immunoblotting
Protein sample was extracted from the homogenates of primary VSMC cultures and immunoprecipitated using STAT3 antibody (SC-482, Santa Cruz), or STAT5 antibody (SC-28685, Santa Cruz), followed by western-blotting using phospho-STAT3 antibody (SC-8059, Santa Cruz), or phospho-STAT5 antibody (716900, Invitrogen) respectively, as previously described [25].
2.7. Cell proliferation
VSMCs from each genotype were isolated and cultured as described [26]. Cell proliferation was assessed on the basis of mitochondrial dehydrogenase activity of the cells using the MTT dye–reduction assay [27]. In brief, after the addition of ligand or vehicle, the cells were incubated with the MTT labeling solution (0.5 mg/mL) at 37°C for 4 hours. The cells were then solubilized in 100 μL of 0.1N HCl and isopropyl alcohol, and shaken for 30 seconds on a plate rotator. Absorbance at 540 nm was measured with the use of a microtiter plate reader with a reference wavelength of 690 nm. Direct cell counts were also carried out to determine the number of viable cells using trypan blue exclusion.
2.8. Cell migration
The migration of VSMCs was determined using a QCM™ 24-well colorimetric cell migration assay kit (Millipore) according to the manufacturer’s instructions. Briefly, the cells were suspended in serum free media (DMEM) at a concentration of 1 × 106 cells/mL. 0.3 mL aliquots of the cell suspension were added to the top chambers of the transwell membranes with 8-μm pores. The lower transwell compartments contained 0.5 mL of DMEM containing various migration factors including Ang II (10−7 M), FBS (10%) or platelet-derived growth factor (PDGF-BB) (20 ng/ml). Incubation was continued for 24 hours at 37°C in a CO2 incubator. The adherent cells were then stained, washed, and allowed to air dry. The die was extracted from the cells, and optic density was measured at 560 nm.
2.9. Apoptosis
Annexin V/Propidium iodide staining was employed to determine early apoptosis in the control and the Jak2 null VSMCs. VSMCs were serum-starved overnight and then treated with either vehicle, 10% FBS, 20 ng/ml PDGF or 10−7 M Ang II for 24 hours. 105 cells were re-suspended in 100 μl of 1X binding buffer and apoptotic levels were determined via the FITC Annexin V Apoptosis Detection Kit (BD Pharmingen) following the manufacturer’s instructions and analyzed on a FACSCalibur flow cytometer (Becton Dickinson). Apoptotic cells were also identified using the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) method which specifically labels the 3′-hydroxyl termini of DNA strand breaks. The TUNEL assay was carried out using the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer’s instructions.
2.10. Statistical analysis
All results were expressed as means +/− SEM. Statistical comparison of the different genotypes were performed by unpaired Student’s t test. P values of less than 0.05 were considered statistically significant.
3. Results
3.1. Deletion of VSMC Jak2 prevents Ang II-mediated neointima formation and narrowing of the vascular lumen following injury
Vascular remodeling is a pathologic response to vascular injury characterized by VSMC proliferation, migration, neointima formation, and a narrowing of the vascular lumen [28]. We wanted to determine whether VSMC Jak2-null mice are protected from neointima formation and narrowing of the vascular lumen flowing vascular injury. For this, the right carotid arteries of control (SM22αCre(−);Jak2fl/fl) and VSMC Jak2-null (SM22αCre(+);Jak2fl/fl) mice were subjected to iron chloride-induced vascular injury with simultaneous Ang II infusion. Left carotid arteries were exposed via blunt dissection, but not subjected to iron chloride-induced injury and thus served as contralateral controls. Iron chloride-induced vascular injury is a well established vascular injury model which has been mostly used in thrombosis studies [29–31]. It causes de-endothelialization of the artery resulting in neointima formation due to erythrocyte hemolysis and hemoglobin oxidation [32]. Furthermore, iron chloride-induced arterial injury increases oxidative stress in the vessel which is an important component of all the risk factors leading to neointima formation including smoking, hypertension and vascular surgery.
Mice were euthanized 7 and 14 days after injury. H&E representative sections in Figure 1A show a clear increase in thickness of the neointima in the injured arteries of the control mice at day 7 and 14, but neither in the VSMC Jak2-null mice nor any of the uninjured contralateral arteries. Using computer assisted quantitative morphometric analysis, we found that compared to the non-injured contralateral carotid arteries, vascular injury induced significant increases in the intima/media ratio in the control mice at day 7 and 14, which was lacking in the VSMC Jak2 null mice (Figure 1B). As a consequence, there was a significant narrowing on the carotid artery lumen in the injured arteries from the control mice while the VSMC Jak2-null mice were protected from this deleterious effect (Figure 1C). These collective results suggest that deletion of VSMC Jak2 prevents neointima formation and the subsequent narrowing of the vessel following vascular injury.
Figure 1.

Deletion of VSMC Jak2 prevents Ang II-mediated neointima formation and narrowing of the vascular lumen following injury. A, H&E stained sections showing neointima formation in control mice. The green dotted lines separate the intima from media. B, Intima/media ratio. C, Lumen area. *p<0.05 vs. contralateral control, **p<0.01 vs. contralateral control.
3.2. Deletion of VSMC Jak2 prevents Ang II-mediated vascular fibrosis following injury
Vascular fibrosis is an important component of vascular injury response [33]. Thus, we quantified extracellular matrix (ECM) deposition using trichrome-blue staining of sections from iron chloride-injured control and Jak2 null mouse carotid arteries 7 and 14 days after injury. Representative sections indicated that the density of trichrome blue staining was significantly reduced in the Jak2 null mice at both time points when compared to the control group (Figure 2A) and quantitative analysis of all sections found this difference to be significant (Figure 2B). Overall, these results suggest that deletion of VSMC Jak2 reduces Ang II-induced ECM deposition following vascular injury.
Figure 2.

Deletion of VSMC Jak2 prevents Ang II-mediated fibrosis and loss of SMA following injury. A, Trichrome-blue stained sections showing increased collagen density in control mice. B, Quantification of trichrome-blue staining. C, Immunohistochemistry of SMA. D, Quantification of SMA staining. *p<0.05 vs. contralateral non-injured artery.
3.3. Deletion of VSMC Jak2 prevents the loss of smooth muscle α-actin in response to Ang II-mediated vascular injury
Vascular injury is known to reduce the expression of contractile markers within VSMC [1, 2]. We hypothesized that the Jak2-null mice would be protected from the injury-induced loss of smooth muscle α-actin (SMA). Immunohistochemistry of injured carotid sections showed that there was a significant loss of SMA immunoreactivity (red stain) at both day 7 and day 14 in the control mice when compared to the contralateral carotid artery (Figure 2C). However, SMA staining was preserved in the injured carotid arteries in the Jak2 null mice, and was comparable to that observed in their contralateral arteries. Quantitative analysis of all sections again found this difference between the two genotypes to be significant (Figure 2D). As such, these results suggest that VSMC Jak2-null mice are protected from the loss of the contractile phenotype that occurs following vascular injury.
3.4. Deletion of VSMC Jak2 prevents neointima formation by inhibiting cell proliferation and inducing apoptosis
We hypothesized that a mechanism whereby deletion of VSMC Jak2 prevents neointima formation is via reduced cellular proliferation. To determine this, the levels of the proliferative marker, Ki-67, were measured in sections from each condition. Representative sections indicated an increase in the number of Ki-67 positive nuclei (brown stain) in the injured arteries of the control mice at day 7 and 14, but not in the VSMC Jak2 null-mice (Figure 3A). Quantification of Ki-67 positive nuclei across all sections found that when compared to the non-injured contralateral arteries, injury caused significant increases in the number of Ki-67 positive cells in the control mice at day 7 and 14, but this was lacking in the Jak2-null mice (Figure 3B). These results suggest that deletion of VSMC Jak2 prevents neointima formation by inhibiting cell proliferation.
Figure 3.

Deletion of VSMC Jak2 inhibits cell proliferation and induces apoptosis. A, Representative sections showing immunohistochemistry of Ki-67. B, Quantification of the number of Ki-67-positive nuclei. C, Representative sections showing immunohistochemistry of TUNEL. D, Quantification of the number of TUNEL positive cells. *p<0.05 vs. contralateral non-injured control.
Previous studies have shown that induction of apoptosis is a possible mechanism for the prevention of neointima formation [14, 34]. Therefore, we wanted to determine whether the reduced neointima formation observed in the VSMC Jak2-null mice correlates with increased apoptosis. Shown are representative TUNEL stained sections (Figure 3C; brown stained cells indicated by red arrows) and the average number of TUNEL positive cells plotted as a function of treatment group (Figure 3D). There was no significant difference in the number of TUNEL positive cells observed in injured carotid arteries taken from the control group when compared to the non-injured, contralateral arteries. In contrast, there was a significant increase in the number of apoptotic cells 7 days after injury in the Jak2-null mice and this returned to near baseline levels by day 14. Altogether, the results in Figure 3 suggest that VSMC Jak2 deletion prevents neointima formation by inhibiting cell proliferation and inducing apoptosis in VSMCs.
3.5. VSMC Jak2 induces neointima formation by increasing phosphorylation of Jak2 and STAT5
Immunohistochemistry was carried out in order to determine the relative levels of Jak2, phospho-Jak2, and phospho-STAT5 (a downstream target of Jak2) in the injured and non-injured carotid arteries of both the control and VSMC Jak2-null mice (Figure 4). With respect to the control mice, we found that there were readily detectable levels (brown stain) of Jak2, phospho-Jak2 and phospho-STAT5 in the contralateral arteries and vascular injury increased this staining pattern both in the media and neointima. With respect to the VSMC Jak2-null mice, there was little to no staining for Jak2, phospho-Jak2, and phospho-STAT5 both in the contralateral control and the injured arteries. These results demonstrate that the reduced neointima formation observed in the VSMC Jak2-null mice correlates with reduced phosphorylation of Jak2 and STAT5 in VSMC.
Figure 4.
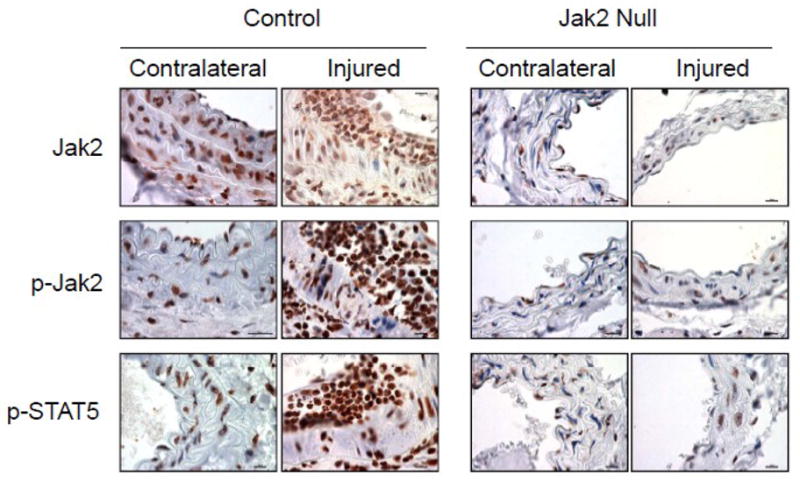
VSMC Jak2 induces neointima formation by increasing phosphorylation of Jak2 and STAT5. Immunohistochemistry of representative sections showing relative expression of Jak2, phospho-Jak2 and phospho-STAT5 in injured and non-injured carotid arteries of the control and the Jak2-null mice.
3.6. VSMC Jak2 mediates Ang II-induced cell proliferation and migration
In order to determine whether the Jak2 dependent effects observed in vivo are specific for Ang II per se, we isolated VSMC from control and VSMC Jak2-null mice and cultured them ex vivo. The cells were subsequently treated with either vehicle, 10% FBS, platelet derived growth factor (PDGF), or Ang II, and then evaluated for cell proliferation and cell migration properties. For the control cells, FBS, PDGF, and Ang II all induced robust cell proliferation as measured by MTT (Figure 5A). For the VSMC Jak2 null cells, FBS and PDGF induced robust cells proliferation while Ang II did not (Figure 5B), To confirm these results using a different approach, we repeated the study, but instead determined cell proliferation via a direct counting of cells. We similarly observed that FBS, PDGF, and Ang II all induced robust proliferation of the control cells (Figure 5C), but only FBS and PDGF induced proliferation of the VSMC Jak2-null cells (Figure 5D). Together, these results suggest that Jak2 is rate-limiting in Ang II-induced VSMC proliferation.
Figure 5.

VSMC Jak2 mediates Ang II-induced cell proliferation and migration. VSMC were treated with 10% FBS, 20 ng/ml PDGF or 10−7 M Ang II as indicated. Resulting cell proliferation of the control cells (A) and VSMC Jak2 null cells (B) is shown using the MTT cell proliferation assay. Cell proliferation using trypan blue exclusion for control (C) and VSMC Jak2 null (D) cells. E, Cell migration in these same cells. *p<0.05 vs. control. Shown are mean +/− SEM for three independent experiments, each run in triplicate.
Next, we wanted to determine the role of Jak2 in VSMC migration in response to these same stimuli. We found that there was no significant difference in cell migration between the genotypes when the cells were treated with FBS or PDGF (Figure 5E). However, when the cells were treated with Ang II, cell migration in the Jak2-null cells was significantly attenuated when compared to the control cells (Figure 5E), suggesting that Jak2 mediates Ang II induced cell migration. Collectively, the data in Figure 5 correlate the specific loss of Jak2 with significantly impaired Ang II-mediated VSMC proliferation and migration.
3.7. VSMC Jak2 is required for Ang II-mediated cell survival
The in vivo immunohistochemistry data indicate that Jak2 promotes cell survival as cells that lack Jak2 are more prone to apoptosis after injury (Figures 4C and 4D). Here, we wanted to determine whether this Jak2-dependent event was specific for Ang II. For this, primary cultures of VSMC from control and VSMC Jak2-null mice were serum starved overnight. The following day, the cells were treated with the indicated ligands and early apoptosis was measured 24 hours later. We found that in the control cells, ~10% of all cells treated with vehicle control solution were in early apoptosis (Figure 6A). Treatment with FBS, PDGF, and Ang II promoted cell survival as indicated by the lower levels of apoptosis in these cells. When the same experiment was performed in the VSMC Jak2-null cells, the ability of Ang II to decrease the levels of apoptosis was lost, thereby indicating a critical role for Jak2 in Ang II-mediated cell survival. To demonstrate this using an alternate approach, the cells were treated in the same way, but this time apoptosis levels were measured via TUNEL stain, an indicator of both early and late apoptosis (Figure 6B). We similarly found an essential requirement for Jak2 in promoting Ang II-mediated cell survival as evidenced by the inability of the null cells to decrease the levels of apoptosis in response to Ang II. As such, these data correlate the reduced neointimal formation observed in the VSMC Jak2-null genotype with increased apoptosis and this is Ang II specific.
Figure 6.

Jak2 is required for Ang II-mediated VSMC survival. After overnight serum starvation, VSMC were treated with vehicle control solution, 10% FBS, 20 ng/ml PDGF or 10−7 M Ang II as indicated. 24 hours later, apoptosis levels were measured via annexin V/propidium iodide selection (A) or via TUNEL staining (B). *p<0.05 versus vehicle treated, #p<0.05 versus control genotype. Shown are mean +/− SEM for three independent experiments, each run in triplicate.
3.8. VSMC Jak2 deletion is associated with reduced Ang II-mediated activation of STAT3 and STAT5
We next wanted to determine whether the activation of the Jak2/STAT signaling pathway is also specific for Ang II. For this, we analyzed the relative abilities of FBS, PDGF and Ang II to induce phosphorylation of STAT3 and STAT5; both of which are mitogenic proteins and are direct downstream signaling targets of Jak2. With respect to STAT3, ligand induced phosphorylation of STAT3 was significantly reduced in the null cells when compared to the control cells for all conditions (Figure 7A and 7B). These results suggest that activation of STAT3 is primarily mediated by Jak2 since the absence of Jak2 renders the cells incapable of phosphorylating STAT3 under any condition. In addition, we found that within the control cells, the Ang II-induced phosphorylation of STAT3 was significantly increased, when compared to the other stimulating ligands. With respect to STAT5, the phospho-STAT5 levels were not significantly different in the FBS and PDGF treated control cells when compared to vehicle treated cells, but were significantly different in the Ang II treated cells (Figure 6C and 6D). Furthermore, there was no significant difference in ligand induced phosphorylation of STAT5 amongst the Jak2-null cells (Figure 7C and 7D). As such, these results suggest that Jak2 is the primary mediator of Ang II-induced phosphorylation of STAT5, and that activation of the Jak2/STAT5 pathway is specific for Ang II.
Figure 7.

VSMC Jak2 deletion reduces Ang II-mediated activation of STAT3 and STAT5. A, Representative blots of phospho-STAT3 and STAT3. B, Phospho-STAT3 quantification. C, Representative blots of phospho-STAT5 and STAT5. D, Phospho-STAT5 quantification. *p<0.05 vs. ligand-treated control cells, #p<0.05 vs. vehicle-treated control cells, &p<0.05 vs. Ang II treated control cells. Shown are mean +/− SEM for three independent experiments.
4. Discussion
Neointima formation is a pathologic consequence of vascular endothelial damage which often results from various risk factors including percutaneous vascular surgery procedures. We have investigated the impact of VSMC deletion of Jak2 on the prevention of Ang II-mediated neointima formation following vascular injury in mice. This study identifies Jak2 as a critical mediator of the pathological processes involved in Ang II-mediated neointima formation. To our knowledge, this is the first report to establish a causal relationship between Jak2 and Ang II-mediated neointima formation following vascular injury.
Following endothelial damage, the process of neointima formation starts with an initiation of VSMC proliferation and migration into the intima, followed by a sustained proliferation of VSMC in the neointima [27, 35]. This compromises the vascular lumen, and limits blood supply to distal areas of the vessel. Ang II plays a key role in mediating the processes involved in the pathogenesis of neointima formation following vascular injury [36]. However, the specific mechanisms involved in Ang II-mediated neointima formation remain largely unidentified. Ang II induces tyrosine-phosphorylation and activation of Jak2 in rat VSMC resulting in activation of its downstream signaling substrates, the STATs [16]. Subsequent studies showed that the Ang II-induced activation of Jak2 results in proliferation of cultured VSMC [37]. We found that not only did VSMC Jak2 deletion prevent Ang II-induced neointima formation, but it also prevented the pathological phenotypic alterations induced by vascular injury including vascular fibrosis and loss of the contractile marker, SMA. The mechanisms by which VSMC Jak2 deletion prevents neointima formation include inhibition of cell proliferation and migration. Furthermore, we found that Ang II-induced activation of the proliferative proteins, STAT3 and STAT5, is impaired in the VSMC Jak2-null cells. The Jak2-mediated growth promoting and migratory effects are specific for Ang II since other ligands such as FBS and PDGF did not lead to differential effects on cell proliferation and migration in the control and the Jak2-null cells. From these data, we conclude that Jak2 plays a rate liming role in the causation of Ang II-induced neointima formation following vascular injury in vivo.
It is well established that Ang II plays a key role in mediating neointima formation following vascular injury [28]. This provided us with an excellent model of examining the role of VSMC Jak2 deletion in an environment in which neointima formation following injury is exasperated by chronic Ang II infusion. Although studies have shown that the Jak2 signaling pathway couples to the AT1-R resulting in VSMC proliferation in vitro [7], the only study to date examining the relationship between Jak2 and neointima formation in vivo reported that Jak2 is only transiently expressed during the process of neointima formation [13]. The same study also reported that treatment of vascular strips with Ang II ex vivo enhances phosphorylation of Jak2 [13]. Therefore, until now, the pathophysiological link between Jak2 and Ang II-mediated neointima formation has been only correlative and no study has implicated Jak2 in playing a causative role in Ang II-mediated vascular remodeling in vivo.
Placing the Cre recombinase cDNA under the control of tissue specific promoters has allowed for the conditional deletion of genes such as Jak2. However, previous work has clearly demonstrated that the pattern of Cre expression in the targeted tissue is a mosaicism [38, 39]. In other words, most cells will express Cre, but some will not. The reason(s) for this is not fully understood, but it is thought that it may be due to modifying events such as epigenetic silencing of the transgene within a given cell [40, 41]. Hence, the gene targeted for deletion, in this case Jak2, is not removed from the cell. Quantitative examinations of our anti-Jak2 immunohistochemistry (Figure 4) as well as Rosa26/LacZ expression patterns (data not shown) indicate that Jak2 is deleted from ~95% of all VSMC in the carotid arteries of these mice. This degree of deletion is clearly enough to impair Jak2 function as determined by the significantly reduced VSMC migration, VSMC proliferation, neo-intimal formation, and preservation of contractile markers, when compared to controls. Given that Jak2 has been implicated in a number of cardiovascular diseases including hypertension, heart failure, and diabetes [42–45], we now have an excellent model for the future examination of the role of VSMC derived Jak2 in these and other disorders.
In conclusion, the present study demonstrated for the first time that Jak2 plays a central role in the causation of Ang II-induced neointima formation following vascular injury in vivo. Therefore, inhibition of Jak2 may provide a potential prophylactic therapeutic strategy for prevention of neointima formation and progression following angioplasty and/or vascular stenting.
Acknowledgments
We thank the University of Florida Pathology Core Facility for their help in histological processing of tissues. This work was supported by a National Institutes of Health Award (R01-HL67277), an American Heart Association Grant-in-Aid Award (#0855361E), and an American Heart Association Pre-Doctoral Research Fellowship Award (10PRE4310065).
Footnotes
Disclosures
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Owens GK, Vernon SM, Madsen CS. Molecular regulation of smooth muscle cell differentiation. J Hypertens Suppl. 1996 Dec;14(5):S55–64. [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004 Jul;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 3.Sobue K, Hayashi K, Nishida W. Expressional regulation of smooth muscle cell-specific genes in association with phenotypic modulation. Mol Cell Biochem. 1999 Jan;190(1–2):105–18. [PubMed] [Google Scholar]
- 4.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998 May 1;93(3):385–95. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 5.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998 May 1;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 6.Delhommeau F, Pisani DF, James C, Casadevall N, Constantinescu S, Vainchenker W. Oncogenic mechanisms in myeloproliferative disorders. Cell Mol Life Sci. 2006 Dec;63(24):2939–53. doi: 10.1007/s00018-006-6272-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirabo A, Sayeski PP. Jak2 Tyrosine Kinase: A Potential Therapeutic Target for AT1 Receptor Mediated Cardiovascular Disease. Pharmaceuticals. 2010 November 9;3(11):3478–93. [Google Scholar]
- 8.Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988 Apr;62(4):749–56. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- 9.Berk BC, Vekshtein V, Gordon HM, Tsuda T. Angiotensin II-stimulated protein synthesis in cultured vascular smooth muscle cells. Hypertension. 1989 Apr;13(4):305–14. doi: 10.1161/01.hyp.13.4.305. [DOI] [PubMed] [Google Scholar]
- 10.Daemen MJ, Lombardi DM, Bosman FT, Schwartz SM. Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ Res. 1991 Feb;68(2):450–6. doi: 10.1161/01.res.68.2.450. [DOI] [PubMed] [Google Scholar]
- 11.Su EJ, Lombardi DM, Wiener J, Daemen MJ, Reidy MA, Schwartz SM. Mitogenic effect of angiotensin II on rat carotid arteries and type II or III mesenteric microvessels but not type I mesenteric microvessels is mediated by endogenous basic fibroblast growth factor. Circ Res. 1998 Feb 23;82(3):321–7. doi: 10.1161/01.res.82.3.321. [DOI] [PubMed] [Google Scholar]
- 12.Xi XP, Graf K, Goetze S, Fleck E, Hsueh WA, Law RE. Central role of the MAPK pathway in ang II-mediated DNA synthesis and migration in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999 Jan;19(1):73–82. doi: 10.1161/01.atv.19.1.73. [DOI] [PubMed] [Google Scholar]
- 13.Seki Y, Kai H, Shibata R, Nagata T, Yasukawa H, Yoshimura A, et al. Role of the JAK/STAT pathway in rat carotid artery remodeling after vascular injury. Circ Res. 2000 Jul 7;87(1):12–8. doi: 10.1161/01.res.87.1.12. [DOI] [PubMed] [Google Scholar]
- 14.Shibata R, Kai H, Seki Y, Kato S, Wada Y, Hanakawa Y, et al. Inhibition of STAT3 prevents neointima formation by inhibiting proliferation and promoting apoptosis of neointimal smooth muscle cells. Hum Gene Ther. 2003 May 1;14(7):601–10. doi: 10.1089/104303403321618128. [DOI] [PubMed] [Google Scholar]
- 15.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996 Feb 15;379(6566):645–8. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 16.Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature. 1995 May 18;375(6528):247–50. doi: 10.1038/375247a0. [DOI] [PubMed] [Google Scholar]
- 17.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996 Feb 9;84(3):331–4. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 18.Schieffer B, Bernstein KE, Marrero MB. The role of tyrosine phosphorylation in angiotensin II mediated intracellular signaling and cell growth. J Mol Med. 1996 Feb;74(2):85–91. doi: 10.1007/BF00196783. [DOI] [PubMed] [Google Scholar]
- 19.Krempler A, Qi Y, Triplett AA, Zhu J, Rui H, Wagner KU. Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis. 2004 Sep;40(1):52–7. doi: 10.1002/gene.20063. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Miano JM, Mercer B, Olson EN. Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J Cell Biol. 1996 Mar;132(5):849–59. doi: 10.1083/jcb.132.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci U S A. 1999 Apr 27;96(9):5037–42. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Xu L. An optimized murine model of ferric chloride-induced arterial thrombosis for thrombosis research. Thromb Res. 2005;115(1–2):95–100. doi: 10.1016/j.thromres.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000 Jun;105(11):1605–12. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G, Wu J, Sun C, Qi M, Hang C, Gong Y, et al. Potential role of JAK2 in cerebral vasospasm after experimental subarachnoid hemorrhage. Brain Res. 2008 Jun 12;1214:136–44. doi: 10.1016/j.brainres.2008.03.085. [DOI] [PubMed] [Google Scholar]
- 25.Sayeski PP, Ali MS, Harp JB, Marrero MB, Bernstein KE. Phosphorylation of p130Cas by angiotensin II is dependent on c-Src, intracellular Ca2+, and protein kinase C. Circ Res. 1998 Jun 29;82(12):1279–88. doi: 10.1161/01.res.82.12.1279. [DOI] [PubMed] [Google Scholar]
- 26.Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23(4):185–8. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- 27.Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986 May 22;89(2):271–7. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz SM, Reidy MA, O’Brien ER. Assessment of factors important in atherosclerotic occlusion and restenosis. Thromb Haemost. 1995 Jul;74(1):541–51. [PubMed] [Google Scholar]
- 29.Wang X, Xu L. An optimized murine model of ferric chloride-induced arterial thrombosis for thrombosis research. Thromb Res. 2005;115(1–2):95–100. doi: 10.1016/j.thromres.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 30.HajMohammadi S, Enjyoji K, Princivalle M, Christi P, Lech M, Beeler D, et al. Normal levels of anticoagulant heparan sulfate are not essential for normal hemostasis. J Clin Invest. 2003 Apr;111(7):989–99. doi: 10.1172/JCI15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schafer K, Muller K, Hecke A, Mounier E, Goebel J, Loskutoff DJ, et al. Enhanced thrombosis in atherosclerosis-prone mice is associated with increased arterial expression of plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003 Nov 1;23(11):2097–103. doi: 10.1161/01.ATV.0000097766.36623.DF. [DOI] [PubMed] [Google Scholar]
- 32.Woollard KJ, Sturgeon S, Chin-Dusting JP, Salem HH, Jackson SP. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J Biol Chem. 2009 May 8;284(19):13110–8. doi: 10.1074/jbc.M809095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakabayashi K, Suzuki H, Sato T, Iso Y, Katagiri T, Takeyama Y. Eplerenone suppresses neointimal formation after coronary stent implantation in swine. Int J Cardiol. 2006 Feb 15;107(2):260–6. doi: 10.1016/j.ijcard.2005.03.078. [DOI] [PubMed] [Google Scholar]
- 34.Shibata R, Kai H, Seki Y, Kato S, Morimatsu M, Kaibuchi K, et al. Role of Rho-associated kinase in neointima formation after vascular injury. Circulation. 2001 Jan 16;103(2):284–9. doi: 10.1161/01.cir.103.2.284. [DOI] [PubMed] [Google Scholar]
- 35.Davis C, Fischer J, Ley K, Sarembock IJ. The role of inflammation in vascular injury and repair. J Thromb Haemost. 2003 Aug;1(8):1699–709. doi: 10.1046/j.1538-7836.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 36.Laporte S, Escher E. Neointima formation after vascular injury is angiotensin II mediated. Biochem Biophys Res Commun. 1992 Sep 30;187(3):1510–6. doi: 10.1016/0006-291x(92)90473-x. [DOI] [PubMed] [Google Scholar]
- 37.Marrero MB, Schieffer B, Li B, Sun J, Harp JB, Ling BN. Role of Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase cascades in angiotensin II- and platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 1997 Sep 26;272(39):24684–90. doi: 10.1074/jbc.272.39.24684. [DOI] [PubMed] [Google Scholar]
- 38.Palmiter RD, Wilkie TM, Chen HY, Brinster RL. Transmission distortion and mosaicism in an unusual transgenic mouse pedigree. Cell. 1984 Apr;36(4):869–77. doi: 10.1016/0092-8674(84)90036-9. [DOI] [PubMed] [Google Scholar]
- 39.Wilkie TM, Brinster RL, Palmiter RD. Germline and somatic mosaicism in transgenic mice. Developmental biology. 1986 Nov;118(1):9–18. doi: 10.1016/0012-1606(86)90068-0. [DOI] [PubMed] [Google Scholar]
- 40.Long MA, Rossi FM. Silencing inhibits Cre-mediated recombination of the Z/AP and Z/EG reporters in adult cells. PloS one. 2009;4(5):e5435. doi: 10.1371/journal.pone.0005435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calero-Nieto FJ, Bert AG, Cockerill PN. Transcription-dependent silencing of inducible convergent transgenes in transgenic mice. Epigenetics & chromatin. 2010 Jan 19;3(1):3. doi: 10.1186/1756-8935-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grote K, Luchtefeld M, Schieffer B. JANUS under stress--role of JAK/STAT signaling pathway in vascular diseases. Vascular pharmacology. 2005 Nov;43(5):357–63. doi: 10.1016/j.vph.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 43.Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010 Feb;16(2):183–90. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- 44.Lata K, Madiraju N, Levitt L. JAK2 mutations and coronary ischemia. N Engl J Med. 2010 Jul 22;363(4):396–7. doi: 10.1056/NEJMc1003478. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Li Y, Liu H, Liu S, Liu Q, Wang XM, et al. Peroxynitrite mediates glomerular lesion of diabetic rat via JAK/STAT signaling pathway. Journal of endocrinological investigation. 2009 Nov;32(10):844–51. doi: 10.1007/BF03345756. [DOI] [PubMed] [Google Scholar]