

Abstract
This review focuses on conceptual and methodological advances in our understanding and characterization of the conformational heterogeneity of proteins. Focusing on X-ray crystallography, we describe how polysterism, the interconversion of pre-existing conformational substates, has traditionally been analyzed by comparing independent crystal structures or multiple chains within a single crystal asymmetric unit. In contrast, recent studies have focused on mining electron density maps to reveal previously ‘hidden’ minor conformational substates. Functional tests of the importance of minor states suggest that evolutionary selection shapes the entire conformational landscape, including uniquely configured conformational substates, the relative distribution of these substates, and the speed at which the protein can interconvert between them. An increased focus on polysterism may shape the way protein structure and function is studied in the coming years.
Keywords: Polysterism, Conformational substates, X-ray crystallography, NMR, Evolution, Protein dynamics, Catalysis
Introduction
The idea that proteins fluctuate between many structures of an ensemble is well established [1]. In contrast to the ensemble view, protein X-ray crystallography generally follows a convention, established with myoglobin and hemoglobin in the middle of the last century [2, 3], to describe the model built into the electron density map in the singular form: “we have solved the structure of protein X”. The solved structure generally represents the best fit of a single polypeptide model to the electron density generated by the diffraction pattern of ~1015 molecules in the crystal lattice during seconds to hours of data collection. The protein molecules within a crystal populate a ‘conformational landscape’ dominated by nearly iso-energetic and conformationally similar substates [4]. However, the refinement process, with few exceptions [5–7], is designed to converge on the single structure.
Here we review how crystallography can be used to reveal conformational substates that interconvert independent of any conformational changes induced by ligand binding or other perturbations. We refer to pre-existing conformational diversity as polysterism (as in [8, 9]), making a semantic distinction between structural diversity (polysterism) and sequence diversity (polymorphism). Early investigations of polysterism that used crystallography and complementary solution methods are highlighted before we focus on several recent studies that have provided new insight into functionally important polysterism, often from a single electron density map. These studies reveal a general theme: the minor substates of the conformational landscape often resemble the structures that play important roles in ligand binding, enzyme catalytic cycles, or evolutionary transitions. Thus, characterizing the polysterism of a single protein can yield insight by identifying minor conformations that may be sampled during evolution or during a reaction cycle.
Crystalline enzymes are often active
Despite the stable lattice arrangement necessary for X-ray diffraction, many enzyme crystals are capable of catalyzing chemical reaction [10, 11]. However, their activity is often reduced compared to the free enzyme in a crowded solution [12, 13]. Because the crowding of a typical protein crystal lattice is similar to the macromolecular crowding in a biological cellular environment [14–16], the reduction in catalytic efficiency likely originates from specific conformational restrictions of the crystal lattice. These observations provided some of the first indications that the ability to transition between structurally distinct conformational substates may be a functionally important property of proteins.
Crystallography gives endpoints for an induced fit view
The importance of structurally distinct substates in enzyme function was first promoted through Koshland’s [17] induced fit hypothesis, which proposed that proteins move between discrete structures as a result of interaction with a ligand. Induced fit theory, which was originally based on the necessity for substrate-specific catalytically competent structure of hexokinase to allow activity with glucose and prevent activity with other potential substrates such as water, was generalized to most enzymes and receptors. Koshland’s predictions were later supported by clear structural differences between ligand-bound and unbound crystal structures of carboxypeptidase and hexokinase [18–20], providing a structural corroboration for the importance of conformational rearrangements during the catalytic cycle of some enzymes. These structural studies established that solving independent crystal structures with different ligands is a powerful tool that can be used to observe the end-points of conformational change. The importance of conformational change in enzyme function was buttressed by solution experiments such as volume changes calculated from centrifugation and small-angle X-ray scattering experiments [18, 20].
Many conformational substates are present in a protein crystal
Despite an initial emphasis on the single-structure view of proteins, polysterism has been widely appreciated from the beginnings of structure–function studies. For instance, polysterism was a fundamental aspect of the allosteric model proposed by Monod, Wyman and Changeaux (MWC) [21] in 1965, in which allosteric effectors were proposed to function through changing the pre-existing distribution of active or inactive conformational substates within a population of protein molecules. Polysterism was also an important part of statistical mechanical descriptions of protein conformation and function [22, 23], while solvent interaction was predicted to introduce a heterogeneity in proteins at an early stage [4].
Although a role for intrinsic conformational diversity was recognized conceptually, the structural evidence to support it was initially slight. In the early stages of the field of protein crystallography, when high-resolution diffraction was relatively rare, polysterism was often not observable, even if it was present. More importantly, crystallization, by its very nature, favors ordered aggregation of molecules that are homogeneous in composition and conformation, establishing the direct relationship between crystalline order and diffraction. Indeed, large regions of disordered solvent molecules were recognized as a likely origin of X-ray diffraction resolution limits [14]. These observations introduced a paradox—in order to resolve functional conformational heterogeneity, we need high-resolution X-ray diffraction, but intrinsic heterogeneity limits diffraction.
To resolve this apparent contradiction, the pioneering studies of Frauenfelder [24] used an indirect readout of heterogeneity at moderate resolutions: atomic B-factors (also called Debye–Waller factors, temperature factors, atomic displacement parameters, and thermal parameters). The B-factor fits the decay of electron density surrounding the modeled mean position of an atom. Using atomic B-factors, they calculated the magnitudes of the spatial distribution of protein atomic movements without resolving independent discrete conformations. Frauenfelder and Petsko used crystals of myoglobin and variable diffraction temperatures to discover differences between the mobility of core and surface atoms. They observed that the magnitude of motion varied according to the distance from the central haem-group. Building upon this observation, they produced a model of a rigid core surrounded by semi-liquid regions to the outside, and suggested a possible ‘dynamic’ pathway for substrate and product diffusion. This analysis suggested that conformational substates, particularly for side chains, exist within the crystal and that temperature can affect the magnitude of motion within these substates. Concurrently, Phillips and coworkers [25] demonstrated a conservation of the distribution of B-factors between homologous proteins crystallized in different space groups. Since the homologous proteins crystallized in different lattice arrangements, their results suggested that B-factor distributions are, at least partially, independent of the crystallization process. Additionally, the conservation of high B-factors at the entrance to the active site suggested that the protein mobility might have been maintained throughout evolution and play a significant part in the enzymatic activity. These groundbreaking papers provided structural support for the role of polysterism and heralded a new view within protein crystallography: there are interconverting structures within the crystal, revealed by deeply considering experimental data, beyond the singular model of the protein.
Evidence for conformational substates from solution
While crystallography has remained popular for independently revealing interconverting structures, substantial evidence for the importance of polysterism has come from solution methods. In particular, nuclear magnetic resonance (NMR) relaxation-based methods have opened up the field of protein dynamics at a wide range of time scales [26]. Order parameters of fast (ps–ns) time scale dynamics can lead to degenerate models of motion [27]. These measurements are limited to backbone amides and specific side-chain residues and do not provide any information about coupling or correlated motion between sites [28]. Intermediate (μs–ms) time scale motions revealed by CPMG (Carr Purcell Meiboom Gill, R2) or R1ρ relaxation dispersion methods yield information about two- or three-state differences in chemical shift, population, and exchange rates [29]. However, since distinct states are inferred due to differences in chemical shift, many types of motion can be invoked to explain the signal. Further, movement of surrounding residues, rather than the residue being probed, can drive the chemical shift differences. The simultaneous fitting of relative population, exchange rates, and chemical shifts is generally underdetermined for an individual residue. This is normally overcome by implying a collective motion for a group of residues and jointly constraining their relative populations and interconversion rates [26]. NMR relaxation methods can identify groups of residues that likely experience exchange between states, but these methods do not provide a reference to a causal structural mechanism of the dynamic signal.
The power of these techniques was demonstrated in experiments that probed the allosteric behavior of the signaling protein nitrogen regulatory protein C (NtrC). Relaxation methods demonstrated that phosphorylation affects a population shift between two pre-existing conformations, providing strong validation of the MWC model of allostery [30]. Mutational perturbations to the structural transition pathway predicted by molecular dynamics simulations confirmed that phosphorylation stabilizes the minor conformation of the unphosphorylated state [31]. Later in this review, we describe three enzymes where different classes of structural rearrangements have been probed by relaxation methods and how crystallography can be leveraged to provide a structural interpretation of the dynamic signal.
Connecting polysterism to enzyme function
Much of the effort in protein structure–function studies over the past decades has focused on defining structural folds and static active site chemistries. Considerably less energy has been devoted to defining the role of polysterism and conformational change. This allocation of effort is understandable since studying the full conformational landscape of an enzyme before understanding its catalytic mechanism and structural fold would not provide particularly useful information. Another reason for the comparative neglect of polysterism in the literature is that experimentally characterizing scarcely populated states of the ensemble remains difficult and that linking these minor conformations to function is even more challenging [32, 33].
However, several recent developments have lead to the increased study of the role of polysterism in enzyme function. Our understanding of active site chemistry has now developed to the level where functional active sites can be designed de novo [34, 35]. Concurrently, many technical advances in crystalline and solution structural studies have made studying dynamics or polysterism feasible for a majority of proteins [36]. This has meant that simply describing a structure and putative catalytic mechanism is now only the beginning of many structure–function studies. This review examines ‘protein structural dynamics’ by focusing on the detection of protein polysterism and its relevance to the equilibrium steps of protein function and evolution. The dynamic, or time-resolved, aspect and the role of conformational change in the chemical step of catalysis are beyond the scope of the review and have recently been reviewed elsewhere [32, 33]. The challenge over the coming years will involve understanding how conformational substates, and the dynamic transitions between them, function during all steps of the catalytic cycle of an enzyme. We will now discuss six recent examples where this challenge has been answered.
Dihydrofolate reductase; polysterism through a catalytic cycle
Dihydrofolate reductase (DHFR) catalyzes transfer of hydrogen from NADPH–dihydrofolate to yield tetrahydrofolate, a required step in the synthesis of purines and some amino acids. This central role in metabolism also makes DHFR an attractive anti-cancer and anti-bacterial drug target. Induced fit interpretations of DHFR conformational changes observed by crystallography at different states of the catalytic cycle by Kraut and coworkers [37, 38], led to an increased understanding of the catalytic mechanism. Subsequent NMR experiments showed, with reference to the crystal structures, that diverse conformational fluctuations, including hydrophobic core rotamer exchange [39] and loop motions [40], are pre-existing in each functional state and may help to drive the transitions during catalytic cycle [41]. The linear agreement of high-energy chemical shifts from relaxation dispersion experiments and ground-state chemical shifts of the neighboring steps of the reaction cycle implies that polysterism helps to bias the enzyme conformation along the reaction trajectory (Fig. 1a). Furthermore, the agreement between the rate constants of the reaction and conformational sampling supports the idea that protein motions, rather than active site chemistry, limits turnover.
Fig. 1.

Catalytically important motions detected by NMR relaxation experiments can arise through different structural mechanisms. a Polysterism revealed by comparison of NMR chemical shifts with reference to independent crystal structures in DHFR (1RC4 and 1RB2). The Met20 loop of DHFR samples a minor conformational substate (shown in orange) when bound to the substrates NADP+ and THF. The minor state chemical shift is correlated with the chemical shift of the major conformational substate for the immediately preceding state of the catalytic cycle. b Independent crystal structures of AdK in the apo/open state (blue 2RH5) and nucleotide-bound/closed state (yellow, with nucleotide in red 2RGX) reveal that subdomain lid closure underlies the NMR relaxation signal. In an orthogonal view, independent chains from one crystallographic asymmetric unit (blue, light blue, and cyan 2RH5) reveal that the transition path towards the closed state (yellow) is populated even when no nucleotide is bound. Residues on mobile loops are shown in stick and sphere to provide a visual landmark of the polysterism. c In the room-temperature structure of CypA (3K0N), interconverting side-chain conformations give rise to an NMR relaxation dispersion signal. The minor (orange) and major (red) conformational substates extending from the core (left) to the active site Arg55 (right) are shown within electron density mesh. Examining the electron density at low levels (light blue 0.3σ) below normal contour (dark blue 1σ) reveals the minor conformational substate
The dynamic properties of DHFR have lead to new therapeutic and protein engineering strategies. A small molecule that breaks the coupling of dynamic features acts as an efficient inhibitor of the enzyme [42]. The fusion of a PAS domain to a specific surface site of DHFR allosterically couples dynamic loop regions and a network of internal residues that promote catalysis [43]. This strategy generated a novel allosteric circuit that allowed for light/dark-control of DHFR enzymatic activity [44]. Domain fusions that bias the conformations of flexible active site loops may represent a general strategy for engineering allosteric control of enzyme function. For other enzymes, the challenge is to define polysteric regions and evolutionarily conserved sites that can be used to mechanically couple allostery, conformational flexibility, and enzyme function.
Antibodies; polysterism leads to binding plasticity
The work on antibody specificity by Tawfik and colleagues [45] has provided clear evidence of functionally relevant polysterism, through the use of crystallography and fast-kinetic analysis of pre-equilibrium states. Crystallographic analysis of catalytic antibodies raised against a transition-state analogue of carboxylesters indicated that the structure of the ligand-free form of the antibody was essentially identical to the structure of the antibody-TS analogue complex. These results led to the conclusion that catalytic antibodies most likely follow a simple lock-and-key mechanism in ligand binding [46, 47]. However, pre-steady-state kinetic experiments revealed that the antibodies exist in a pre-equilibrium between distinct conformational substates and that ligand binding induces an equilibrium shift to the bound state conformation [48]. Moreover, the crystallization conditions were found to bias the conformational distribution of the apo-enzyme towards the bound-like state, explaining why only this configuration was observed by X-ray diffraction.
The model that ligand binding stabilized selected conformations of a pre-existing equilibrium was further tested through analysis of another antibody, SPE7. SPE7 was raised against a small molecule hapten (2,4 dinitrophenyl) and also found to bind the protein antigen (Trx-Shear3) [49, 50]. Pre-steady-state kinetics again established a pre-equilibrium consisting primarily of two conformations that could be altered through the addition of hapten or protein antigen. The kinetic data was supported by crystal structures of the two major pre-equilibrium states and of antibody:hapten and antibody:protein complexes. As shown in Fig. 2, the major (AB1) and minor (AB2) ligand-free substates respectively resemble the protein (AB4) and hapten (AB3) bound structures. These similarities are most apparent in the light chain of the antibody, where a cleft at which the hapten binds is formed by Y34, W93, and N96 in AB2/AB3, but is largely absent in AB1/AB4 as a result of different conformations of these side chains. Notably, despite this selection of pre-existing conformational substates by hapten- or protein-binding, the final bound structures still display significant differences with the unbound states. This suggests that some induction of conformational change still occurs. This aspect was subsequently addressed in greater detail when the interactions between SPE7 and haptens with high and low affinity were tested. Both high- and low-affinity haptens were observed to form identical transition complexes with similar affinity, yet only the high-affinity hapten formed hydrogen bonds with previously unexposed parts of the antigen to allow the final bound form to be ‘locked in’ [51]. These examples demonstrate that ligand binding selectivity (and promiscuity) can originate from inherent conformational polysterism and that both selection of pre-existing states and induced conformational change can play a role in protein–ligand interaction.
Fig. 2.

Hapten or protein binding selects pre-existing conformations of the antibody SPE7. Two distinct conformations of unbound SPE7 (AB1: 1OAQ; AB2: 1OCW) have been characterized in which part of the binding site consisting of Y34, W93 and N96 of the light chain are in different conformations. Owing to the orientation of W96, the binding site of the major conformation, AB1, is considerably ‘flatter’ than AB2. Ligand binding selects from these pre-existing substates, with protein-binding (AB4: 1OAZ) maintaining the AB1-like conformation of these residues and hapten-binding (DNP-Serine; AB3: 1OAU) maintaining an AB2-like conformation
Acetylcholinesterase; polysterism from simulations and crystallography
Acetylcholinesterase (AChE) has been studied in great detail owing to its essential role in nerve signal transduction [52] and its inhibition by pharmaceuticals [53] and both natural and synthetic toxins [54, 55]. It is one of nature’s most efficient enzymes, with high turnover (k cat) values ~105 s−1, and k cat/K M values (~109 s−1 M−1) that approach the diffusion limit [56]. When the crystal structure of AChE was first solved in 1991, the unexpected observation that its active site was located at the base of a very deep and narrow gorge [57] seemed at odds with the magnitude of its kinetic parameters and indicated that conformational change might play an essential role in substrate and product diffusion to/from the active site. In addition to ‘breathing’ of the active site cleft [58], a subtle conformational rearrangement of side chains occurs during the catalytic cycle of AChE that promotes efficient catalysis and substrate and product diffusion. These discoveries have emerged from a series of X-ray crystallography experiments and molecular dynamics simulations. Three amino acids have been found to be prominently polysteric: F330, W279, and W84 (Fig. 3).
Fig. 3.
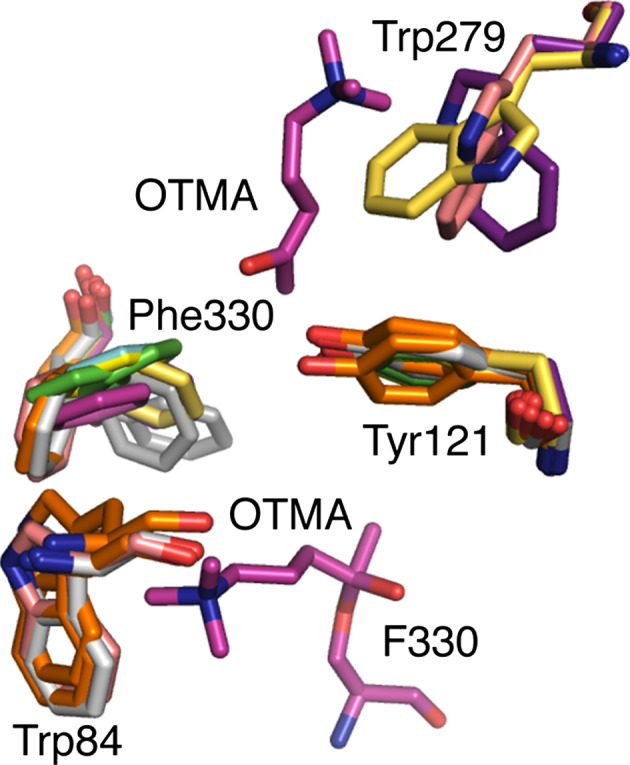
Polysterism in the active site and peripheral site of acetylcholinesterase. The gateway to the active site from the peripheral anionic site in AChE is formed by F330 and Y121. The positioning of substrate in the active site and peripheral anionic site is shown by the substrate analog OTMA from the structure 2C5F. Several structures of AChE are overlayed showing the conformational flexibility of F330. These include: 2C4H (green), 2C58 (cyan), 2C5F (magenta), 2C5G (light yellow), 2V96 (light grey), 2V98 (orange), 1EA5 (pink), 2CEK (grey), 1ACJ (dark yellow). Polysterism in the ‘backdoor residue of the active site in shown by the structures 1EA5, 2V98, and 2V96. Polysterism of W279 at the peripheral anionic site is shown by 2CKM (purple), 1EA5 and 1ACJ
Colletier et al. [59] studied the migration of acetylcholine into the active site using substrate analogues and exploiting substrate inhibition. They observed that in native crystal structures, the cross section between F330 and Y121, what is known as the active site ‘gate’ is only 5 Å; less than the diameter of the quaternary group of the substrate acetylcholine. A series of crystal structures were acquired in which different rotameric states of F330 were observed to drastically change the size of the active site gate. These conformational changes were found both to control access to the active site by fluctuating between open and closed conformations and to provide cationic interactions with the substrate during the chemical step in the catalytic cycle. Different rotameric states of W279, located in the peripheral anionic site, have also been observed as a result of ligand binding, in particular with bi-functional inhibitors that bind at both the peripheral anionic site and the active site [60]. This polysterism and its role in ligand binding has been referred to as “dynamic selectivity” by McCammon and colleagues [61] who observed its effects through molecular dynamics (MD) simulations. Finally, the active site residue W84 may fluctuate through rotameric states that allow product release through a “backdoor”, which would eliminate competition with incoming substrate for entrance to the active site through the narrow gorge [62]. While structural comparisons have revealed heterogeneity in this region through static [55, 63], and kinetic [64], crystallography experiments and molecular dynamics simulations [67], mutagenesis studies have been inconclusive [65, 66].
The polysterism discussed above was observed through analysis of many separate crystal structures; it was not known whether these conformations existed in the absence of ligands. To address this question, Xu et al. [67, 68] performed a series of molecular dynamics simulations and compared the sampled geometries with the many apo-enzyme and ligand bound structures of AChE available in the literature. They found that the apo-enzyme continuously samples almost all of the side-chain conformations observed to result from ligand binding. These results suggest that, in AChE at least, conformational fluctuations provide access to a polysteric landscape of distinct conformational substates that possess unique catalytic properties tailored to specific steps along the catalytic cycle and have different ligand affinities.
Adenylate kinase; subdomain polysterism from a single crystal
Adenylate kinase (AdK) maintains the balance of cellular energy by catalyzing the interconversion of adenine nucleotides. Alignment of the apo and nucleotide-bound crystal structures of AdK revealed a very large conformational transition [69]. The apo-structure resembles an open hand with the finger and thumb lids (Fig. 1b). In structures of the enzyme bound to its nucleotide substrates, the two lids close down around the active site providing a favorable electrostatic environment for phospho-transfer. NMR studies showed that the conformational lid rearrangement, rather than the chemical phospho-transfer step, limits the rate of the catalytic cycle in homologous AdKs [70]. Given the importance of lid flexibility, several studies have examined how motion is induced by substrate.
Surprisingly, convincing evidence that lid rearrangement is pre-existing in the apo-enzyme came from comparisons of independent chains within a single crystal (Fig. 1b) [71]. In crystals obtained by Henzler-Wildman et al., three independent copies of apo-AdK each experience a different crystal-packing environment. The changes in crystal contacts allowed the two lids to rotate about several hinges. The phenomenon of hinge-bending in multiple chains from a single crystal was first observed in a mutant T4 lysozyme [72]. For AdK, the alignment of the independent copies suggested a concerted movement of the lids towards the closed conformations even in the absence of substrate. Adk represents an example of polysterism observed through multiple chains within a single crystal. The conformational heterogeneity observed in the crystal resembles the solution trajectory predicted by computational analysis of crystal structures of the Adk in different nucleotide-bound states.
A comprehensive suite of single-molecule FRET, NMR, and computational studies further confirmed the relevance of crystalline polysterism in AdK. These studies concluded that the enzyme could harvest the directionality of pre-existing motions during a catalytic cycle [71, 73]. This result builds upon the theme that substrate binding does not induce entirely new structures; rather, binding shifts the populations of substates that pre-exist in the ensemble. Although this work defined the apo-state energy landscape, many detailed mechanistic questions remain about how the lid movements are controlled. Particularly, questions remain as to whether lid closing involves local unfolding [74] and the ratio of populations of the open and closed forms in different nucleotide states [75]. AdK remains an excellent system for examining the relevance of enzyme conformational heterogeneity to the catalytic cycle. Polysterism, from multiple independent chains in the same crystal, was key to unraveling this relationship in AdK. However, molecular dynamics simulations and NMR experiments provide the necessary time-scale resolution to augment the functional importance of the pre-existing conformational substates.
Cyclophilin A; polysterism in electron density at different temperatures
Cyclophilin A (CypA), catalyzes the isomerization of proline residues in proteins, which can regulate protein folding, signaling, and molecular assembly. CypA, like AdK and DHFR, undergoes motions detectable by NMR during catalysis that correspond to the catalytic turnover rate [76]. Furthermore, motions in CypA are pre-existing in the active site, hydrophobic core, and loop regions whether or not substrate is bound [77]. However, unlike AdK or DHFR, comparison of X-ray structures did not suggest a mechanism for the conformational diversity in the active site and core regions detected by NMR. In other systems, large backbone movements of loops or hinge-based subdomain reorientations provide the chemical shift differences detected by relaxation dispersion experiments. Although dynamics of the loop regions of CypA arise because of this conventional motion, the active site and core motions are surprisingly caused by a coupled network of interconverting side-chain substates [78] (Fig. 1c).
The minor state side-chain conformations of apo-CypA were discovered by using the program Ringer [5] to sample low electron density levels in a map obtained from data collected at room temperature. Interestingly, conventionally frozen crystals only displayed evidence of the major state. The observation of these substates confirms a link between crystal data collection temperature and substate population [79]. Due to the limited number of enzymes where structures are available at multiple temperatures, it is unclear whether room temperature data collection universally leads to a more accurate description of conformational substates. For example, in alpha-lytic protease, higher resolution cryogenic data collection enabled a more complete description of the conformational landscape than a room temperature structure [80].
A remote mutation designed to test the importance of the side-chain substates in CypA not only flipped the population of the substates, but also caused a parallel decrease in the rate of conformational interconversions measured by NMR and the rate of catalysis [78]. This and other mutations tested with the HIV capsid as a substrate [81] suggest that the catalytic cycle of CypA is limited by conformational transitions that are populated in the apo-state. Interestingly, evolution has harvested CypA polysterism as a mechanism for species restriction in HIV. Elegant structural and thermodynamic studies revealed the mechanism that targets a retrotransposed mutant CypA in rhesus macaque monkeys to bind specifically to HIV-2 capsids [82]. Two mutations, located in the loop regions identified as polysteric by NMR relaxation dispersion analysis, dramatically reshape the dominant conformation of the binding pocket. This study further showed a correspondence between loop conformation, binding selectivity, and sensitivity to viral infection. Since wild-type CypA binds the capsid of both HIV-1 and HIV-2, the conformation stabilized by the mutations is likely present in the wild-type ensemble. Thus, CypA represent an example of how polysterism, revealed through comparison of multiple crystal structures, can be used in the evolutionary arms race between host and pathogen by sculpting loop conformations to determine binding specificity. The binding selectivity imparted by loop motion complements the network of residues extending from the core to the active site that undergo side-chain rearrangements that are necessary for turnover [78].
Bacterial phosphotriesterase; polysterism promotes evolution
The bacterial phosphotriesterase (PTE) has attracted considerable attention as a result of its rapid evolution of extremely high catalytic efficiency for the breakdown of human-made pesticides such as paraoxon. Raushel and coworkers [83–85] have established that the catalytic cycle is comprised of separate steps involving formation of the Michaelis complex, the chemical step and product release and that for reactive substrates, such as paraoxon, the slowest rate is product release or conformational change. Recently different states along the reaction coordinate have been captured in crystallo, providing new structural insight [11, 86].
Although the enzyme:substrate and enzyme:product complexes of PTE are consistent with a highly efficient chemical step in the catalytic cycle, it was observed that, as in AChE [59], the entrance to the active site was smaller in diameter than the cross section of the substrate/product [87]. This observation pointed to an important role for conformational change in the catalytic cycle. To investigate this in more detail, a series of crystal structures of mutants of PTE were solved in which the active site was observed to exist in equilibrium between ‘open’ and ‘closed’ conformations that are ideally configured for non-chemical (substrate and product diffusion) and chemical (catalysis) steps in the catalytic cycle, respectively (Fig. 4). Different mutations were found to affect the distribution of open and closed states, and through this the rate of product release. It should be noted that the observation and building of these minor geometries was performed through human inspection of electron density maps. As discussed in a later section, recent technical developments have enabled detection of much more, previously ‘hidden’ polysterism, automatically. This study characterized the conformational landscape of the PTEs and linked these states to different steps in its function, but more importantly demonstrated that mutations accrued through evolution can alter activity by modulating the energy barriers, and thus the accessibility, of different conformational substates required for function.
Fig. 4.
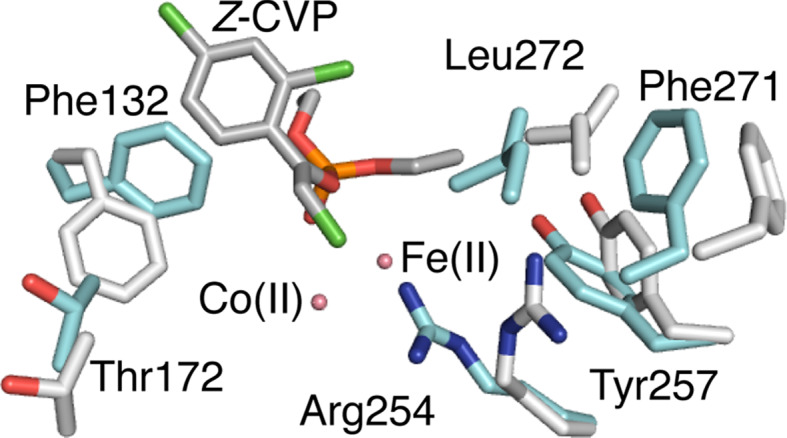
Major and minor conformations of the bacterial phosphotriesterase. The major ‘closed’ conformation of PTE is shown in cyan, while the minor ‘open’ conformation is shown in grey (3A4J). Both conformations were modeled into the electron density of an apo-enzyme crystal. The substrate Z-chlorfenvinphos is shown docked based on known enzyme:substrate complexes (2R1N). It can be seen that in the ‘closed’ conformation, several side-chains adopt rotamers that extend into the active site, greatly reducing its volume. One of these, F132, is shown to sterically clash with Z-chlorfenvinphos in its dominant conformation but to allow binding in its minor ‘open’ conformation
A similar story is seen in the role of polysterism in promiscuous, or non-native, PTE activity. Previous work has shown that the slow turnover of the pesticide chlorfenvinphos was principally a result of the active site gorge of the enzyme being too narrow to accommodate the substrate [88]. Mutation of a phenylalanine to an alanine opened the active site gorge and resulted in a 500-fold increase in catalytic efficiency. The question remained: how does the enzyme catalyze hydrolysis of the chlorfenvinphos at all when there is no possible binding mode? The description of the multi-state equilibrium model of the phosphotriesterase provides an explanation [87]: the primary residue that blocks chlorfenvinphos binding (F132) is observed to adopt a minor conformation in which its ring is flipped out of the active site in such a way as to allow productive chlorfenvinphos binding. This therefore provides a straightforward example of the hypothesis put forward by Tokuriki and Tawfik [36], which posited that conformational diversity can provide important starting points for evolution. This also demonstrates that polysterism can allow promiscuous activities to be catalyzed by minor conformational substates that may not be used for the analogous step in the native catalytic cycle.
Glucocorticoid receptor; polysterism in allosteric signaling
The importance of minor states revealed by weak electron density extends beyond catalysis. For example, the allosteric effect of DNA binding to the glucocorticoid receptor (GR) manifested in alternate conformations, observed in weak electron density, that modulate the responsiveness to hormone treatment (Fig. 5) [89]. A series of crystal structures of GR bound to different DNA sequences revealed polysterism in the lever arm. These structures demonstrated the existence of conformational substates in this region through comparisons between structures and also within individual electron density maps. Allosteric modulation of GR conformational substates alters the recruitment of the transcriptional machinery and leads to different transcriptional activation, independent of the effects of binding affinity to the DNA sequence. These studies highlight the critical role that crystallography will continue to play in characterizing the importance of minor conformational substates in many aspects of protein function, from catalysis to signaling.
Fig. 5.

DNA binding allosterically modulates lever arm conformations and the transcriptional activation of glucocorticoid receptor. a The glucocorticoid receptor bound to the Sgk-sequence DNA (3G9P) adopts a dimeric structure with polysterism in the lever arm. b The electron density (dark blue 1σ, light blue 0.3σ) for the chain A (yellow) lever arm is well fit and suggests only small amplitude motion around the major conformation. c In contrast, despite originating from the same map, the electron density for the lever arm of chain B (black) for this area reveals potential polysterism. Although the lever arm is modeled into a different conformation, there is much unexplained density at low levels. d The conformations of the lever arms of chain A (yellow) and chain B (black) represent the major structure difference between the two chains. Low levels of electron density (light blue 0.3σ) shown from the area of chain B suggest that the conformation modeled for chain A may represent a component of the ensemble for both chains
Technical challenges and recent advances
Given the importance of conformational substates in evolution and the catalytic cycles of enzymes, the question remains: what are the best ways to reveal these critical minor states? Clearly the best description of the native ensemble comes from a combination of methods, often including NMR relaxation techniques, and experimental tests of the importance of the observed polysterism. Here, we have reviewed several ways that protein crystallography can provide insight into spatial distribution of conformational substates. For example, structural changes can be revealed through independent crystal structures (DHFR, Antibodies, AChE), comparisons of multiple chains in the same crystal (AdK), or by examining the electron density for minor conformations (CypA, PTE, GR).
Recently developed crystallographic refinement methods aim to describe the ensemble populated in the crystal. These techniques complement the traditional B-factor calculation, which gives an indication of the small-amplitude flexibility around a ground state. Although there is often a concordance between B-factors and fast small-amplitude harmonic motions in solution [90], several alternative methods have been proposed to explore more dramatic motions that can be difficult to interpret in “messy” electron density. For example, automated rebuilding and refinement techniques can create an “ensemble of independent models” [91, 92]. This family of models is generally a fixed number of independent models that do not simultaneously contribute to the refinement process. These methods have been used to suggest that many models can equally explain the diffraction data [92]. A systematic investigation of an “ensemble of models” created with synthetic data revealed that independently refined models represent the uncertainty inherent in a single-model description of the possible conformational space populated in the crystal [91].
To more directly model the multiple structures populated in the crystal, many “multi-copy ensemble” refinement methods have been developed [7]. In “multi-copy ensemble” procedures, a fixed number of interdependent models are simultaneously refined [7]. For certain anharmonic polysteric regions, this procedure can lead to a much better description of the electron density. However, given the limited number of experimental observations in crystallographic data, for many residues it may not be necessary to introduce the added parameters needed to refine locally similar regions for the multiple copies of the ensemble.
At high resolution, it may be more advantageous for many residues to be modeled as in a single conformation with anisotropic B-factors rather than as a part of a “multi-copy ensemble”. For other residues, multiple alternative conformations may be needed to fit the electron density. This is traditionally accomplished through manual inspection of electron density maps, which often leads to an under-estimation of the extent of polysterism [5]. An algorithm, qFit, that implements such a strategy in an automated fashion has recently been implemented [6]. qFit generates a “multi-conformer model” that is generally a single model. However, for select residues, alternative backbone and side-chain conformations are modeled into the electron density. This method gives qualitatively similar results to iterative electron density sampling and manual rebuilding and can capture complex coupled motions that extend over many residues [93].
The automation provided by qFit augments existing strategies for discovering and modeling alternative conformations. For example, electron density sampling as implemented in Ringer [5], reveals that many alternative conformations exist at electron density levels that have been traditionally considered noise by human model builders. Ringer samples electron density in dihedral space and discovers peaks indicative of unmodeled alternative conformations. These alternative conformations can be built after manual inspection of the electron density map and further refined for occupancy by phenix.refine [94]. As seen in PTE and CypA, alternative conformations, revealed at low levels of electron density (generally 0.3–1σ), can be critical for enzyme function.
Another means by which crystallography can access protein polysterism is the analysis of thermal diffuse scattering [95, 96]. Diffuse scattering is the remaining X-ray intensity that is recorded in diffraction images once the Bragg reflections are subtracted. While analysis of the Bragg reflections can give accurate estimates of the individual atomic mean squared displacements and alternate conformations, the diffuse scattering can provide valuable insight into isotropic liquid like displacements [97] and strongly correlated rigid body movements of groups of atoms [98]. This can reveal how particular groups of atoms are displaced together as rigid bodies. Techniques that predict low frequency correlated rigid body movements, such as principal component analysis of molecular dynamics simulations or normal mode analysis of elastic network models, can be validated by analysis of diffuse scatter [99, 100]. This theoretical–experimental cross validation approach can also serve to support or exclude other models of correlated motion used in refinement, such as translation-libation-screw (TLS) refinement [101, 102]. Whether analyzing diffuse or Bragg scatter, access to primary image data would greatly increase the ability to re-examine biological conclusions. Although deposition of scaled reflection data, which permits re-calculation of electron density maps for analysis by Ringer and other software, is now standard for almost all journals, image data are rarely deposited [103]. A major limitation to deposition of image data is storage space, but several institutions, structural genomics consortia, and synchrotrons have started voluntary image deposition. As interpretation of atomic displacements becomes more important to our understanding of protein function, our reliance on the re-analysis of primary data is likely to increase in coming years.
Conclusions
Polysterism has contributed to our understanding of how new functions evolve from existing functions. Many studies have now demonstrated that an ensemble of interconverting structures, rather than a single structure, exerts the function of the protein. Thus, evolutionary selection does not optimize only an active site or the stability of a single structure. Rather, it shapes an entire conformational landscape comprising many sub-structures, their relative populations, and their rates of interconversion. Because mutations may affect the dominant structure modeled in a typical crystal structure or “hidden” minor conformations, polysterism must be carefully considered when considering the role of remote mutations on both catalysis and allostery [104]. If we can accurately describe the conformational landscape of proteins, we might be able to better predict the nature and conformations of transitions between the dominant structures as a protein is perturbed. Realizing this dream has implications for understanding protein evolution and designing better enzymes and inhibitors [105].
Acknowledgments
JSF was funded by an EMBO Short-term Fellowship while this article was conceived. CJJ is supported by a Marie Curie International Incoming Fellowship. We acknowledge Dan Tawfik for providing a stimulating environment. We also thank Tom Alber, David Wemmer, and Martin Weik for discussions. All figures in this review were generated with PyMol (http://pymol.org); we dedicate this article to the legacy of PyMol’s creator Warren DeLano.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- NtrC
Nitrogen regulatory protein C
- AchE
Aacetylcholinesterase
- AdK
Adenylate kinase
- CypA
Cyclophilin A
- DHFR
Dihydrofolate reductase
- PTE
Phosphotriesterase
- MWC
Monod, Wyman and Changeaux (model of allostery)
- GR
Glucocorticoid receptor
References
- 1.Hilser VJ, Garcia-Moreno EB, Oas TG, Kapp G, Whitten ST. A statistical thermodynamic model of the protein ensemble. Chem Rev. 2006;106:1545–1558. doi: 10.1021/cr040423+. [DOI] [PubMed] [Google Scholar]
- 2.Kendrew JC, Dickerson RE, Strandberg BE, Hart RG, Davies DR, Phillips DC, Shore VC. Structure of myoglobin: a three-dimensional Fourier synthesis at 2 Å resolution. Nature. 1960;185:422–427. doi: 10.1038/185422a0. [DOI] [PubMed] [Google Scholar]
- 3.Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North AC. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by X-ray analysis. Nature. 1960;185:416–422. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 4.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 5.Lang PT, Ng HL, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T. Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Sci. 2010;19:1420–1431. doi: 10.1002/pro.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van den Bedem H, Dhanik A, Latombe JC, Deacon AM. Modeling discrete heterogeneity in X-ray diffraction data by fitting multi-conformers. Acta Crystallogr D Biol Crystallogr. 2009;65:1107–1117. doi: 10.1107/S0907444909030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levin EJ, Kondrashov DA, Wesenberg GE, Phillips GN., Jr Ensemble refinement of protein crystal structures: validation and application. Structure. 2007;15:1040–1052. doi: 10.1016/j.str.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlicker C, Fokina O, Kloft N, Grune T, Becker S, Sheldrick GM, Forchhammer K. Structural analysis of the PP2C phosphatase tPphA from Thermosynechococcus elongatus: a flexible flap subdomain controls access to the catalytic site. J Mol Biol. 2008;376:570–581. doi: 10.1016/j.jmb.2007.11.097. [DOI] [PubMed] [Google Scholar]
- 9.Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, Matsumoto K, Yamamoto N, Sugimoto T, Yoshikawa Y, Kumasaka T, Yamamoto M, Tamura A, Kataoka T. Structural basis for conformational dynamics of GTP-bound Ras protein. J Biol Chem. 2010;285:22696–22705. doi: 10.1074/jbc.M110.125161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci USA. 2003;100:3895–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson CJ, Foo JL, Kim HK, Carr PD, Liu JW, Salem G, Ollis DL. In crystallo capture of a Michaelis complex and product-binding modes of a bacterial phosphotriesterase. J Mol Biol. 2008;375:1189–1196. doi: 10.1016/j.jmb.2007.10.061. [DOI] [PubMed] [Google Scholar]
- 12.Lipscomb WN. Enzymatic activities of carobxypeptidase A’s in solution and in crystals. Proc Natl Acad Sci USA. 1973;70:3797–3801. doi: 10.1073/pnas.70.12.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alter GM, Leussing DL, Neurath H, Vallee BL. Kinetic properties of carboxypeptidase B in solutions and crystals. Biochemistry. 1977;16:3663–3668. doi: 10.1021/bi00635a024. [DOI] [PubMed] [Google Scholar]
- 14.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 15.Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli . J Mol Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-V. [DOI] [PubMed] [Google Scholar]
- 16.Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. doi: 10.1016/S0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 17.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett WS, Jr, Steitz TA. Glucose-induced conformational change in yeast hexokinase. Proc Natl Acad Sci USA. 1978;75:4848–4852. doi: 10.1073/pnas.75.10.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fletterick RJ, Bates DJ, Steitz TA. The structure of a yeast hexokinase monomer and its complexes with substrates at 2.7-Å resolution. Proc Natl Acad Sci USA. 1975;72:38–42. doi: 10.1073/pnas.72.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steitz TA, Ludwig ML, Quiocho FA, Lipscomb WN. The structure of carboxypepidase A. V. Studies of enzyme–substrate and enzyme–inhibitor complexes at 6-Å resolution. J Biol Chem. 1967;242:4662–4668. [PubMed] [Google Scholar]
- 21.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 22.Cooper A, Dryden DT. Allostery without conformational change. A plausible model. Eur Biophys J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 23.Karplus M, Ichiye T, Pettitt BM. Configurational entropy of native proteins. Biophys J. 1987;52:1083–1085. doi: 10.1016/S0006-3495(87)83303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frauenfelder H, Petsko GA, Tsernoglou D. Temperature-dependent X-ray diffraction as a probe of protein structural dynamics. Nature. 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 25.Artymiuk PJ, Blake CC, Grace DE, Oatley SJ, Phillips DC, Sternberg MJ. Crystallographic studies of the dynamic properties of lysozyme. Nature. 1979;280:563–568. doi: 10.1038/280563a0. [DOI] [PubMed] [Google Scholar]
- 26.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 27.Li DW, Bruschweiler R. A dictionary for protein side-chain entropies from NMR order parameters. J Am Chem Soc. 2009;131:7226–7227. doi: 10.1021/ja902477s. [DOI] [PubMed] [Google Scholar]
- 28.Trbovic N, Cho JH, Abel R, Friesner RA, Rance M, Palmer AG., 3rd Protein side-chain dynamics and residual conformational entropy. J Am Chem Soc. 2009;131:615–622. doi: 10.1021/ja806475k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loria JP, Berlow RB, Watt ED. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- 30.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 31.Gardino AK, Villali J, Kivenson A, Lei M, Liu CF, Steindel P, Eisenmesser EZ, Labeikovsky W, Wolf-Watz M, Clarkson MW, Kern D. Transient non-native hydrogen bonds promote activation of a signaling protein. Cell. 2009;139:1109–1118. doi: 10.1016/j.cell.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagel ZD, Klinman JP. A 21st century revisionist’s view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 33.Kamerlin SC, Warshel A. At the dawn of the 21st century: is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 35.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, 3rd, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–207. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 37.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 38.Bystroff C, Kraut J. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry. 1991;30:2227–2239. doi: 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- 39.Schnell JR, Dyson HJ, Wright PE. Effect of cofactor binding and loop conformation on side chain methyl dynamics in dihydrofolate reductase. Biochemistry. 2004;43:374–383. doi: 10.1021/bi035464z. [DOI] [PubMed] [Google Scholar]
- 40.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 41.Boehr DD, McElheny D, Dyson HJ, Wright PE. Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc Natl Acad Sci USA. 2010;107:1373–1378. doi: 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauldin RV, Carroll MJ, Lee AL. Dynamic dysfunction in dihydrofolate reductase results from antifolate drug binding: modulation of dynamics within a structural state. Structure. 2009;17:386–394. doi: 10.1016/j.str.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci USA. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, Ranganathan R. Surface sites for engineering allosteric control in proteins. Science. 2008;322:438–442. doi: 10.1126/science.1159052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tawfik DS, Green BS, Chap R, Sela M, Eshhar Z. catELISA: a facile general route to catalytic antibodies. Proc Natl Acad Sci USA. 1993;90:373–377. doi: 10.1073/pnas.90.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charbonnier JB, Golinelli-Pimpaneau B, Gigant B, Tawfik DS, Chap R, Schindler DG, Kim SH, Green BS, Eshhar Z, Knossow M. Structural convergence in the active sites of a family of catalytic antibodies. Science. 1997;275:1140–1142. doi: 10.1126/science.275.5303.1140. [DOI] [PubMed] [Google Scholar]
- 47.Gigant B, Charbonnier JB, Eshhar Z, Green BS, Knossow M. X-ray structures of a hydrolytic antibody and of complexes elucidate catalytic pathway from substrate binding and transition state stabilization through water attack and product release. Proc Natl Acad Sci USA. 1997;94:7857–7861. doi: 10.1073/pnas.94.15.7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindner AB, Eshhar Z, Tawfik DS. Conformational changes affect binding and catalysis by ester-hydrolysing antibodies. J Mol Biol. 1999;285:421–430. doi: 10.1006/jmbi.1998.2309. [DOI] [PubMed] [Google Scholar]
- 49.Eshhar Z, Ofarim M, Waks T. Generation of hybridomas secreting murine reaginic antibodies of anti-DNP specificity. J Immunol. 1980;124:775–780. [PubMed] [Google Scholar]
- 50.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 51.James LC, Tawfik DS. Structure and kinetics of a transient antibody binding intermediate reveal a kinetic discrimination mechanism in antigen recognition. Proc Natl Acad Sci USA. 2005;102:12730–12735. doi: 10.1073/pnas.0500909102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silman I, Sussman JL. Acetylcholinesterase: how is structure related to function? Chem Biol Interact. 2008;175:3–10. doi: 10.1016/j.cbi.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 53.Greenblatt HM, Dvir H, Silman I, Sussman JL. Acetylcholinesterase: a multifaceted target for structure-based drug design of anticholinesterase agents for the treatment of Alzheimer’s disease. J Mol Neurosci. 2003;20:369–383. doi: 10.1385/JMN:20:3:369. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez-Ithurralde D, Silveira R, Barbeito L, Dajas F. Fasciculin, a powerful anticholinesterase polypeptide from Dendroaspis angusticeps venom. Neurochem Int. 1983;5:267–274. doi: 10.1016/0197-0186(83)90028-1. [DOI] [PubMed] [Google Scholar]
- 55.Barthold CL, Schier JG. Organic phosphorus compounds–nerve agents. Crit Care Clin. 2005;21:673–689. doi: 10.1016/j.ccc.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 56.Radic Z, Pickering NA, Vellom DC, Camp S, Taylor P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry. 1993;32:12074–12084. doi: 10.1021/bi00096a018. [DOI] [PubMed] [Google Scholar]
- 57.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 58.Tai K, Shen T, Borjesson U, Philippopoulos M, McCammon JA. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys J. 2001;81:715–724. doi: 10.1016/S0006-3495(01)75736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colletier JP, Fournier D, Greenblatt HM, Stojan J, Sussman JL, Zaccai G, Silman I, Weik M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006;25:2746–2756. doi: 10.1038/sj.emboj.7601175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colletier JP, Sanson B, Nachon F, Gabellieri E, Fattorusso C, Campiani G, Weik M. Conformational flexibility in the peripheral site of Torpedo californica acetylcholinesterase revealed by the complex structure with a bifunctional inhibitor. J Am Chem Soc. 2006;128:4526–4527. doi: 10.1021/ja058683b. [DOI] [PubMed] [Google Scholar]
- 61.Shen T, Tai K, Henchman RH, McCammon JA. Molecular dynamics of acetylcholinesterase. Acc Chem Res. 2002;35:332–340. doi: 10.1021/ar010025i. [DOI] [PubMed] [Google Scholar]
- 62.Gilson MK, Straatsma TP, McCammon JA, Ripoll DR, Faerman CH, Axelsen PH, Silman I, Sussman JL. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science. 1994;263:1276–1278. doi: 10.1126/science.8122110. [DOI] [PubMed] [Google Scholar]
- 63.Nachon F, Stojan J, Fournier D. Insights into substrate and product traffic in the Drosophila melanogaster acetylcholinesterase active site gorge by enlarging a back channel. FEBS J. 2008;275:2659–2664. doi: 10.1111/j.1742-4658.2008.06413.x. [DOI] [PubMed] [Google Scholar]
- 64.Colletier JP, Royant A, Specht A, Sanson B, Nachon F, Masson P, Zaccai G, Sussman JL, Goeldner M, Silman I, Bourgeois D, Weik M. Use of a ‘caged’ analogue to study the traffic of choline within acetylcholinesterase by kinetic crystallography. Acta Crystallogr D Biol Crystallogr. 2007;63:1115–1128. doi: 10.1107/S0907444907044472. [DOI] [PubMed] [Google Scholar]
- 65.Faerman C, Ripoll D, Bon S, Le Feuvre Y, Morel N, Massoulie J, Sussman JL, Silman I. Site-directed mutants designed to test back-door hypotheses of acetylcholinesterase function. FEBS Lett. 1996;386:65–71. doi: 10.1016/0014-5793(96)00374-2. [DOI] [PubMed] [Google Scholar]
- 66.Kronman C, Ordentlich A, Barak D, Velan B, Shafferman A. The “back door” hypothesis for product clearance in acetylcholinesterase challenged by site-directed mutagenesis. J Biol Chem. 1994;269:27819–27822. [PubMed] [Google Scholar]
- 67.Xu Y, Colletier JP, Jiang H, Silman I, Sussman JL, Weik M. Induced-fit or preexisting equilibrium dynamics? Lessons from protein crystallography and MD simulations on acetylcholinesterase and implications for structure-based drug design. Protein Sci. 2008;17:601–605. doi: 10.1110/ps.083453808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu Y, Colletier JP, Weik M, Jiang H, Moult J, Silman I, Sussman JL. Flexibility of aromatic residues in the active-site gorge of acetylcholinesterase: X-ray versus molecular dynamics. Biophys J. 2008;95:2500–2511. doi: 10.1529/biophysj.108.129601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulz GE, Muller CW, Diederichs K. Induced-fit movements in adenylate kinases. J Mol Biol. 1990;213:627–630. doi: 10.1016/S0022-2836(05)80250-5. [DOI] [PubMed] [Google Scholar]
- 70.Wolf-Watz M, Thai V, Henzler-Wildman K, Hadjipavlou G, Eisenmesser EZ, Kern D. Linkage between dynamics and catalysis in a thermophilic–mesophilic enzyme pair. Nat Struct Mol Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 71.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, Hubner CG, Kern D. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 72.Faber HR, Matthews BW. A mutant T4 lysozyme displays five different crystal conformations. Nature. 1990;348:263–266. doi: 10.1038/348263a0. [DOI] [PubMed] [Google Scholar]
- 73.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 74.Whitford PC, Onuchic JN, Wolynes PG. Energy landscape along an enzymatic reaction trajectory: hinges or cracks? HFSP J. 2008;2:61–64. doi: 10.2976/1.2894846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hanson JA, Duderstadt K, Watkins LP, Bhattacharyya S, Brokaw J, Chu JW, Yang H. Illuminating the mechanistic roles of enzyme conformational dynamics. Proc Natl Acad Sci USA. 2007;104:18055–18060. doi: 10.1073/pnas.0708600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 77.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 78.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Halle B. Biomolecular cryocrystallography: structural changes during flash-cooling. Proc Natl Acad Sci USA. 2004;101:4793–4798. doi: 10.1073/pnas.0308315101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rader SD, Agard DA. Conformational substates in enzyme mechanism: the 120 K structure of alpha-lytic protease at 1.5-Å resolution. Protein Sci. 1997;6:1375–1386. doi: 10.1002/pro.5560060701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bosco DA, Eisenmesser EZ, Clarkson MW, Wolf-Watz M, Labeikovsky W, Millet O, Kern D. Dissecting the microscopic steps of the cyclophilin A enzymatic cycle on the biological substrate HIV capsid by NMR. J Mol Biol. 2010;403(5):723–738. doi: 10.1016/j.jmb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 82.Price AJ, Marzetta F, Lammers M, Ylinen LM, Schaller T, Wilson SJ, Towers GJ, James LC. Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat Struct Mol Biol. 2009;16:1036–1042. doi: 10.1038/nsmb.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caldwell SR, Newcomb JR, Schlecht KA, Raushel FM. Limits of diffusion in the hydrolysis of substrates by the phosphotriesterase from Pseudomonas diminuta . Biochemistry. 1991;30:7438–7444. doi: 10.1021/bi00244a010. [DOI] [PubMed] [Google Scholar]
- 84.Caldwell SR, Raushel FM, Weiss PM, Cleland WW. Transition-state structures for enzymatic and alkaline phosphotriester hydrolysis. Biochemistry. 1991;30:7444–7450. doi: 10.1021/bi00244a011. [DOI] [PubMed] [Google Scholar]
- 85.Hong SB, Raushel FM. Metal–substrate interactions facilitate the catalytic activity of the bacterial phosphotriesterase. Biochemistry. 1996;35:10904–10912. doi: 10.1021/bi960663m. [DOI] [PubMed] [Google Scholar]
- 86.Jackson C, Kim HK, Carr PD, Liu JW, Ollis DL. The structure of an enzyme–product complex reveals the critical role of a terminal hydroxide nucleophile in the bacterial phosphotriesterase mechanism. Biochim Biophys Acta. 2005;1752:56–64. doi: 10.1016/j.bbapap.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 87.Jackson CJ, Foo JL, Tokuriki N, Afriat L, Carr PD, Kim HK, Schenk G, Tawfik DS, Ollis DL. Conformational sampling, catalysis, and evolution of the bacterial phosphotriesterase. Proc Natl Acad Sci USA. 2009;106:21631–21636. doi: 10.1073/pnas.0907548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jackson CJ, Weir K, Herlt A, Khurana J, Sutherland TD, Horne I, Easton C, Russell RJ, Scott C, Oakeshott JG. Structure-based rational design of a phosphotriesterase. Appl Environ Microbiol. 2009;75:5153–5156. doi: 10.1128/AEM.00629-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clore GM, Schwieters CD. Concordance of residual dipolar couplings, backbone order parameters and crystallographic B-factors for a small alpha/beta protein: a unified picture of high probability, fast atomic motions in proteins. J Mol Biol. 2006;355:879–886. doi: 10.1016/j.jmb.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 91.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Adams PD, Moriarty NW, Zwart P, Read RJ, Turk D, Hung LW. Interpretation of ensembles created by multiple iterative rebuilding of macromolecular models. Acta Crystallogr D Biol Crystallogr. 2007;63:597–610. doi: 10.1107/S0907444907009791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.DePristo MA, de Bakker PI, Blundell TL. Heterogeneity and inaccuracy in protein structures solved by X-ray crystallography. Structure. 2004;12:831–838. doi: 10.1016/j.str.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 93.Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 94.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wall ME. Methods and software for diffuse X-ray scattering from protein crystals. Methods Mol Biol. 2009;544:269–279. doi: 10.1007/978-1-59745-483-4_17. [DOI] [PubMed] [Google Scholar]
- 96.Clarage JB, Phillips GN., Jr Analysis of diffuse scattering and relation to molecular motion. Methods Enzymol. 1997;277:407–432. doi: 10.1016/S0076-6879(97)77023-X. [DOI] [PubMed] [Google Scholar]
- 97.Wall ME, Ealick SE, Gruner SM. Three-dimensional diffuse X-ray scattering from crystals of Staphylococcal nuclease. Proc Natl Acad Sci USA. 1997;94:6180–6184. doi: 10.1073/pnas.94.12.6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Doucet J, Benoit JP. Molecular dynamics studied by analysis of the X-ray diffuse scattering from lysozyme crystals. Nature. 1987;325:643–646. doi: 10.1038/325643a0. [DOI] [PubMed] [Google Scholar]
- 99.Meinhold L, Smith JC. Protein dynamics from X-ray crystallography: anisotropic, global motion in diffuse scattering patterns. Proteins. 2007;66:941–953. doi: 10.1002/prot.21246. [DOI] [PubMed] [Google Scholar]
- 100.Riccardi D, Cui Q, Phillips GN., Jr Evaluating elastic network models of crystalline biological molecules with temperature factors, correlated motions, and diffuse X-ray scattering. Biophys J. 2010;99:2616–2625. doi: 10.1016/j.bpj.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moore PB. On the relationship between diffraction patterns and motions in macromolecular crystals. Structure. 2009;17:1307–1315. doi: 10.1016/j.str.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 102.Perez J, Faure P, Benoit JP. Molecular rigid-body displacements in a tetragonal lysozyme crystal confirmed by X-ray diffuse scattering. Acta Crystallogr D Biol Crystallogr. 1996;52:722–729. doi: 10.1107/S0907444996002594. [DOI] [PubMed] [Google Scholar]
- 103.Androulakis S, Schmidberger J, Bate MA, DeGori R, Beitz A, Keong C, Cameron B, McGowan S, Porter CJ, Harrison A, Hunter J, Martin JL, Kobe B, Dobson RC, Parker MW, Whisstock JC, Gray J, Treloar A, Groenewegen D, Dickson N, Buckle AM. Federated repositories of X-ray diffraction images. Acta Crystallogr D Biol Crystallogr. 2008;D64:810–814. doi: 10.1107/S0907444908015540. [DOI] [PubMed] [Google Scholar]
- 104.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 105.Friedland GD, Kortemme T. Designing ensembles in conformational and sequence space to characterize and engineer proteins. Curr Opin Struct Biol. 2010;20:377–384. doi: 10.1016/j.sbi.2010.02.004. [DOI] [PubMed] [Google Scholar]