

Abstract
Since 1909, the cancer immunosurveillance concept has undergone four distinct eras. These include a general acceptance during 1957-1974, an abandonment during 1974-1996, resurrection during 1996-2001 in the form of an elegant theory of tumour immunoediting proposed by Robert Schreiber, and a retreat since 2006. Recently, in the Journal of Pathology, Ciampricotti et al reported an elegant experimental model designed by establishing RAG2−/−/MMTV-NeuT mice. Using this, they demonstrated that the development and metastasis of HER-2/neu-positive spontaneous mammary carcinoma were not altered by the presence or absence of the adaptive immune system. Their fascinating results are a call to revisit controversial reports as to an effective role of the adaptive immune system in tumour inhibition versus tumour promotion or tolerance in the development of spontaneous carcinomas. Ciampricotti and colleagues present a strong case for revising our ideas of cancer immunoediting and appreciating the complexity of the interaction between cancer and the immune system.
Keywords: tumour immunosurveillance, tumour immunoediting, breast cancer, tumour escape, tolerance
The concept of cancer immunosurveillance had a chequered history before it was developed into a cancer immunoediting theory by Robert Schreiber [1]. Recently, Ciampricotti and colleagues [2] provided evidence that challenged the cancer immunoediting theory by establishing RAG2−/−/MMTV-NeuT transgenic mice and revealing that adaptive immune response is not involved in promoting or inhibiting spontaneous tumour development. They also demonstrated that the inflammatory tumour microenvironment is not associated with adaptive immune responses. It can be argued that their experimental system was similar to human cancer, where tumours develop spontaneously. The RAG2−/−/MMTV-NeuT transgenic mouse model of spontaneous breast carcinoma used in their studies may be more clincally relevant than those reports based on transplantable tumour models. Consideration of the history of tumour immunology shows that although the majority of experiments were conducted on transplanted tumour models or chemically induced cancers, there were also spontaneous tumour models that supported the tumour immunosurveillance concept. For instance, the surgeon Bradley Coley (1862–1936) demonstrated that bacterial infection may induce immune responses that could protect patients from cancer. Promising results of the ‘Coley vaccine’ in inducing regression of established tumours provided a key cornerstone of the early history of tumour immunology [3].
The concept that the immune system can recognize and eliminate nascent malignant cells was first conceived in 1909 by Paul Erlich [4] and was later formulated into the cancer immunosurveillance theory by Burnet and Thomas in the late 1950s [5,6]. The theory was mainly supported by the immune-mediated rejection of transplanted tumours induced by chemical carcinogens or viruses in syngeneic mice [7,8]. However, studies in animals with impaired immune systems produced discordant results as to the susceptibility of these mice to spontaneous or chemically induced tumours [9]. In fact, the higher incidence of cancer in immunocompromised hosts was attributed to their inability to clear transforming infectious agents, rather than a lack of the tumour immunosurveillance. The concept of cancer immunosurveillance was later largely abandoned because of the observation that athymic nude mice did not show an increased incidence of spontaneous or chemically induced tumours compared to wild-type animals [10,11]. Surprisingly, Prehn et al proposed that the immune system can promote tumour growth [12]. Retreat from the cancer immunosurveillance concept was then reflected in Thomas's statement that currently available experimental animals cannot show the existence of tumour immunosurveillance [13].
During the 1990s it became evident that nude mice had NK cells and leaky T and B cell function, which could represent some degree of immunosurveillance. Using a different titre of MCA revealed that nude and SCID mice were more susceptible to tumour formation than wild-type (WT) mice [14,15]. During 1994–1998, two key findings raised interest in cancer immunosurveillance theory. First, it was demonstrated that endogenous IFN-γ could protect the host against transplanted and chemically induced tumours [16,17] and spontaneous tumours [18,19]. The second key finding was a greater sensitivity of perforin−/− mice to MCA-induced tumours compared with their WT counterparts [20–23]. RAG2-deficient mice were also more sensitive to MCA-induced sarcomas than WT mice [24]. A higher incidence of virally-induced cancers, such as non-Hodgkin's lymphoma, Kaposi's sarcoma and carcinomas of the skin and genitourinary region, were also reported in immunocompromised or immunodeficient patients [25–29]. An increased incidence of spontaneous cancer after organ transplantation has been reported [30–33], which could support the equilibrium phase of the immunoediting. These observations revived cancer immunosurveillance theory until early 1990s, yet the key question remained: why do cancers occur in immunocompetent individuals?
To answer this question, Robert Schreiber proposed the term ‘cancer immunoediting’ in order to broadly describe the dual host-protecting and tumour-sculpting actions of the immune system that not only surveil for, and eliminate, nascent malignant cells but also shape neoplastic disease through equilibrium and escape mechanisms [1]. On the other hand, Willimsky and Blankenstein challenged the immunoediting concept by demonstrating that immunogenic tumours expressing SV40 T antigen did not escape their recognition but rather induced tolerance in the transgenic mice. They showed that tumours were immunogenic and were rejected by the transgenic mice after vaccination but before spontaneous tumour formation [34]. Spontaneous tumours could suppress effector function of an anti-tumour immune response, so that even vaccination could not induce rejection of transplanted tumours in mice bearing spontaneous tumours (Figure 1, pathway 3). This possibility was supported by the observation that young transgenic mice could also reject some tumour cell lines that were established from spontaneous tumour cells [34]. There was no experiment conducted in the spontaneous tumour-bearing mice to distinguish tumour-induced immune suppression from tolerance after the vaccination.
Figure 1.
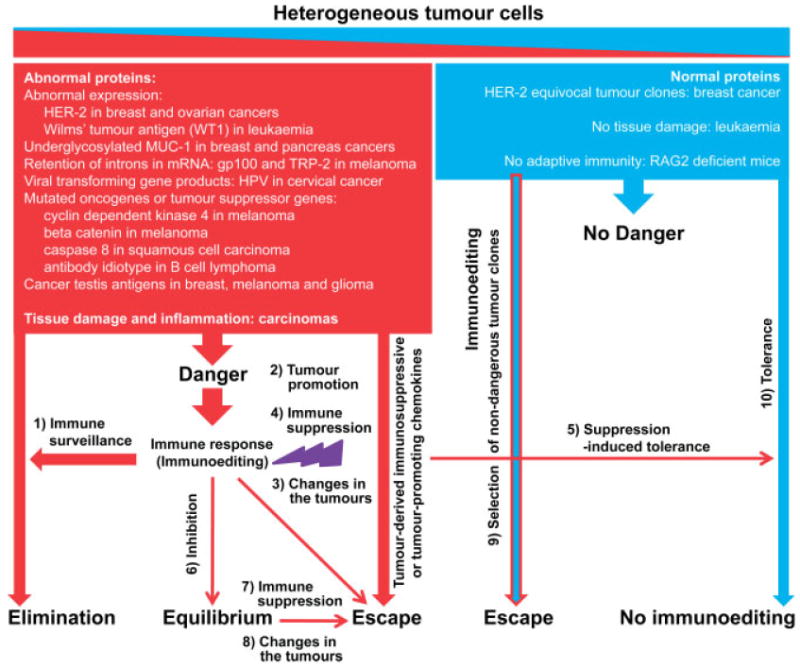
A unified theory of cancer immune complexity. Tumours comprised of a heterogenous population of cells that expands from clones that send danger signals to the immune system (red) to clones that do not appear to be dangerous to the immune system (blue). The balance between these two clones determines the state of tumour immunosurveillance in the host. There are 10 possible pathways depicted here, which describes the interplay between tumour immunoediting and tolerance.
In line with the immune tolerance model, Ciampricotti et al have now demonstrated a similar rate of spontaneous tumour formation in WT NeuT and RAG2−/−/MMTV-NeuT mice [2]. They showed that genetic elimination of the adaptive immune system did not alter the latency, multiplicity, outgrowth and phenotype of mammary tumours. They also reported an increased accumulation of CD11b+ Gr1+ MDSC in the tumour-bearing mice. Such phenomenon has also been reported in the FVBN202 transgenic mouse model of neu-positive mammary carcinoma by our group [35,36] and by others [37]. Therefore, tumour growth may be due to an increased accumulation of MDSC preceding spontaneous mammary tumour formation in WT NeuT mice, which could suppress immunosurveillance (Figure 1, pathway 3) and push the tumours toward a situation in which tolerance is mimicked (Figure 1, pathways 9 and 10).
The data presented by Ciampricotti et al. [2] are in fact an evolution in our understanding of tumour immunology. Although the results apparently contradict the experimental data that support the tumour immunoediting theory, putting all the reports into a unfied picture leads to the notion that tumour cells and cells of the immune system interact in a complex network that is not simply a one-way road (Figure 1). According to this model, tumours are heterogeneous because of the genetic instability and therefore each tumour cell may communicate differently with cells of the adaptive immune system. Whereas dangerous clones (red colour) elicit immune responses that may trigger tumour immunosurveillance, non-dangerous clones (blue colour) fail to do so and may favour the immune tolerance. The clonal composition of the tumours determines the balance between immunoediting and tolerance. However, most tumours, including HER-2/neu over-expressing carcinomas, melanoma or SV40 T antigen, contain a higher frequency of dangerous clones. Therefore, they are expected to fall into the immunoediting category. Even in such cases, the quality of the immune response determines the outcome. While the specific type of immune response may facilitate spontaneous regression of melanoma or reduce the incidence of cancer in immunocompetent hosts compared to immunodeficient individuals (pathway 1), other immune response types, particularly M2 macrophages, may exhibit tumour-promoting effects (pathway 2) or induce epigenetic changes in the tumours and result in tumour escape and relapse (pathway 3). It has been reported that IFN-γ-producing T cells simultaneously induce apoptosis and antigen loss in the tumour cells, which results in initial tumour rejection but eventual tumour relapse [38–40].
Tumour clones that express immunosuppressive chemokines or receptors may suppress the immunoediting process (pathway 4) and lead to the state of immune tolerance (pathway 5). Moderate effector cells of the immune system or less invasive tumour clones may result in the state of tumour inhibition and equilibrium phase (pathway 6). These clones may form invasive cancer in elderly or immunocompromised patients (pathway 7) or may undergo epigenetic changes as a result of a chronic immune pressure and relapse (pathway 8). Tumours may also be selected under the immune pressure, such that less dangerous clones may survive and form cancer (pathway 9). Cancers with a dominant population of non-dangerous clones may appear silent to the immune system and result in tolerance (pathway 10). Some pathways depicted in Figure 1 have already been reported [40–42]. It can be argued that tumours utilize complex pathways rather than a single pathway in order to survive in an immunologically hostile microenvironment. The discordant results in the field also imply the existence of different pathways, which may be complementary rather than conflicting. Therefore, rather than focusing on a single pathway, we need to understand the complexity of these pathways. The proposed unified theory of cancer immune complexity is an effort toward a better understanding of the interaction between cells of the immune system and cancer cells.
Although cancer immunoediting theory can explain a number of observations in cancer patients, it has mainly been developed based on the murine model. The overall structure of the immune system in mice and humans is quite similar [43]. Therefore, results obtained from the murine model indicate the need for understanding the diverse interactions between tumour cells and cells of the host immune system in humans. However, taking into consideration the differences between the mouse model and humans, the tumour immunoediting concept may not provide a comprehensive understanding of tumour immunology in humans. For example, there are several differences in FcR expression between mice and humans [44,45]. Given that FcR represents a link between the adaptive immune system and the innate immune system, such differences could regulate different responses. In addition, the mutation of key signalling molecules in T cells has markedly different effects in mice and humans. Deletion or mutation in common γ-chain cytokine receptors results in severe immunological defects, which differ between human and mouse XSCID [46,47]. Therefore, we propose to develop a unified theory of tumour immune complexity that appreciates the differences among species. Understanding the diversity of interactions between malignant cells and the immune system in humans would require an improvement in our immunomonitoring tools so as to determine the extent to which research results from the murine model can be applied to humans.
Acknowledgments
This work was supported by NIH (Grant No. R01 CA104757) and the Virginia Commonwealth University (VCU) Presidential Research Incentive Program (Grant No. 145820), both to MH Manjili. Animal studies conducted by my group and outlined in the present paper have been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at VCU.
Footnotes
Invited commentary for Ciampricotti M et al. Development of metastatic HER2+ breast cancer is independent of the adaptive immune system.
No conflicts of interest were declared.
References
- 1.Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Ciampricotti M, Vrijland K, Hau CS, et al. Development of metastatic HER2+ breast cancer is independent of the adaptive immune system. J Pathol. 2011;224:56–66. doi: 10.1002/path.2837. [DOI] [PubMed] [Google Scholar]
- 3.Coley WB. The treatment of malignant tumors by repeated innoculations of erysipelas: with a report of ten original cases. Am J Med Sci. 1893;10:487–511. [PubMed] [Google Scholar]
- 4.Ehrlich P. Über den jetzigen stand der karzinomforschung. Ned Tijdschr Geneeskd. 1909;5:273–290. [Google Scholar]
- 5.Burnet FM. Cancer—a biological approach. Br Med J. 1957;1:841–847. doi: 10.1136/bmj.1.5023.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas L. Delayed hypersensitivity in health and disease. In: Lawrence HS, editor. Cellular and Humoral Aspects of the Hypersensitive States. Hoeber-Harper; New York: 1959. pp. 529–532. [Google Scholar]
- 7.Old LJ, Boyse EA. Immunology of experimental tumors. Annu Rev Med. 1964;15:167–186. doi: 10.1146/annurev.me.15.020164.001123. [DOI] [PubMed] [Google Scholar]
- 8.Klein G. Tumor antigens. Annu Rev Microbiol. 1966;20:223–252. doi: 10.1146/annurev.mi.20.100166.001255. [DOI] [PubMed] [Google Scholar]
- 9.Stutman O. Immunodepression and malignancy. Adv Cancer Res. 1975;22:261–422. doi: 10.1016/s0065-230x(08)60179-7. [DOI] [PubMed] [Google Scholar]
- 10.Rygaard J, Povlsen CO. The mouse mutant nude does not develop spontaneous tumors. An argument against immunological surveillance. Acta Pathol Microbiol Scand Microbiol Immunol. 1974;82:99–106. doi: 10.1111/j.1699-0463.1974.tb02299.x. [DOI] [PubMed] [Google Scholar]
- 11.Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science. 1974;183:534–536. doi: 10.1126/science.183.4124.534. [DOI] [PubMed] [Google Scholar]
- 12.Prehn RT. Perspectives on oncogenesis: does immunity stimulate or inhibit neoplasia? J Reticuloendothel Soc. 1970;10:1–16. [PubMed] [Google Scholar]
- 13.Thomas L. On immunosurveillance in human cancer. Yale J Biol Med. 1982;55:329–333. [PMC free article] [PubMed] [Google Scholar]
- 14.Engel AM, Svane IM, Mouritsen S, et al. Methylcholantrene-induced sarcomas in nude mice have short induction times and relatively slow levels of surface MHC class I expression. Acta Pathol Microbiol Immunol Scand. 1996;104:629–639. doi: 10.1111/j.1699-0463.1996.tb04923.x. [DOI] [PubMed] [Google Scholar]
- 15.Engel AM, Svane IM, Rygaard J, et al. MCA sarcomas induced in scid mice are more immunogenic than MCA sarcomas induced in congenic, immunocompetent mice. Scand J Immunol. 1997;45:436–470. doi: 10.1046/j.1365-3083.1997.d01-419.x. [DOI] [PubMed] [Google Scholar]
- 16.Dighe AS, Richards E, Old LJ, et al. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN-γ receptors. Immunity. 1994;1:447–456. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon-γ-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smyth MJ, Thia KY, Street SE, et al. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–760. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Street SE, Trapani JA, MacGregor D, et al. Suppression of lymphoma and epithelial malignancies effected by IFN-γ. J Exp Med. 2000;196:129–134. doi: 10.1084/jem.20020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Street SE, Cretney E, Smyth MJ. Perforin and IFN-γ activities independently control tumor initiation, growth, and metastasis. Blood. 2001;97:192–197. doi: 10.1182/blood.v97.1.192. [DOI] [PubMed] [Google Scholar]
- 21.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 22.van den Broek ME, Ka¨gi D, Ossendorp F, et al. Decreased tumor surveillance in perforin-deficient mice. J Exp Med. 1996;184:1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smyth MJ, Thia KY, Street SE, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shankaran V, Ikeda H, Bruce AT, et al. IFN-γ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 25.Gatti RA, Good RA. Occurrence of malignancy in immunodeficiency diseases. A literature review. Cancer. 1971;28:89–98. doi: 10.1002/1097-0142(197107)28:1<89::aid-cncr2820280117>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 26.Penn I. Posttransplant malignancies. Transpl Proc. 1999;31:1260–1262. doi: 10.1016/s0041-1345(98)01987-3. [DOI] [PubMed] [Google Scholar]
- 27.Birkeland SA, Storm HH, Lamm LU, et al. Cancer risk after renal transplantation in the Nordic countries, 1964–1986. Int J Cancer. 1995;60:183–189. doi: 10.1002/ijc.2910600209. [DOI] [PubMed] [Google Scholar]
- 28.Sheil AGR. Cancer after transplantation. In: Morris PJ, editor. Kidney Transplantation. Saunders; Philadelphia, PA: 2001. pp. 558–570. [Google Scholar]
- 29.Boshoff C, Weiss R. AIDS-related malignancies. Nat Rev Cancer. 2002;2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 30.Sheil AG. Cancer after transplantation. World J Surg. 1986;10:389–396. doi: 10.1007/BF01655298. [DOI] [PubMed] [Google Scholar]
- 31.Penn I. Malignant melanoma in organ allograft recipients. Transplantation. 1996;61:274–278. doi: 10.1097/00007890-199601270-00019. [DOI] [PubMed] [Google Scholar]
- 32.Penn I. Sarcomas in organ allograft recipients. Transplantation. 1995;60:1485–1491. doi: 10.1097/00007890-199560120-00020. [DOI] [PubMed] [Google Scholar]
- 33.Pham SM, Kormos RL, Landreneau RJ, et al. Solid tumors after heart transplantation: lethality of lung cancer. Ann Thorac Surg. 1995;60:1623–1626. doi: 10.1016/0003-4975(95)00120-4. [DOI] [PubMed] [Google Scholar]
- 34.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 35.Kmieciak M, Morales JK, Morales J, et al. Danger signals and nonself entity of tumor antigen are both required for eliciting effective immune responses against HER-2/neu positive mammary carcinoma: implications for vaccine design. Cancer Immunol Immunother. 2008;57:1391–1398. doi: 10.1007/s00262-008-0475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worschech A, Kmieciak M, Knutson KL, et al. Signatures associated with rejection or recurrence in HER-2/neu-positive mammary tumors. Cancer Res. 2008;68:2436–2446. doi: 10.1158/0008-5472.CAN-07-6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeuchi N, Hiraoka S, Zhou XY, et al. Anti-HER-2/neu immune responses are induced before the development of clinical tumors but declined following tumorigenesis in HER-2/neu transgenic mice. Cancer Res. 2004;64:7588–7595. doi: 10.1158/0008-5472.CAN-04-1081. [DOI] [PubMed] [Google Scholar]
- 38.Beatty GL, Paterson Y. IFN-γ can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J Immunol. 2000;165:5502–5508. doi: 10.4049/jimmunol.165.10.5502. [DOI] [PubMed] [Google Scholar]
- 39.Le Poole IC, Riker AI, Quevedo ME, et al. Interferon-γ reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. Am J Pathol. 2002;160:521–528. doi: 10.1016/s0002-9440(10)64871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kmieciak M, Knutson KL, Dumur CI, et al. HER-2/neu antigen loss and relapse of mammary carcinoma are actively induced by T cell-mediated anti-tumor immune responses. Eur J Immunol. 2007;37:675–685. doi: 10.1002/eji.200636639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reiman JM, Kmieciak M, Manjili MH, et al. Tumor immunoediting and immunosculpting pathways to cancer progression. Semin Cancer Biol. 200;17:275–287. doi: 10.1016/j.semcancer.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morales JK, Kmieciak M, Graham L, et al. Adoptive transfer of HER2/neu-specific T cells expanded with alternating γ-chain cytokines mediate tumor regression when combined with the depletion of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2009;58:941–953. doi: 10.1007/s00262-008-0609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49–71. doi: 10.1016/s0300-483x(03)00043-x. [DOI] [PubMed] [Google Scholar]
- 44.Monteiro RC, Van De Winkel JG. IgA Fc receptors. Annu Rev Immunol. 2003;21:177–204. doi: 10.1146/annurev.immunol.21.120601.141011. [DOI] [PubMed] [Google Scholar]
- 45.Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 46.Leonard WJ. Type I cytokines and interferons and their receptors. In: Paul WE Jr, editor. Fundamental Immunology. 4th. Lippincott-Raven; Philadelphia, PA: 1999. pp. 741–774. [Google Scholar]
- 47.Fischer A, Cavazzana-Calvo M, De Saint Basile G, et al. Naturally occurring primary deficiencies of the immune system. Annu Rev Immunol. 1997;15:93–124. doi: 10.1146/annurev.immunol.15.1.93. [DOI] [PubMed] [Google Scholar]