

Abstract
We assess the performance of our previously reported structure-based support vector machine target-specific scoring function across 41 targets, 40 among them from the Directory of Useful Decoys (DUD). The area under the curve of receiver characteristic plots (ROC-AUC) revealed that scoring with SVMSP resulted in consistently better enrichment over all targets families and outperforming Glide and other scoring functions, most notably among kinases. In addition, SVM-SP performance showed little variation among protein classes, exhibited excellent performance in a test case using a homology model, and in some cases showed high enrichment even with few structures used to train a model. We put SVM-SP to the test by virtual screening 1,125 compounds against two kinases, EGFR and CaMKII. Among the top 25 EGFR compounds, three compounds (1–3) inhibited kinase activity in vitro with IC50 of 58, 2, and 10 μM. In cell culture, compounds 1–3 inhibited non-small cell lung carcinoma (H1299) cancer cell proliferation with similar IC50 values for compound 3. For CaMKII, one compound inhibited kinase activity in a dose-dependent manner among 20 tested with an IC50 of 48 μM. These results are encouraging given that our in-house library consists of compounds that emerged from virtual screening of other targets with pockets that are different from typical ATP binding sites found in kinases. In light of the importance of kinases in chemical biology, these findings could have implications in future efforts to identify chemical probes of kinases within the human kinome.
INTRODUCTION
The objective of virtual screening1 methods is the identification of compounds that bind and modulate the function of their target. Virtual screening efforts now routinely lead to active compounds.1,2 Typically, 104–106 compounds are screened and about 102 of the highest scoring ligands are acquired for subsequent experimental elimination of false positives. Compound databases like ZINC (http://docking.zinc.org) have significantly facilitated this process.3,4 In virtual screening, these compounds are docked to the target of interest to generate three-dimensional structures of receptor-ligand complexes. Compounds are then rank-ordered in a step known as scoring, which attempts to replicate the trend observed if the compounds were ranked by their experimentally measured binding affinity. Scoring functions can be classified as empirical,5–7 knowledge-based,8 and force field-based.9 Identifying the scoring function method optimal to the particular target of interest is a continuing challenge.10,11 The performance of scoring functions varies significantly depending on the target.12,13 This receptor-dependence is reflected in the constant stream of new scoring functions that are developed in an effort to improve upon previously-derived scoring functions.
Here we expand on a target-specific Support Vector Machine (SVM) scoring method that we have previously reported14 and assess its performance on 41 targets, 40 among them from the Directory of Useful Decoys (DUD) validation set.15 The difference with our earlier approach is that we have used our own pair potentials rather than the DFIRE pair potentials. The DFIRE potentials were obtained from a set of 200 protein-ligand co-crystal structures. However, our dataset consisted of 2,018 co-crystal structures. In addition, the number of features (descriptors) included in a vector was reduced from 190 to 135 by excluding atom pairs that are not observed, such as metal-metal atom pairs for example. The scoring functions are tailored to each target through the use of support vector machine (SVM)16 algorithm that is trained with statistical knowledge-based data obtained from three-dimensional structures of receptor-ligand complexes that originate from a positive and a negative set. In this work, we develop our own pair potentials to serve as features for the derivation of the SVM models using a positive set consisting of x-ray structures of compounds bound to their target, and a negative set consisting of decoy molecules docked to the target for which a scoring function is being developed. This ensures that the resulting SVM model will favor molecules that are native-like and bind to the target rather than molecules that adopt binding modes of inactive compounds (decoys). We further test the method by screening an in-house library of 1,125 compounds in search for inhibitors of a receptor tyrosine kinase, epidermal growth factor receptor (EGFR),17 and a serine/threonine kinase, namely calcium-calmodulin-dependent protein kinase II (CaMKII).18 The top compounds are tested for activity using in vitro assays. Inhibitors of EGFR are further tested in cell culture for inhibition of a non-small cell lung carcinoma (NSCLC) cell line (H1299) proliferation.
Results
Developing an SVM Model
The derivation of a successful SVM model depends on the careful choice of objects that comprise the training set, and the features extracted from these objects. While a number of efforts have employed SVM to rank compounds,19–23 our approach is a significant departure from these methods in several respects. First, our SVM models are trained using three-dimensional structures of protein-ligand complexes. Second the features used to derive the SVM algorithms consist of pair potentials obtained following the approach used to derive knowledge-based statistical scoring function. Third, our method combines tailoring of scoring functions with machine learning.
The process of generating an SVM model begins with the creation of a training dataset that consists of a negative and a positive set. Features extracted from each of these sets train the SVM algorithm. In the context of structure-based molecular design, negative and positive sets are defined as a collection of inactive and active compounds bound to their target, respectively. SVM represents each complex as an N-dimensional vector in space, with N corresponding to the number of features. During training, the SVM algorithm will attempt to find a hyperplane that separates these points in hyperspace.
As mentioned above, our approach is unique as we define training set objects as three dimensional structures of receptor-ligand complexes. To select features, we rationalized that pair potentials that are the basis of common knowledge-based statistical functions such as PMF8 and DFIRE24 are the best option, as they are able to capture the nuances of the receptor-ligand interaction as we reported previously.14 In this work, however, as described in the Supporting Information, we derived our own pair potentials. The selection of pair potentials as features offers a number of advantages, such as obviating the need to compute physico-chemical terms like solvation, entropy and enthalpy terms, which can be time-consuming and result in large errors.
The next step in deriving an SVM model is to create the two sets of objects that will constitute the training set. We focus on the training set as a means to tailor the SVM algorithm to each individual target. In the context of developing target-specific scoring functions to distinguish between active and decoy molecules, an ideal positive set would consist of a priori known active molecules, while the negative set would consist of decoy molecules bound to the target of interest. While obtaining structures of decoy molecules bound to a receptor can be achieved in a straightforward manner with molecular docking, it is typically not the case that a sufficiently large set of three dimensional structures of active compounds bound to the target of interest is available. In an effort to tailor the scoring function to its target, we defined the positive set to consist of a collection of structures that shared strong homology to the target protein. The SVM models derived with this approach are referred as SVM-SP. Further efforts to tailor the scoring functions to their receptors led us to dock 5,000 decoy molecules were docked onto the target receptor as described in more detail in the Supporting Information. We have found that 5,000 decoy molecules resulted in best performance. The expectation is that the algorithm will be trained to disfavor molecules that adopt decoy-like binding modes to the target.
Assessing Performance of SVM-SP
The Directory of Useful Decoys (DUD) provides a useful list of targets along with a library containing a set of known ligands and decoy molecules for validation of the SVM scoring methods. The ratio of decoy to ligand for each target is 36. A tool that is commonly used to assess the performance of a scoring function is the receiver operating characteristic (ROC) plot.25 An ROC curve is constructed by ranking the docked complexes, selecting a set of compounds starting from the highest scoring compounds, and counting the number of active compounds. This process is repeated a number of times for a gradually increasing set of compounds selected from the ranked list. In an ROC plot, the farther away the curve is from the diagonal, the better the performance of the scoring function. The area under the ROC curve, which we refer as ROC-AUC, can also be used as a representation of the performance of the scoring function. A perfect scoring function will result in an area under the curve of 1, while a random scoring function will have an ROC-AUC of 0.5.
Following the docking of each set of decoys and ligands to its target within DUD using the program AutoDock426 the resulting complexes were scored with SVM-SP, ChemScore,5 GoldScore,6 PMF,27 X-Score,28 DFIRE29 and AutoDock.26 The Glide program was used for the docking to generate the Glide scores. Figure 1A provides the mean ROC-AUCs for each scoring function obtained over all 40 targets within DUD for SVM-SP scoring and other scoring functions. The results reveal that SVMSP showed best overall performance with a mean ROC-AUC of 0.80, followed by Glide (0.68), ChemScore (0.66), GoldScore (0.64), AutoDock (0.64), PMF (0.58) and DFIRE (0.49). To gain insight into the performance of scoring functions for individual targets, ROC-AUCs of DUD proteins are shown in a color-coded map in Fig. 1B. It is worth mentioning that the better performance observed by SVM-SP is not completely unexpected since the scoring function is tailored to each target. It is possible that better enrichment would have also been observed for other scoring functions had DUD targets been included in their training sets.
Figure 1.

(a) Mean values for ROC-AUC scores and (b) ROC-AUC values for 41, 40 among them from the DUD validation set.
The performance of the scoring functions was also assessed by protein family also shown in Fig. 1A and B. The 41 targets are classified into six categories, namely serine proteases, nuclear hormone receptors, kinases, serine proteases, metalloenzymes, folate enzymes and “other enzymes”. The mean ROC-AUC for each family of proteins is shown in Fig. 1A and B. SVM-SP scoring performs consistently well for each class of proteins with ROC-AUC values 0.75 or greater in five of the six classes. It was also encouraging that the performance of SVM-SP did not vary significantly from one family to another, showing less target dependence than other scoring functions. The lack of variation may be attributed to the customization aspect of SVM-SP.
Among kinases, the enrichment levels of SVM-SP are particularly noteworthy with a mean ROC-AUC of 0.83. Most scoring functions showed mean ROC-AUC values of 0.66 or less, confirming what was previously known, which is that ranking compounds docked to ATP sites in kinases is a challenge. Among the 9 kinases, ROC-AUC values from SVM-SP ranged from 0.69 to a remarkably high 0.98 as shown in Fig. 1B. Among all kinases, highest enrichment was found for EGFR and FGFr1 kinases with ROC-AUC of 0.98 and 0.91, respectively. The lowest ROC-AUC among kinases was 0.69 for CDK2. It is interesting to note that the number of complexes used in the positive set to derive the SVM-SP model for each target was not a factor in performance. For example, enrichment in HSP90 was higher than in CDK2 (0.78 versus 0.68), even though around 450 structures were used to derive SVM-SP for CDK2, compared to 50 for the former (Table S1). Remarkably only 4 structures were used to develop an SVM-SP model for TK, and this enzyme's ROC-AUC score was on par with those of other kinases. Finally, the performance of SVM-SP for the homology model of PDGFRβ is worth noting. The ROC-AUC of SVM-SP for the target is 0.87, compared with 0.63 for Gold and 0.44 for Glide. These results are highly encouraging as they suggest that SVM-SP can perform well even in the absence of a high resolution crystal structure and can be applied to search for inhibitors for the large number of kinases in the human kinome whose structures have yet to be solved.
Among the “other enzymes” class, SVM-SP demonstrated excellent performance with ROCAUC scores greater than 0.9 in four out of 15 enzymes, namely GPB (glycogen phosphorylase β), NA (neuraminidase), PNP (purine nucleoside phosphorylase) and SAHH (S-adenosyl-homocysteine hydrolase). The lowest enrichment was found in ACHE and COX-1 (ROC-AUC = 0.54 and 0.58 respectively).
In sum, it was encouraging that SVM-SP performed better than all scoring functions across all DUD targets, especially among kinases, showed little variation across the protein classes, exhibited excellent performance on a homology model, and performed very well even when trained on a few structures in the positive set.
Virtual Screening, Biochemical Characterization and Assessment in Cell Culture
The particularly good performance of SVM-SP in ranking compounds docked to kinases prompted us to assess whether the scoring function can identify inhibitors of these enzymes in a chemical database. Two kinases were chosen for this purpose, namely the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase, and calcium-calmodulin-dependent protein kinase II (CaMKII), a serinethreonine kinase. The former is an important target in cancer, including currently FDA-approved drugs such as erlotinib. The latter has been suggested as a target for treatment of various neurological disorders.
An in-house library of 1,125 compounds was docked to the ATP binding site of EGFR (PDB code 1M17) using the docking program AutoDock 4 (Details are provided in the Supporting Information). The resulting three-dimensional structures of compounds bound to EGFR were scored with SVM-SP that was developed for the enzyme for the validation work described above. Since our in-house library contained a number of closely related structures, the top 100 compounds were clustered to ensure the structural diversity of compounds selected. A representative structure from each of the top 25 clusters was selected. These 25 compounds were tested for inhibition of EGFR kinase activity at a concentration of 25 μM initially using a FRET-based assay described in detail in the Supporting Information. The concentration of ATP was used near-Km at 11.5 μM and 0.05% BRIJ-35 was used to prevent aggregation. Four compounds were found to inhibit kinase activity of EGFR at a level of 25% or greater. A follow-up concentration-dependent study was conducted, and three compounds showed inhibition in a dose-dependent manner. These compounds ranked 4, 8 and 18, respectively, among the top 25 compounds. The concentration of compound that led to 50% inhibition (IC50) was estimated at 2, 10, and 56 μM for compounds 1–3, respectively (Fig. 2A). The chemical structure of the three compounds is shown in Fig. 2B. A pairwise comparison of these three structures with the 444 EGFR inhibitors from the DUD dataset using the Babel fingerprint resulted in Tanimoto coefficients equal or less than 0.45, suggesting little similarity and arguing that each compound represents a new class of EGFR inhibitors.
Figure 2.

(a) Dose-dependent inhibition of EGFR (compound 1–3) and CaMKII (compound 4); (b) Chemical structure of compounds 1–4. (c) Effect of compound 3 on H1299 cancer cell proliferation. H1299 cells (1,000 cells per well) were seeded in 96-well plate overnight. Indicated compounds or DMSO only (0.1% vol/vol) were added and incubated for 72 h. IC50 value was calculated by Sigmaplot 11.0 software.
A literature search to determine whether these compounds have been previously used as anti-cancer agents revealed that bromo derivative of 1 was tested previously for anti-proliferative activity in carcinoma cells.20 However, the remaining two compounds are not known for their anti-cancer properties. Compound 2 was a hit in a screen for inhibitors of human fatty acid synthase thioesterase. Compound 3 has not been previously reported as biologically active. We assessed these compounds for their effects on cell growth in a highly invasive non-small cell lung carcinoma (NSCLC) cell line (H1299) that expresses EGFR. A colorimetric assay that follows the reduction of (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to purple formazan is performed in a dose-dependent manner over a period of 3 days. While all three compounds inhibited cell proliferation, compound 1 showed incomplete inhibition (Fig. S1A) likely due to resistance mechanism triggered against this compound. Compound 2 appeared to be inhibiting growth completely (Fig. S1B), but the estimated IC50 of 58 μM is significantly larger than the IC50 measured for inhibition of EGFR kinase activity (2 μM). This can be explained by the negative charge that is making the compound less likely to be cell permeable and does not reach its target as effectively. The lack of complete inhibition for compound 1 could be due to resistance mechanism that the cell develops at higher concentration, possibly by up-regulating of efflux pumps.
We put the SMV-SP scoring method further to the test and docked our in-house library of 1,125 compounds to the ATP binding domain for CaMKII (calcium-calmodulin-dependent protein kinase II); a ubiquitous multifunctional serine/threonine protein kinase activated by calcium-calmodulin. The compounds were docked to the ATP binding site of CaMKII (PDB code 2BDW30); the C-Elegans isoform of CaMKII that has high homology to the predominant isoforms of CaMKII expressed in the brain where it is implicated in learning and memory.18 The small molecule-kinase complexes were scored and ranked using an SMV-SP model tailored for CaMKII. The top 100 candidates were clustered to ensure that highly similar compounds were not selected and the top 20 molecules were tested for CaMKII inhibition. Fewer molecules were selected due to the laborious nature of the assay compared to that of EGFR. The small molecules were not pre-incubated with the kinase and therefore likely reflect a dynamic competition between the compound and ATP. CaMKII activity was measured by quantifying 32P incorporation into a peptide substrate (syntide) using a P81 filter assay to separate phosphorylated peptide from unincorporated [32Pγ]-ATP (described in detail in the Supporting Information). Initially, compounds were screened at a concentration of 50 μM. Although three compounds statistically inhibited CaMKII at 50 μM, only one (compound 4) of these was shown to inhibit CaMKII in a dose-dependent manner (Fig. 2B).
The predicted binding modes of 1–3 are compared to erlotinib (EGFR IC50 < 10 nM) in Fig. 3. Erlotinib exploits the entire substrate binding site, including a deep pocket for its 3-ethynylaniline ring, as well as a long cavity for its bis-(methoxyethoxy) substituted quinazoline. All four compounds are predicted to use this same cavity of the ATP binding site: compound 1 through its benzofuran moiety, 2 through the N-acyl substituted 4-chloroanthranilate, and 3 through its dihydroquinoline. It is expected that these compounds are type I kinase inhibitors as they bind strictly to the ATP binding pocket, but conclusive evidence will emerge from future structural or further biochemical studies.
Figure 3.
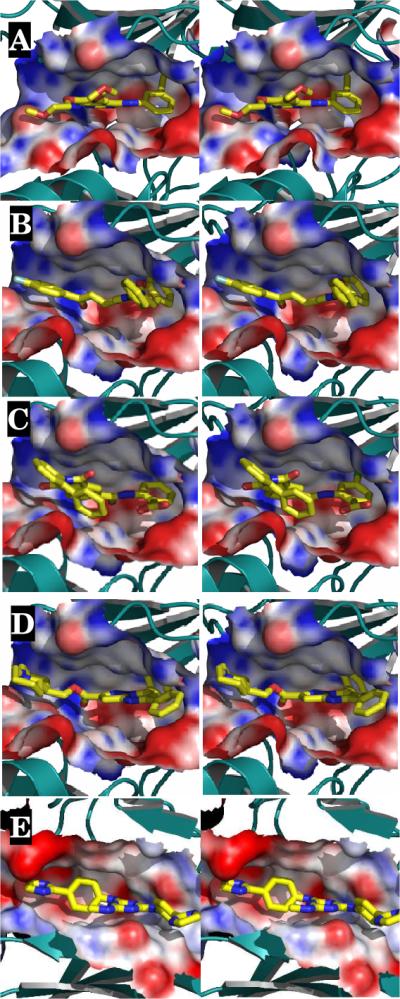
(a) Stereoview of the three-dimensional structures of compounds (A) erlotinib; and compounds (B) 1; (C) 2; (D) 3; and (E) 4. The target is shown in solvent-accessible surface area and color-coded by electrostatic potential. Red, blue and white correspond to negative, positive and neutral charge, respectively.
The effectiveness of our SVM-SP scoring method is strongly suggested by the discovery of three separate μM hits against the EGFR receptor within an in-house library of 1,125 compounds. Considering that our in-house library consisted of compounds selected by a previous virtual screening effort targeted to binding cavities at protein interfaces that are unlike ATP-binding pockets, the discovery of compounds within this set having a good potency is highly encouraging. This approach is one with general value for the virtual screening identification of hit structures for kinases.
Supplementary Material
ACKNOWLEDGMENT
The research was supported by the NIH (CA135380 and CA135380) and the INGEN grant from the Lilly Endowment, Inc (SOM). Computer time on the Big Red supercomputer at Indiana University is funded by the National Science Foundation and by Shared University Research grants from IBM, Inc. to Indiana University. We thank the Lungs for Life for a Fellowship to LL. We are grateful to Jed F. Fisher for his reading of the manuscript and valuable comments.
Footnotes
SUPPORTING INFORMATION. Supporting information constrains computational procedures for the calculations reported in the text.
REFERENCES
- (1).DeGraw AJ, Keiser MJ, Ochocki JD, Shoichet BK, Distefano MD. J. Med. Chem. 2010;53:2464. doi: 10.1021/jm901613f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7455. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kumar BV, Kotla R, Buddiga R, Roy J, Singh SS, Gundla R, Ravikumar M, Sarma JA. J. Mol. Model. 2011;17:151. doi: 10.1007/s00894-010-0701-0. [DOI] [PubMed] [Google Scholar]
- (4).Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC. Nat. Chem. Biol. 2009;5:220. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. J. Comput. Aid. Mol. Des. 1997;11:425. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- (6).Jones G, Willett P, Glen RC, Leach AR, Taylor R. J. Mol. Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- (7).Huey R, Morris GM, Olson AJ, Goodsell DS. J. Comp. Chem. 2007;28:1145. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- (8).Muegge I, Martin YC. J. Med. Chem. 1999;42:791. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- (9).Huang N, Kalyanaraman C, Irwin JJ, Jacobson MP. J. Chem. Inf. Model. 2006;46:243. doi: 10.1021/ci0502855. [DOI] [PubMed] [Google Scholar]
- (10).Jain AN. Curr. Protein Pept. Sc. 2006;7:407. doi: 10.2174/138920306778559395. [DOI] [PubMed] [Google Scholar]
- (11).Shoichet BK. Nature. 2004;432:862. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pham TA, Jain AN. J. Comput. Aid. Mol. Des. 2008;22:269. doi: 10.1007/s10822-008-9174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Seifert MHJ. J. Comput. Aid. Mol. Des. 2009;23:633. doi: 10.1007/s10822-009-9276-1. [DOI] [PubMed] [Google Scholar]
- (14).Li LW, Li J, Khanna M, Jo I, Baird JP, Meroueh SO. ACS Med. Chem. Lett. 2010;1:229. doi: 10.1021/ml100031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Huang N, Shoichet BK, Irwin JJ. J. Med. Chem. 2006;49:6789. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cortes C, Vapnik V. Mach. Learn. 1995;20:273. [Google Scholar]
- (17).Herbst RS. Int. J. Radiat. Oncol. Biol. Phys. 2004;59:21. [Google Scholar]
- (18).Hudmon A, Schulman H. Annu. Rev. Biochem. 2002;71:473. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- (19).Chae MH, Krull F, Lorenzen S, Knapp EW. Proteins. 2010;78:1026. doi: 10.1002/prot.22626. [DOI] [PubMed] [Google Scholar]
- (20).Ballester PJ, Mitchell JB. Bioinformatics. 2010;26:1169. doi: 10.1093/bioinformatics/btq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seifert MH. J. Comput. Aided. Mol. Des. 2009;23:633. doi: 10.1007/s10822-009-9276-1. [DOI] [PubMed] [Google Scholar]
- (22).Martin O, Schomburg D. Proteins. 2008;70:1367. doi: 10.1002/prot.21603. [DOI] [PubMed] [Google Scholar]
- (23).Catana C, Stouten PF. J. Chem. Inf. Model. 2007;47:85. doi: 10.1021/ci600357t. [DOI] [PubMed] [Google Scholar]
- (24).Zhang C, Liu S, Zhu QQ, Zhou YQ. J. Med. Chem. 2005;48:2325. doi: 10.1021/jm049314d. [DOI] [PubMed] [Google Scholar]
- (25).Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO. J. Med. Chem. 2005;48:2534. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- (26).Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J. Comp. Chem. 1998;19:1639. [Google Scholar]
- (27).Muegge I, Martin YC, Hajduk PJ, Fesik SW. J. Med. Chem. 1999;42:2498. doi: 10.1021/jm990073x. [DOI] [PubMed] [Google Scholar]
- (28).Wang R, Lai L, Wang S. J. Comput. Aided Mol. Des. 2002;16:11. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- (29).Zhang C, Liu S, Zhou H, Zhou Y. Protein Sci. 2004;13:400. doi: 10.1110/ps.03348304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Cell. 2005;123:849. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.