

Abstract
Hydrosilation of a variety of ketones and aldehydes using the cationic iridium catalyst, (POCOP)Ir(H)(acetone)+, 1, (POCOP = 2,6-bis(di-tert-butyl phosphinito)phenyl) is reported. With triethyl silane, all but exceptionally bulky ketones undergo quantitative reactions employing 0.5 mol% catalyst in 20-30 min at 25 °C. Hydrosilation of esters and amides results in over-reduction and cleavage of C-O and C-N bonds, respectively. The diastereoselectivity of hydrosilation of 4-tert-butyl cyclohexanone has been examined using numerous silanes and is highly temperature dependent. Using EtMe2SiH, analysis of the ratio of cis:trans hydrosilation products as a function of temperature yields values for ΔΔH‡ (ΔH‡ (trans) - ΔH‡ (cis)) and ΔΔS‡ (ΔS‡ (trans) - ΔS‡(cis)) of -2.5 kcal/mol and -6.9 e.u., respectively. Mechanistic studies show that the ketone complex, (POCOP)Ir(H)(ketone)+, is the catalyst resting state and is in equilibrium with low concentration of the silane complex, (POCOP)Ir(H)(HSiR3)+. The silane complex transfers R3Si+ to ketone forming the oxocarbenium ion, R3SiOCR’2+, which is reduced by the resulting neutral dihydride 3, (POCOP)Ir(H)2, to yield product R3SiOCHR’2 and (POCOP)IrH+ which closes the catalytic cycle.
Introduction
Hydrosilation of carbonyl functionalities is an extensively explored and widely used synthetic methodology.1 This process provides an alternative to hydride reductions of ketones and aldehydes as well as a convenient one-step process for converting these substrates directly to protected alcohols which circumvents the normal two step procedure, reduction to alcohol followed by silyl protection. Several different hydrosilation mechanisms have been shown to operate, dependent on the nature of the catalyst. Late metal catalysts typically proceed through a “Chalk-Harrod” pathway in which the key step involves oxidative addition of the silane to a low valent metal center.2 Early metal catalysts where oxidative addition is disfavored, proceed via sigma bond metathesis mechanisms.3
Recently, high valent metal oxo complexes have been shown to serve as hydrosilation catalysts. Abu-Omar reported that the Re(O)(hoz)2+ (hoz = 2-(2’-hydroxyphenyl)-2-oxazoline) catalyst operates via a sigma bond metathesis mechanism.3m-o Through extensive mechanistic studies Toste and Bergman established that hydrosilations catalyzed by (PPh3)2Re(O)2I occur by a unique mechanism which involves addition of the silane across the Re=O bond, insertion of the carbonyl functionality into the resulting Re-H bond and elimination of the hydrosilation product which closes the cycle and regenerates the Re=O bond.3g-j Piers has reported that Ph3SiH in combination with catalytic amounts of (C6F5)3B achieves hydrosilation of carbonyl compounds.3b,4a-b Surprisingly, although the carbonyl compounds exhibit much higher binding affinities to (C6F5)3B than Ph3SiH, the mechanism involves activation of the silane through coordination to (C6F5)3B, transfer of Ph3Si+ to the carbonyl functionality, and reduction of the resulting Ph3SiOC(R)(R’)+ by (C6F5)3BH- (Scheme 1).
Scheme 1.

We have recently reported reduction of R-X bonds (-X = -Cl, –Br, -I, -OR) using the silane complex 2, a potent R3Si+ donor.5 The mechanism was shown to involve transfer of R3Si+ to X to generate R3Si-X-R+ followed by hydride reduction of this species by the iridium dihydride complex, 3 formed upon silyl transfer (See Scheme 2).
Scheme 2.

The most convenient precatalyst was found to be the stable, crystalline, easily isolated acetone complex, 1. Treatment with excess triethylsilane rapidly generates silane complex 2 in situ and one equivalent of Et3SiOCH(CH3)2, the acetone hydrosilation product (eq. 1). These observations suggested
 |
(1) |
that 2 should function as a hydrosilation catalyst. We report here synthetic and mechanistic details of the catalytic hydrosilation of a variety of carbonyl-containing compounds employing this system which proves to be exceptionally active and highly efficient.
Results and Discussion
Hydrosilation of Carbonyl-Containing Substrates Catalyzed by 1
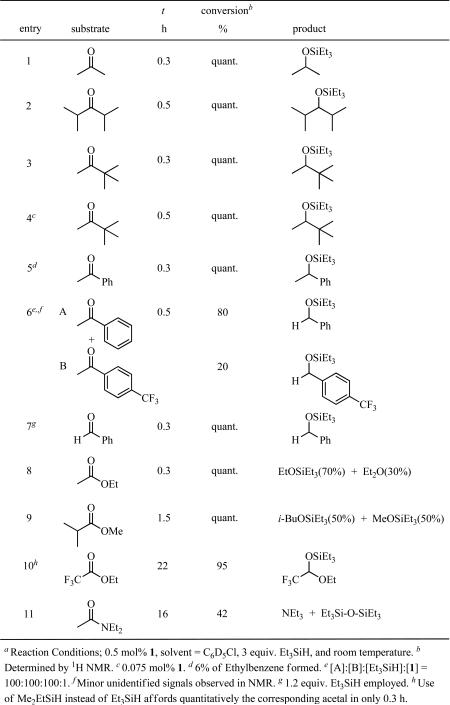
Complex 15a together with Et3SiH initiates hydrosilations of ketones, aldehydes, esters, and amides at room temperature. Results of hydrosilation of several carbonyl-containing substrates are summarized in Table 1. Ketones undergo hydrosilation rapidly to attain quantitative conversions in 0.3- 0.5 h at room temperature (200 TOs) (Table 1, entries 1-6). Hydrosilations of ketones bearing exceptionally bulky alkyl substituents, diisopropyl ketone (entry 2) and methyl tert-butyl ketone (entry 3) are rapidly and quantitatively achieved. The exceptional efficiency of the hydrosilation is illustrated by the quantitative hydrosilation of methyl tert-butyl ketone in 30 min using a 0.075 mol% catalyst loading (1330 TOs, entry 4). Hydrosilation of acetophenone yields 94% of the corresponding silyl alkyl ether and 6% of ethylbenzene, which results from further cleavage of the silyl alkyl ether (entry 5). The competitive hydrosilation reaction between acetophenone and 4’-(trifluoromethyl)acetophenone (entry 6) shows that the activity of the more basic acetophenone is ca. 4 times greater than that of 4’-(trifluoromethyl)acetophenone. Hydrosilation of aldehydes with excess silane often results in secondary cleavage of the resultant silyl alkyl ether.5b However, using just over 1 equiv. of Et3SiH, benzaldehyde undergoes quantitative hydrosilation in 0.3 h to yield the silyl benzyl ether (entry 7).
Table 1.
Hydrosilation of carbonyl functions by 1a
|
Complex 1 catalyzes the hydrosilation of ethyl acetate to afford silyl ethyl ether and diethyl ether in 2:1 ratio (entry 8). This results from formation of the acetal and subsequent cleavage of either of the CO bonds as shown in eq. 2 below.
 |
(2) |
In contrast, the hydrosilation of methyl isobutyrate exhibits only cleavage of the C-OMe bond of the acetal to give only isobutyl silyl ether and methyl silyl ether (entry 9). This selectivity can be attributed to a sterically demanding environment around oxygen in the C--OSiEt3 bond. Introduction of an electron-withdrawing group into the α-position of the ester enables a single hydrosilation even with excess Et3SiH to selectively form the corresponding acetal (entry 10). The preference for single hydrosilation is likely due to the decreased basicity of the acetal formed via initial hydrosilation and the decreased tendency for ionization of the silated acetal.
N,N-Diethyl acetamide is slowly hydrosilated (42% conversion in 16 h) to give Et3N together with the disiloxane (entry 11). After 16 h, no further consumption of the amide is observed. The retarded hydrosilation may result from the deactivation of the electrophilic catalytic Ir species by excess Et3N formed during reaction. The sequence responsible for product formation is shown below (eq. 3) and is similar to that proposed for ester reduction.
| (3) |
Stereochemistry of Hydrosilation of 4-tert-Butyl Cyclohexanone
Reductions of 4-tert-butyl cyclohexanone by various metal hydrides, especially LiAlH4 and NaBH4, have been extensively studied6 and found to preferentially give the trans-alcohol via an axial attack of the hydride (eq. 4, path a). Several explanations have been advanced for this diastereoselectivity including the straightforward proposition that there are unfavorable eclipsing interactions between the axial C-H bonds at C2 and C6 and the incoming hydride reagent during equatorial attack (eq. 4, path b).7
 |
(4) |
The diastereoselectivity of hydrosilations of cyclohexanones has also received attention. Semmelhack has examined the hydrosilation of 4-tert-butyl cyclohexanone by various di- and trialkylsilanes using classical catalysts (PPh3)3RhCl(I) and (PPh3)3RuCl2(II) which operate by the Chalk-Harrod mechanism.2e Bulky triethyl- and triphenyl silanes provide predominantly the more stable trans silyl ether with up to 95% diastereoselectivity, whereas the less bulky diethyl- and diphenyl silanes give the corresponding silyl ethers with a trans/cis ratio of ca. 50:50. On the other hand, hydrosilation by diphenyl silane using (PPh3)4RhH(I) has been reported to give a 84:16 (trans:cis) ratio of diasteromers.2j A triruthenium carbonyl cluster-catalyzed hydrosilation of 4-tert-butyl cyclohexanone affords predominantly the cis-diastereomer with triethyl silane,2l which contrasts with the results of the hydrosilation by the mononuclear Rh or Ru complexes. Most recently, Toste, et. al., have obtained silyl ethers with a high trans selectivity (>96%) using the dioxorhenium(V) catalyst (PPh3)2Re(O)2I which operates by the non-Chalk-Harrod mechanism described above.3j These studies prompted us to examine the diastereoselectivities of hydrosilation of 4-tert-butyl cyclohexanone using catalyst 1 where product ratios depend on axial vs. equatorial attack of iridium dihydride 3 on the silated ketone (see below).
Results of the hydrosilation of 4-tert-butyl cyclohexanone using various silanes and different solvents are shown in Table 2. In chlorobenzene, there is little difference in diastereoselectivity as the bulk of the silane ranges from dimethyl ethyl silane (trans/cis = 69:31) to methyl diethyl silane (74:26) to triethyl silane (68:32) to dimethyl i-propyl silane (76:24). Dimethyl tert-butyl silane and triisopropyl silane are sufficiently unreactive that the reactions must be carried out at 80 °C; however, quantitative conversions can be obtained and result in 87:13 and 51:49 trans/cis product ratios, respectively. In the case of di-tert-butyl silane, selectivity drops to 57:43, but the hydrosilation products are accompanied by a side reaction to form 39% of the silyl enol ether. (Semmelhack et. al., observed the same side product in the hydrosilation using (EtO)3SiH.2e)
Table 2.
Hydrosilation of 4-tert-butyl cyclohexanone with various alkylsilanes catalyzed by 1a
| catalyst | T | t | conversionb | trans/cisb | |||
|---|---|---|---|---|---|---|---|
| entry | mol% | silane | solvent | °C | h | % | % |
| 1 | 1.0 | EtMe2SiH | CD2Cl2 | 22 | 0.3 | quant. | 55 : 45 |
| 2 | 1.0 | EtMe2SiH | C6D5Cl | 22 | 0.3 | quant. | 69 : 31 |
| 3 | 1.0 | EtMe2SiH | toluene-d6 | 22 | 0.3 | quant. | 75 : 25c |
| 4 | 1.0 | Et2MeSiH | C6D5Cl | 22 | 0.3 | quant. | 74 : 26 |
| 5 | 1.0 | Et3SiH | C6D5Cl | 22 | 0.3 | quant. | 68 : 32 |
| 6 | 1.0 | i-PrMe2SiH | C6D5Cl | 22 | 0.3 | quant. | 76 : 24 |
| 7 | 1.0 | t-BuMe2SiH | C6D5Cl | 80 | 2 | quant. | 87 : 13 |
| 8 | 1.0 | (i-Pr)3SiH | C6D5Cl | 80 | 12 | quant. | 51 : 49 |
| 9 | 1.0 | (t-Bu)2SiH2 | C6D5Cl | 22 | 1.5 | quant. | 57 : 43e |
| 10 | 1.39 | EtMe2SiH | CD2Cl2 | -20 | 0.5 | quant. | 82 : 18f |
| 11 | 1.39 | Et3SiH | CD2Cl2 | -20 | 0.5 | quant. | 88 : 12f |
Reaction conditions: 3 equiv. of silane in C6D5Cl (0.3 ml).
Determined by NMR spectroscopy.
1 is not completely soluble in toluene-d8.
39% of di-tert-butyl silyl enol ether is observed.
4 equiv. of silane used.
Solvent effects on diastereoselectivity are also minimal. Hydrosilation with dimethyl ethyl silane in methylene chloride, chlorobenzene and toluene varies from trans/cis = 55:45 to 69:31 to 75:25, respectively. The most dramatic effect on selectivity is seen with variation in reaction temperature. Using dimethyl ethyl silane in methylene chloride, the selectivity increases from 55:45 at 22 °C to 82:18 at -20 °C, while for triethyl silane the selectivity is 68:22 at 22 °C in chlorobenzene and increases to 88:12 at -20 °C in methylene chloride. Temperature effects are examined in more detail in the next section.
Hydrosilation of 4-tert-Butyl Cyclohexanone: Effect of Reaction Temperature on Diastereoselectivity
Although appreciable efforts have been dedicated to the elucidation of factors affecting the diastereoselectivity of metal hydride reductions of 4-tert-butyl cyclohexanone, there have been limited studies of temperature effects on selectivity.8 Thus, we have examined the diastereoselectivity of hydrosilation of 4-tert-butyl cyclohexanone with dimethyl ethyl silane over a wide range of temperatures. These data provide information concerning differences of enthalpies and entropies of activation for the two pathways. The trans/cis product ratio for the hydrosilation of 4-tert-butyl cyclohexanone in CD2Cl2 at various reaction temperatures is shown in Table 3. A plot of ln[cis]/[trans] versus 1/T made using the data in Table 3 provides a good linear regression with R2 = 0.993 as shown in Figure 1. By using the relationship shown in eq. 5 (derived from the Erying equation), ΔΔH‡ (ΔH‡ (trans) - ΔH‡ (cis)) and ΔΔS‡ (ΔS‡ (trans) - ΔS‡(cis)) were calculated to be -2.5 kcal/mol and -6.9 e.u., respectively.
| (5) |
Table 3.
Hydrosilation of 4-tert-butyl cyclohexanone with EtMe2SiH at various reaction temperaturesa
| T | t | conversionb | trans/cis ratiob | |
|---|---|---|---|---|
| entry | °C | h | % | % |
| 1 | -60 | 17 | quant. | 92 : 8 |
| 2 | -30 | 3 | quant. | 86 : 14 |
| 3 | 0 | 1 | quant. | 77 : 23 |
| 4 | 17 | 3 | quant. | 70 : 30 |
Reaction conditions; 4 equiv. of EtMe2SiH, 1.39 mol% of 1 in CD2Cl2 (1 ml).
Determined by 1H NMR spectroscopy.
Figure 1.

Plot of ln[cis]/[trans] vs. 1/T using the data from Table 3.
The stereo-determining step is attack of 3 on the silated ketone (eq. 6, see below for mechanistic considerations) and thus axial attack is strongly favored enthalpically over equatorial attack but strongly disfavored entropically. Since this is a bimolecular reaction, both activation entropies are no doubt negative, so axial attack must exhibit a more negative entropy of activation relative to equatorial attack. The reasons for these significant differences are not clear.
 |
(6) |
It is noteworthy that the iridium-catalyzed hydrosilation of 4-tert-butyl cyclohexanone has considerably larger differences in the activation parameters relative to reductions by LiAlH4 and NaBH4 (For LiAlH4, ΔΔH‡ = -0.8 kcal/mol and ΔΔS‡ = 2.0 e.u.).8d In these cases the temperature dependence of the product ratio is small and ΔΔS‡ is small and positive in contrast to a large negative value for the hydrosilation.
Hydrosilation of Alkyl-Substituted Cyclohexanone Derivatives Catalyzed by 1
We have examined the hydrosilations of other alkyl-substituted cyclohexanone derivatives to probe the diastereoselectivities in these cases. Results of hydrosilation of 3,3,5-trimethyl cyclohexanone, 2-methyl cyclohexanone, 2-tert-butyl cyclohexanone, and camphor with dimethyl ethyl silane are summarized in Table 4.
Table 4.
Hydrosilations of alkyl-substituted cyclohexanone derivatives catalyzed by 1a
| T | t | conversionb | trans/cis ratiob | |||
|---|---|---|---|---|---|---|
| entry | ketone | silane | °C | h | % | % |
| 1 | 3,3,5-trimethylcyclohexanone | EtMe2SiH | 22 | 0.3 | quant. | 86 : 14 |
| 2 | 2-methylcyclohexanone | EtMe2SiH | 22 | 0.5 | quant. | 29 : 71 |
| 3 | 2-tert-butylcyclohexanone | EtMe2SiH | 22 | 0.5 | quant. | 24 : 76 |
| 4 | camphor | EtMe2SiH | 0 | 2 | quant. | 21 : 79c |
Reaction conditions; 0.5 mol% of 1 and 3 equiv. of silane in C6D5Cl.
Determined by NMR spectroscopy.
exo/endo ratio.
3,3,5-Trimethyl cyclohexanone undergoes quantitative hydrosilation with dimethyl ethyl silane in 0.3 h to give the silyl alkyl ether with 86% of trans diastereoselectivity (entry 1). The preference for trans stereochemistry can be attributed to blocking of the axial approach of the bulky iridium dihydride by the axial methyl group at C3. (Trans selectivity ranging from 52% to 83% is seen in hydride reductions of 3,3,5-trimethyl cyclohexanone).6h,i,n,p 2-Alkylcyclohexanone derivatives possessing large equatorial alkyl substituents are reported to inhibit axial attack of metal hydrides.6a,e,l,m For example, reduction of 2-methyl cyclohexanone by LiAlH4 affords alcohols with a trans : cis selectivity of ca. 73:27. However, when the steric bulk of the C2 substituent is increased to tert-butyl, axial approach is somewhat inhibited to yield product alcohols with slight cis selectivity (50%-64%). The hydrosilation of 2-methyl cyclohexanone by 1 at 22 °C proceeds rapidly to give the silyl alkyl ether with 71% of cis selectivity (entry 2). The use of 2-tert-butyl cyclohexanone in the hydrosilation leads to a slight increase in the cis selectivity to 76% (entry 3). The steric impact of 2-substituents is significantly greater in these cases compared to the metal reductions. The hydrosilation of camphor at 0 °C yields the exo and endo isomers in 21:79 ratio, respectively (entry 4). In the (Ph3P)4RhH-catalyzed hydrosilation of camphor with diphenyl silane the opposite selectivity is observed with an exo:endo product ratio of 64:36 reported.2j
Mechanistic Investigation of the Hydrosilation of Ketones
Based on earlier mechanistic investigations of silane reductions of alkyl halides and alkyl ethers using iridium complex 1, our working hypothesis concerning the catalytic mechanism of hydrosilation of ketones is shown in Scheme 3 and illustrated with acetone. Binding triethyl silane to the electrophilic iridium center in 2 renders this complex a potent Et3Si+ donor which can transfer Et3Si+ to acetone forming the oxocarbenium ion, 4, and iridium dihydride, 3. The dihydride, earlier established as a good hydride donor,5b reacts with 4 to produce hydrosilated product, 5, and the cationic hydride, 6, which then reenters the catalytic cycle.
Scheme 3.

Proposed catalytic cycle for the iridium-catalyzed hydrosilation of acetone with triethyl silane
Low temperature reactions were carried out to probe the details of this cycle. 13C labeled acetone, (CH3) 132CO) was employed to allow monitoring by 13C as well as 1H and 31P NMR spectroscopy. Initially, the equilibrium between 1 and 2 was probed, as shown in Scheme 4. Reaction of 1 (containing unlabeled bound acetone) with triethyl silane (4 equiv.) in CD2Cl2 at 25 °C rapidly produced 1 equiv. of Et3SiOCHMe2 (1H NMR) and silane complex 2 (21%), CD2Cl2 complex, 6 (48%), and cationic trihydride, 7 (31%) (Scheme 4A). The complexes were identified by 31P NMR shifts. The trihydride results from reaction of 2 with traces of water.9 This solution was cooled to -70 °C and (CH3)2*CO (2 equiv) was added. Acetone immediately displaces silane from 2 and CD2Cl2 from 6 to give a solution containing acetone complex 1* (ca. 64%) and trihydride 7 (ca. 34%) and traces of solvent complex, 6 (Scheme 4B). 1H NMR spectroscopy was used to determine the ratio of free acetone (δ2.13, doublet, J13C-H = 5 Hz) to bound acetone (δ2.57, doublet, J13C-H = 5 Hz) as 64 : 36. No silane complex can be easily detected (< 0.5%) and no hydrosilation product is formed. The hydrosilation product of (CH3)2*CO, Et3SiO*CHMe2 can be distinguished from Et3SiOCHMe2 since the CH resonance at δ3.99 is split into a doublet with J13C-H = 138.5 Hz. These results imply that the equilibrium between 1 and 2, not surprisingly, lies strongly to the left and that equilibrium is established rapidly relative to product formation.
Scheme 4.

To quantitatively assess Keq, 16 equiv. of Et3SiH were added to this solution (Scheme 4C). Even under these conditions no silane complex, 2, could be detected by 31P NMR spectroscopy. Upon warming this solution in the NMR probe to -50 °C, catalytic hydrosilation begins as shown by the appearance in the 1H NMR spectrum of Et3SiO*CHMe2. As acetone is consumed and the ratio of Et3SiH to free acetone further increases, a point is reached where finally a sufficient quantity of silane complex is formed that the equilibrium constant can be measured. After 10 min at -50 °C, the ratio of free acetone to Et3SiH is 1:18.9 (1H NMR) and the ratio of acetone complex, 1, to silane complex, 2, is 37.5:1 (31P NMR) which yields an equilibrium constant of 1.4 × 10-3 at -50 °C (eq. 7)10:
| (7) |
While these experiments establish a rapid equilibrium between 1 and 2 during catalysis with the ketone complex as the nearly exclusive resting state, they do not provide a decision as to whether step I, silation of acetone, or step II, reduction of 4 by iridium dihydride, 3, is turnover-limiting (Scheme 5). Either scenario predicts that the turnover frequency is zero-order in ketone and first-order in Et3SiH. Indeed that has been confirmed for reduction of 4-tert-butyl cyclohexanone at -60 °C under typical catalytic conditions. Figure 2 (left) shows a typical plot of the initial rate of disappearance of 4-tert-butyl cyclohexanone (1.06 M) using Et3SiH (0.639 M) and complex 1 (6.4 mM) in CD2Cl2. Figure 2 (right) shows a plot of the initial rates of disappearance of 4-tert-butyl cyclohexanone at various concentrations of 4-tert-butyl cyclohexanone using 1.87 M Et3SiH and 6.4 mM 1 in CD2Cl2 at -60 °C. This plot establishes that the turnover frequency is zero-order in ketone. Figure 3 shows a plot of the initial rates of disappearance 4-tert-butyl cyclohexanone (0.32-1.06 M) at various concentrations of Et3SiH using complex 1 (6.4 mM) in CD2Cl2 (-60 °C). This plot shows clearly that the turnover frequency is first-order in silane.
Scheme 5.

Free energy diagram for the iridium-catalyzed hydrosilation of acetone with triethyl silane
Figure 2.

(Left) Plot of concentration of 4-tert-butyl cyclohexanones [ketone] vs. time for the hydrosilation of 4-tert-butyl cyclohexanone catalyzed by 1 at -60 °C. (Right) Plot of the initial rate, Vi of 4-tert-butyl cyclohexanone vs. 4-tert-butyl cyclohexanone concentration at -60 °C.
Figure 3.

Plot of the initial rate, Vi of disappearance of 4-tert-butyl cyclohexanone vs. Et3SiH concentration at -60 °C.
Jian et al., has reported the Ir-catalyzed reduction of alkyl halides by Et3SiH, and proposed a quite similar catalytic cycle to Scheme 3 based on the kinetic data, although, as here, they were not able to distinguish whether the silation of alkyl halides, RX, or the transfer of hydride to Et3SiXR+ was the turnover limiting step.5a
Conclusions
Iridium complex 1 is a highly active, long-lived catalyst for hydrosilation of a variety of ketones and aldehydes with trialkyl silanes to afford the corresponding silyl alkyl ethers in excellent yields. Highly hindered ketones such as diisopropyl ketone are effective substrates. The majority of the cases studied here have used Et3SiH as the silane, but several other silanes including the bulky (i-Pr)3SiH have been shown to be effective with cyclohexanone. Hydrosilation of esters leads to “over reduction” and cleavage of C-O bonds. Similarly, diethyl acetamide reacts to yield triethyl amine. The diastereoselectivity of the hydrosilation of 4-tert-butyl cyclohexanone with 1 and EtMe2SiH shows unprecedented temperature dependence. Analysis of product ratios as a function of temperature yields values for ΔΔH‡ (ΔH‡ (trans) - ΔH‡ (cis)) and ΔΔS‡ (ΔS‡ (trans) - ΔS‡(cis)) of -2.5 kcal/mol and -6.9 e.u., respectively.
A mechanistic study of the reaction showed that the ketone and silane complexes are in rapid equilibrium relative to the rate of catalytic hydrosilation and that ketone complex is strongly favored and can be viewed as the resting state. Catalysis ensues by silation of ketone by the cationic silane complex followed by reduction of this species by the resultant iridium dihydride. Low temperature mechanistic studies revealed that the ketone complex is the dominant resting state in rapid equilibrium with silane complex, 2. The turnover-limiting step in this catalytic cycle is either silation of ketone by 2, or the reduction of resultant oxocarbenium ion via a hydride transfer from iridium dihydride, 3. The turnover frequency is first-order in silane and zero-order in ketone in accord with this proposal. This mechanism is similar to that proposed by Piers for hydrosilation of ketones using (C6F5)3B/Ph3SiH where the silane is activated by (C6F5)3B and transfers Ph3Si+ to ketone.4a
Experimental Section
General Procedures
All manipulations were carried out using standard Schlenk, high-vacuum and glovebox techniques. Argon and nitrogen were purified by passing through columns of BASF R3-11 catalyst (Chemalog) and 4 Å molecular sieves. THF was distilled under a nitrogen atmosphere from sodium benzophenone ketyl prior to use. Pentane, methylene chloride and toluene were passed through columns of activated alumina11 and degassed by either freeze-pump-thaw methods or by purging with argon. Benzene and acetone were dried with 4 Å molecular sieves and degassed by freeze-pump-thaw methods. Silanes were dried with LiAlH4 or 4 Å molecular sieves and vacuum transferred into a sealed flask. All of the other substrates purchased from Sigma-Aldrich were dried with either K2CO3 or 4 Å molecular sieves and vacuum transferred into a sealed flask for the substrate with boiling point less than ca. 110 °C, except that 4-tert-butyl cyclohexanone was purified by sublimation at ca. 40 °C. Deuterated solvents (CD2Cl2, C6D5CD3, C6D5Cl) for NMR were dried with CaH2 or 4 Å molecular sieves and vacuum transferred into a sealed flask. NMR spectra were recorded on Bruker spectrometers (DRX-400, AVANCE-400, AMX-300 and DRX-500). 1H and 13C NMR spectra were referenced to solvent peaks. Ph3C[B(C6F5)4],12 (POCOP)Ir(H)2,13 and [(POCOP)Ir(H)(acetone)]+[B(C6F5)4]-, 15 were prepared according to published procedures.
General Procedure for the Hydrosilation of Substrates with Et3 SiH in C6D5Cl
Et3SiH (0.48 ml, 3.00 mmol, 3 equiv.) was added to a solution of 1 (6.7 mg, 0.005 mmol, 0.5 mol%) in C6D5Cl (0.3 ml) in a medium-walled J. Young NMR tube and the contents were well shaken. The substrate (1.00 mmol, 1 equiv.) was then added and the reaction was allowed to stand at room temperature. The progress was followed by NMR spectroscopy. With the exception of diethyl acetamide (entry 11, Table 1) conversions are quantitative and no starting material remains at the end of the reaction. NMR data for the hydrosilated products in Table 1 are available in Supporting Information.
General Procedure for the Hydrosilation of 4-tert-Butyl Cyclohexanone with Various Silanes in C6D5Cl
Silane (3.00 mmol, 3 equiv.) was added to a solution of 1 (6.7 mg, 0.005 mmol, 0.5 mol%) in C6D5Cl (0.3 ml) in a medium-walled J. Young NMR tube and the contents were well shaken. 4-tert-Butylcyclohexanone (154 mg, 1.00 mmol, 1 equiv.) was then added and the reaction was allowed to stand at room temperature for the specified time. The reaction mixture was then analyzed by NMR spectroscopy. NMR data for the hydrosilated products in Table 2 are available in Supporting Information.
General Procedure for the Hydrosilation of Alkyl-Substituted Cyclohexanone Derivatives with EtMe2SiH in C6D5Cl
Silane (0.40 ml, 3.00 mmol, 3 equiv.) was added to a solution of 1 (6.7 mg, 0.005 mmol, 0.5 mol%) in C6D5Cl (0.3 ml) in a medium-walled J. Young NMR tube and the contents were well shaken. Alkyl-substituted cyclohexanone derivatives (1.00 mmol, 1 equiv.) were then added and the reaction mixtures were allowed to stand at room temperature or 0 °C for a specific time. The reaction mixtures were then analyzed by NMR spectroscopy. NMR data for the hydrosilated products in Table 4 are available in Supporting Information.
General Procedure for Kineic Studies (CD2Cl2
Et3SiH was added to a solution of 1 (6.7 mg, 0.005 mmol) in CD2Cl2 in a medium-walled J. Young NMR tube and the solution was well shaken. The NMR tube was placed in a bath at -100 °C to freeze the solution, and then 4-tert-butyl cyclohexanone in CD2Cl2 (0.32-0.98 M) was added on the top of the frozen solution at -100 °C. After briefly shaking, the NMR tube was quickly placed in the NMR probe pre-cooled to -70 °C. The reaction was allowed to warm -60 °C and the ratio of 4-tert-butyl cyclohexanone to the silyl ether product was monitored with respect to time by 1H NMR. The data were analyzed using the method of initial rates and the initial reduction rates were obtained from the linear portion of the concentration vs. time curve in the early stage of the reaction (Figure 2, 3).
Low Temperature Reaction of 1, Et3SiH, and acetone-2-13C, -70°C to -50 °C
To the solution of 1 (10.05 mg, 0.0075 mmol) in CD2Cl2 in a medium-walled J. Young NMR tube was added 4 equiv. of Et3SiH (4.8 μl, 0.03 mmol), and the solution was stirred at 22 °C for 30 min. The solution was cooled to -70 °C and 1 equiv. of acetone-2-13C was added from a stock solution of acetone-2-13C in CD2Cl2. After briefly shaking, the NMR tube was quickly placed in the NMR probe pre-cooled to -80 °C. The reaction was allowed to warm to -70 °C and monitored by NMR spectroscopy. After NMR experiments at -70 °C, 16 equiv. of Et3SiH was additionally added to the solution while keeping temperature at -70 °C, followed by warming the solution up to -50 °C. The hydrosilation of acetone-2-13C with Et3SiH began at -50 °C and the concentration of all species were monitored by NMR spectroscopy to determine the equilibrium constant between 1 and 2 (Scheme 6).5a,b
Scheme 6.

Equilibrium between 1 and 2 at -50 °C
Supplementary Material
Acknowledgment
We gratefully acknowledge funding by the National Institutes of Health (Grant No. GM 28939).
References
- 1.Reviews on hydrosilation: Ojima I, Li Z, Zhu J. In: The Chemistry of Organic Silicon Compounds. Rappoport Z, Apeloig Y, editors. Vol. 2. John Wiley & Sons; New York: 1998. Nishiyama H, Itoh K. In: Catalytic Asymmetric Synthesis. Ojima I, editor. Wiley-VCH; New York: 2000. Marciniec B, Guliňski J. J. Organomet. Chem. 1993;446:15.
- 2.Chalk AJ, Harrod JF. J. Am. Chem. Soc. 1965;87:16.Ojima I, Nihonyanagi M, Nagai Y. J. Chem. Soc., Chem. Commun. 1972:938.Ojima I, Nihonyanagi M, Kogure T, Kumagai M, Horiuchi S, Nakatsugawa K, Nagai Y. J. Organomet. Chem. 1975;94:449.Kobayashi M, Koyama T, Ogura K, Seto S, Ritter FJ, Br□ggemann-Rotgans IEM. J. Am. Chem. Soc. 1980;102:6602.Semmelhack MF, Misra RN. J. Org. Chem. 1982;47:2469.Ojima I, Kogure T. Organometallics. 1982;1:1390.Brunner H, Becker R, Riepl G. Organometallics. 1984;3:1354.Brunner H, Kurzinger A. J. Organomet. Chem. 1988;346:413.. A catalytic reductive silation of 4-tert-butyl cyclohexanone using 0.012% Zn(OSO2CH3)2 in combination with lithium hydride and chloro trimethyl silane gives predominantly the less stable reduction product with 72% of cis-selectivity:Ohkuma T, Hashiguchi S, Noyori R. J. Org. Chem. 1994;59:217.Zheng GZ, Chan TH. Organometallics. 1995;14:70.Wright ME, Cochran BB. Organometallics. 1996;15:317.Nagashima H, Suzuki A, Iura T, Ryu K, Matsubara K. Organometallics. 2000;19:3579.Tao B, Fu GC. Angew. Chem. Int. Ed. 2002;41:3892. doi: 10.1002/1521-3773(20021018)41:20<3892::AID-ANIE3892>3.0.CO;2-A.Gade LH, Cesar V, Bellemin-Laponnaz S. Angew. Chem. Int. Ed. 2004;43:1014. doi: 10.1002/anie.200353133.
- 3.a Glaser PB, Tilley TD. J. Am. Chem. Soc. 2003;125:13640. doi: 10.1021/ja037620v. [DOI] [PubMed] [Google Scholar]; b Parks DJ, Blackwell JM, Piers WE. J. Org. Chem. 2000;65:3090. doi: 10.1021/jo991828a. [DOI] [PubMed] [Google Scholar]; c Caseri W, Pregosin PS. Organometallics. 1988;7:1373. [Google Scholar]; d Hanna PK, Gregg BT, Cutler AR. Organometallics. 1991;10:31. [Google Scholar]; e Cavanaugh MD, Gregg BT, Cutler AR. Organometallics. 1996;15:2764. [Google Scholar]; f Gregg BT, Cutler AR. J. Am. Chem. Soc. 1996;118:10069. [Google Scholar]; g Kennedy-Smith JJ, Nolin KA, Gunterman HP, Toste FD. J. Am. Chem. Soc. 2003;125:4056. doi: 10.1021/ja029498q. [DOI] [PubMed] [Google Scholar]; h Nolin KA, Ahn RW, Toste FD. J. Am. Chem. Soc. 2005;127:17168. doi: 10.1021/ja050831a. [DOI] [PubMed] [Google Scholar]; i Blanc A, Toste FD. Angew. Chem. Int. Ed. 2006;45:2096. doi: 10.1002/anie.200503852. [DOI] [PubMed] [Google Scholar]; j Nolin KA, Krumper JR, Pluth MD, Bergman RG, Toste FD. J. Am. Chem. Soc. 2007;129:14684. doi: 10.1021/ja074477n. [DOI] [PubMed] [Google Scholar]; k Fernandes AC, Fernandes R, Romao CC, Royo B. Chem. Commun. 2005:213. doi: 10.1039/b414145h. [DOI] [PubMed] [Google Scholar]; l Royo B, Romao CC. J. Mol. Catal. A: Chem. 2005;236:107. [Google Scholar]; m Ison EA, Trivedi ER, Corbin EA, Abu-Omar MM. J. Am. Chem. Soc. 2005;127:15374. doi: 10.1021/ja055704t. [DOI] [PubMed] [Google Scholar]; n Ison EA, Cessarich JE, Du GD, Franwick PE, Abu-Omar MM. Inorg. Chem. 2006;45:2385. doi: 10.1021/ic0520768. [DOI] [PubMed] [Google Scholar]; o Du GD, Franwick PE, Abu-Omar MM. J. Am. Chem. Soc. 2007;129:5180. doi: 10.1021/ja068872+. [DOI] [PubMed] [Google Scholar]; p Carter MB, Schiott B, Guiterrez A, Buchwald SL. J. Am. Chem. Soc. 1994;116:11667. [Google Scholar]; q Yun J, Buchwald SL. J. Am. Chem. Soc. 1999;121:5640. [Google Scholar]
- 4.a Parks DJ, Piers WE. J. Am. Chem. Soc. 1996;118:9440. [Google Scholar]; b Parks DJ, Piers WE, Parvez M, Atencio R, Zaworotko MJ. Organometallics. 1998;17:1369. [Google Scholar]
- 5.a Yang J, Brookhart M. J. Am. Chem. Soc. 2007;129:12656. doi: 10.1021/ja075725i. [DOI] [PubMed] [Google Scholar]; b Yang J, White PS, Brookhart M. J. Am. Chem. Soc. 2008;130:17509. doi: 10.1021/ja806419h. [DOI] [PubMed] [Google Scholar]; c Yang J, White PS, Schauer C, Brookhart M. Angew. Chem. Int. Ed. 2008;47:4141. doi: 10.1002/anie.200705359. [DOI] [PubMed] [Google Scholar]; d Yang J, Brookhart M. Adv. Synth. Catal. 2009;351:175. [Google Scholar]
- 6.a Noyce DS, Denney DB. J. Am. Chem. Soc. 1950;72:5743. [Google Scholar]; b Cram DJ, Abd Elhafez FA. 1952;74:5828. ibid. [Google Scholar]; c Barton DHR. J. Chem. Soc. 1953:1027. [Google Scholar]; d Schlesinger HI, Brown HC, Hoekstra HR, Rapp LR. J. Am. Chem. Soc. 1953;75:199. [Google Scholar]; e Dauben WG, Fonken GJ, Noyce DS. J. Am. Chem. Soc. 1956;78:2579. [Google Scholar]; f Dauben WG, Bozak RE. J. Org. Chem. 1959;24:1596. [Google Scholar]; g Wheeler DMS, Huffman JW. Experientia. 1960;16:516. [Google Scholar]; h Haubenstock H, Eliel EL. J. Am. Chem. Soc. 1962;84:2363. [Google Scholar]; i Haubenstock H, Eliel EL. J. Am. Chem. Soc. 1962;84:2368. [Google Scholar]; j House HO, Babad H, Toothill RB, Noltes AW. J. Org. Chem. 1962;27:4141. [Google Scholar]; k Richer JC. 1965;30:324. ibid. [Google Scholar]; l Brown HC, Deck HR. J. Am. Chem. Soc. 1965;87:5620. [Google Scholar]; m Ashby EC, Sevenair JP, Dobbs FR. J. Org. Chem. 1971;36:197. [Google Scholar]; n Eliel EL, Senda Y. Tetrahedron. 1970;26:2411. [Google Scholar]; o Wigfield DC, Phelps DJ. J. Org. Chem. 1976;41:2396. [Google Scholar]; p McMahon RJ, Wiegers KE, Smith SG. J. Org. Chem. 1981;46:99. [Google Scholar]
- 7.a Coxon JM, Houk KN, Luibrand RT. J. Org. Chem. 1995;60:418. [Google Scholar]; b Li H, le Noble WJ. Recl. Trav. Chim. Pays-Bas. 1992;111:199. [Google Scholar]; c Cherest M, Felkin H, Prudent N. Tetrahedron Lett. 1968:2199. [Google Scholar]; d Cherest M, Felkin H. Tetrahedron Lett. 1968:2205. [Google Scholar]; e Cherest M, Felkin H. Tetrahedron Lett. 1973:4307. [Google Scholar]
- 8.a Lansbury PT, Macleay RE. J. Org. Chem. 1963;28:1940. [Google Scholar]; b Wigfield DC, Phelps DJ. J. Org. Chem. 1976;41:2396. [Google Scholar]; c Ashby EC, Boone JR. J. Org. Chem. 1976;41:2890. [Google Scholar]; d Rosenberg RE, Vilardo JS. Tetrahedron Lett. 1996;37:2185. [Google Scholar]
- 9.The trihydride is inert and is carried through the next sequence of reactions.
- 10.The reaction of acetone-2-13C (3 equiv.) with a solution containing 2, 6, and 7 in a ratio of 0.68:0.12:0.19 in the presence of excess triethyl silane (90 equiv.) at -70 °C leads to hydrosilation (36% conversion) in 5 min to afford Et3SiO-13CHMe2, 5. 1H and 31P NMR spectra at -70 °C exhibit the proton resonances for triethyl silane and free acetone-2-13C, and the phosphorus resonances of iridium species 1 (61%) and 2 (9%) as well as 6 (2%) and 7 (28%). Based on these NMR data, the equilibrium constant, K (-70 °C) between 1 and 2 can be calculated to be 1.8 × 10-3 in agreement with the value of 1.4 × 10-3 obtained at -50 °C.
- 11.a Alaimo PJ, Peters DW, Arnold J, Bergman RG. J. Chem. Educ. 2001;78:64. [Google Scholar]; b Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 12.Scott VJ, Çelenligil-Çetin R, Ozerov OV. J. Am. Chem. Soc. 2005;127:2852. doi: 10.1021/ja0426138. [DOI] [PubMed] [Google Scholar]
- 13.Goettker-Schnetmann I, White P, Brookhart M. Organometallics. 2004;23:1766. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.