

Abstract
Increasing evidence suggests that processes termed epithelial-mesenchymal transitions (EMTs) play a key role in therapeutic resistance, tumor recurrence, and metastatic progression. NF-κB signaling has been previously identified as an important pathway in the regulation of EMT in a mouse model of tumor progression. However, it remains unclear whether there is a broad requirement for this pathway to govern EMT and what the relative contribution of IKK family members acting as upstream NF-κB activators is toward promoting EMT and metastasis. To address this question, we have used a novel, small-molecule inhibitor of IκB kinase 2 (IKK2/IKKβ), termed BI 5700. We investigated the role of IKK2 in a number of mouse models of EMT, including TGFβ-induced EMT in the mammary epithelial cell line EpRas, CT26 colon carcinoma cells, and 4T1 mammary carcinoma cells. The latter model was also used to evaluate in vivo activities of BI 5700.We found that BI 5700 inhibits IKK2 with an IC50 of 9 nM and was highly selective as compared to other IKK family members (IKK1, IKKε, and TBK1) and other kinases. BI 5700 effectively blocks NF-κB activity in EpRas cells and prevents TGFβ-induced EMT. In addition, BI 5700 reverts EMT in mesenchymal CT26 cells and prevents EMT in the 4T1 model. Oral application of BI 5700 significantly interferes with metastasis after mammary fat-pad injection of 4T1 cells, yielding fewer, smaller, and more differentiated metastases as compared to vehicle-treated control animals. We conclude that IKK2 is a key regulator of both the induction and maintenance of EMT in a panel of mouse tumor progression models and that the IKK2 inhibitor BI 5700 constitutes a promising candidate for the treatment of metastatic cancers.
Keywords: epithelial-mesenchymal transition, metastasis, IKK2, NF-κB, chemical inhibitor
Introduction
Carcinoma metastasis is the primary cause of death for most cancer patients.1 While several major mechanisms in carcinoma initiation have been uncovered,2 mechanisms and signaling pathways in metastasis are only now beginning to emerge.3,4 The invasion-metastasis cascade involves localized invasion by primary tumor cells, intravasation, translocation through blood vessels to distant sites, extravasation, and finally the formation of a progressively growing lesion at a secondary site termed colonization.4 Evidence is accumulating that epithelial cancer cells adopt embryonic transcription programs during the invasive phase of metastasis, which enable them to convert into cells with mesenchymal properties.5-7 These epithelial-mesenchymal transitions (EMTs) are characterized by variably reduced intercellular adhesion, loss of planar and apical-basal polarity, and acquisition of mesenchymal features, such as enhanced migratory activity, invasiveness, and an increased resistance to apoptosis. A series of EMT-inducing transcription factors, notably Snail, Slug, Zeb1, SIP1, and Twist, have been implicated in this transition, mainly by their ability to act as repressors of E-cadherin, loss of which is recognized as a hallmark of EMT.8 A large number of pathways, including TGFβ signaling and the Ras pathway, appear to collaborate to orchestrate this process.9 An increasing body of evidence demonstrates that EMT can occur in tumor cells during cancer progression,5,7 and a recent study suggests that EMT can induce mammary cancer cells to adopt stem cell–like characteristics.10
Importantly, chronic inflammation is increasingly considered as an essential process during cancer progression,11-14 in line with recently detected roles of the NF-κB pathway in inflammation-associated cancer and tumor progression.15-18 NF-κB transcription factors bind to DNA as heterodimers or homodimers that consist of 5 possible subunits (RelA/p65, c-Rel, RelB, p50, and p52). In unstimulated cells, NF-κB complexes are retained in the cytoplasm in an inactive form due to the action of specific inhibitor proteins (IκBs), most notably IκBα. In the canonical NF-κB signaling pathway, NF-κB nuclear translocation is controlled by upstream signaling events initiated primarily by proinflammatory stimuli, resulting in activation of the IKK complex that consists of 3 subunits termed IKK1 (IKKα), IKK2 (IKKβ), and NEMO (IKKγ). This kinase complex phosphorylates IκBα, which results in its ubiquitination and subsequent proteasomal degradation. Free NF-κB translocates to the nucleus to enable and regulate transcription of target genes linked to inflammation and cancer.17
We recently showed that activation of the NF-κB signaling pathway by different approaches caused epithelial-mesenchymal transition9,15 in the well-characterized, combined in vitro/in vivo EpH4/Ep-Ras model for cancer progression and metastasis.19,20 Inhibition of the NF-κB pathway through a constitutively active, nonphosphorylatable version of IκBα overexpressed in EpRas cells interfered with their TGFβ-induced induction of EMT and blocked their metastatic capacity upon tail-vein injection in mice.15 Recent studies have further implicated this pathway in EMT and metastasis and have begun to link NF-κB to other key regulators of EMT. For example, inhibition of NF-κB activity reduced the invasive phenotype driven by the NF-κB c-Rel subunit in mammary tumor cells induced by the mutagen 7,12-dimethylbenz(α)anthracene.21 In addition, the NF-κB subunit RelB was responsible for promoting the more invasive phenotype of estrogen receptor (ER)α-negative or low breast cancers, and Bcl-2 was identified as an important downstream mediator of RelB, both suppressing apoptosis and inducing EMT.22 Furthermore, evidence is mounting that NF-κB plays a key role in activating EMT-inducing transcription factors, such as Snail,18 providing a mechanistic explanation for the NF-κB pathway in inflammation-induced EMT and metastasis.
While all 4 IKKs and IKK-related kinases (IKK1, IKK2, TBK1, IKKε) have been linked with tumorigenesis,23,24 their relative contribution in the induction and maintenance of EMT has not been addressed. Here, we describe BI 5700, a novel selective small-molecule inhibitor of IKK2, which effectively interfered with EMT in EpRas cells and in metastasizing CA-IKK2-EpRas cells. In addition, BI 5700 can also revert EMT in metastasizing mouse colon carcinoma cells (CT26)25 and prevent EMT in murine mammary carcinoma cells able to metastasize from orthotopic tumor sites (4T1).26 We show that in contrast to the nonmetastatic or weakly metastatic cell types of this cell series,26 4T1 cells expressed high NF-κB pathway activity, and their metastatic capacity was effectively blocked by inhibiting NF-κB signaling, using the IκBα superrepressor or the IKK2 inhibitor BI 5700. Oral application of BI 5700 significantly interfered with metastasis after orthotopic injection of 4T1 cells, yielding fewer, smaller, and more differentiated metastases as seen in control animals. In contrast, mammary gland tumor formation was only weakly inhibited by BI 5700, indicating that among the IKK family members, IKK2 plays a key role in promoting tumor progression and metastasis.
Results
BI 5700 Specifically Inhibits IKK2 and Effectively Blocks NF-κB Activity in EpRas Cells
We identified a novel, selective small-molecule inhibitor of IKK2 belonging to the thienopyridine class, termed BI 5700 (Fig. 1A). This compound blocked IKK2 with a 50% inhibitory concentration (IC50) of 9 nM. Therefore, BI 5700 was >65-fold more potent on IKK2 as compared to IKK1 and FLT3 and more than 100- to 1,000-fold selective versus a wide range of other kinases tested, including the related kinases IKKε and TBK-1 (Supplementary Table S1). Our selectivity data indicate that BI 5700 is more selective for IKK2 versus other IKK family members when compared to many of the previously described IKK inhibitors.29 In HeLa cells, BI 5700 exhibited an IC50 of 290 nM in inhibiting the phosphorylation of IκBα and an IC50 of 430 nM in inhibiting the expression of the NF-κB target gene ICAM-1 (data not shown). To further characterize BI 5700, we determined its antiproliferative activity in a panel of cancer cell lines (Table 1). BI 5700 was not generally cytotoxic and had the strongest effects on cell viability in cancer cell lines with somatic aberrations leading to NF-κB pathway activation, including T-ALL lines harboring constitutive Notch signaling linked to NF-κB activation30-32 and multiple myeloma lines with mutations upstream of IKK/NF-κB.33 In contrast, previously described IKK2 inhibitors BMS-34554134 or BAY-11-708235 were generally cytotoxic at low micromolar levels in all cell lines tested (data not shown), most likely due to off-target effects (data not shown).29
Figure 1.

The IKK2-selective inhibitor BI 5700 inhibits NF-κB activation by TNFα/TGFβ. (A) Chemical structure of BI 5700. (B) EpRas cells were treated (+) or not treated (–) with TNFα (40 ng/mL) for 30 minutes. NF-κB DNA-binding activity was determined by EMSA, using an NF-κB-specific probe (upper panel). An Oct-1-specific probe (lower panel) was used as a loading control. Note dose-dependent inhibition of TNFα-induced NF-κB activity by BI 5700. (C) EpRas cells were stimulated (+) or not stimulated (–) with TGFβ1 (5 ng/mL) for 2 hours without (0) or with BI 5700 added in the amounts indicated and analyzed by EMSA as described in B. (D) EpRas cells were treated (+) or not treated (–) with TNFα as in panel B, but mRNA expression of the NF-κB target gene MCP-1 was analyzed by RT-PCR using β-actin as a loading control (see Materials and Methods).
Table 1.
Antiproliferative Activity of BI 5700 in Cancer Cell Lines
| Cell Line | Cell Type | Unstimulated IC50, µM | Plus TNF-α IC50, µM |
|---|---|---|---|
| No NF-κB pathway activation reported | |||
| HelaS3 | Cervix carcinoma | 48 | 7 |
| MDA-MB 231 | Breast carcinoma | >50 | 13 |
| MCF7 | Breast carcinoma | >50 | 28 |
| EpRas | Breast carcinoma (mouse) | >50 | ND |
| 4T1 | Breast carcinoma (mouse) | >50 | ND |
| NF-κB pathway activation reported | |||
| DND-41 | T-ALL | 7 | 5 |
| BE-13 | T-ALL | 12 | 0.4 |
| CCRF-CEM | T-ALL | 17 | 3 |
| Jurkat | T-ALL | 9 | 3 |
| HDML-2 | Hodgkin lymphoma | 13 | 5 |
| JJN-3 | Multiple myeloma | 9 | 9 |
| L-363 | Multiple myeloma | 11 | 11 |
Note: IC50 values (i.e., compound concentration that inhibits cell viability by 50%) as determined by alamarBlue assay were calculated from several independent experiments for each condition. TNF-α was added at 150 ng/mL. ND = not determined.
Previous work had suggested a critical role of NF-κB activation in epithelial-mesenchymal transition (EMT) using the murine EpRas model system. To address more specifically the ability of BI 5700 to prevent NF-κB-induced EMT, we first determined its effect on NF-κB activity in EpRas cells. BI 5700 blocked both the TNFα- and TGFβ-induced NF-κB DNA-binding activity in EpRas cells (Fig. 1 B and C). As shown in Figure 1D, BI 5700 also inhibited the TNFα-induced expression of MCP-1, a key target gene of TGFβ and NF-κB during EMT.15,36 The IC50 of approximately 1 to 2 µM was consistent across these different cellular assays for NF-κB activity in EpRas cells. Despite the fact that inhibition of NF-κB was effectively inhibited at low micromolar concentrations, cell viability of EpRas cells was not affected up to 50 µM (Table 1). These findings allowed the use of BI 5700 to dissect the role of IKK2 in EMT of EpRas cells and other cellular EMT models without cytotoxic effects obscuring respective phenotypic cellular readouts.
IKK2 Kinase Inhibitor BI 5700 Prevents TGFβ-Induced EMT in EpRas Cells
To determine if and how BI 5700 might affect TGFβ-induced EMT in EpRas cells, we analyzed this process in cells seeded on permeable supports (filters) (Fig. 2 A and C) and in collagen gels (Fig. 2 B and D), treated or not treated with various concentrations of BI 5700, and exposed or not exposed to TGFβ. BI 5700 caused no alterations of epithelial morphology in cells cultivated on porous support (filters), allowing epithelial polarization (Fig. 2A, bottom left). In a more physiological culture system to analyze epithelial cell behavior and plasticity, that is, 3-dimensional serum-free collagen I gel cultures,15 fully polarized EpRas control cells formed tubular and alveolar structures with large lumina (Fig. 1B) that remained unchanged upon treatment with BI 5700 (Fig. 1B, bottom left). Similarly, BI 5700 up to 8 µM neither affected E-cadherin expression at the cell surface nor induced mesenchymal markers (vimentin) (Fig. 2 C and D, middle and bottom left panels). In EpRas control cells not treated with inhibitor, TGFβ induced EMT as expected, causing the cells to adopt a fibroblastoid morphology and to form disorganized strands and chords of cells with fibroblastoid appearance (Fig. 2 A and B, top panels). These cells had lost E-cadherin expression and strongly expressed vimentin (Fig. 2 C and D, top panels). β-actin changed from a cortical plasma membrane localization (not shown) to a cytoplasmic, sometimes stress fiber–like localization (Fig. 2D, top middle right).
Figure 2.
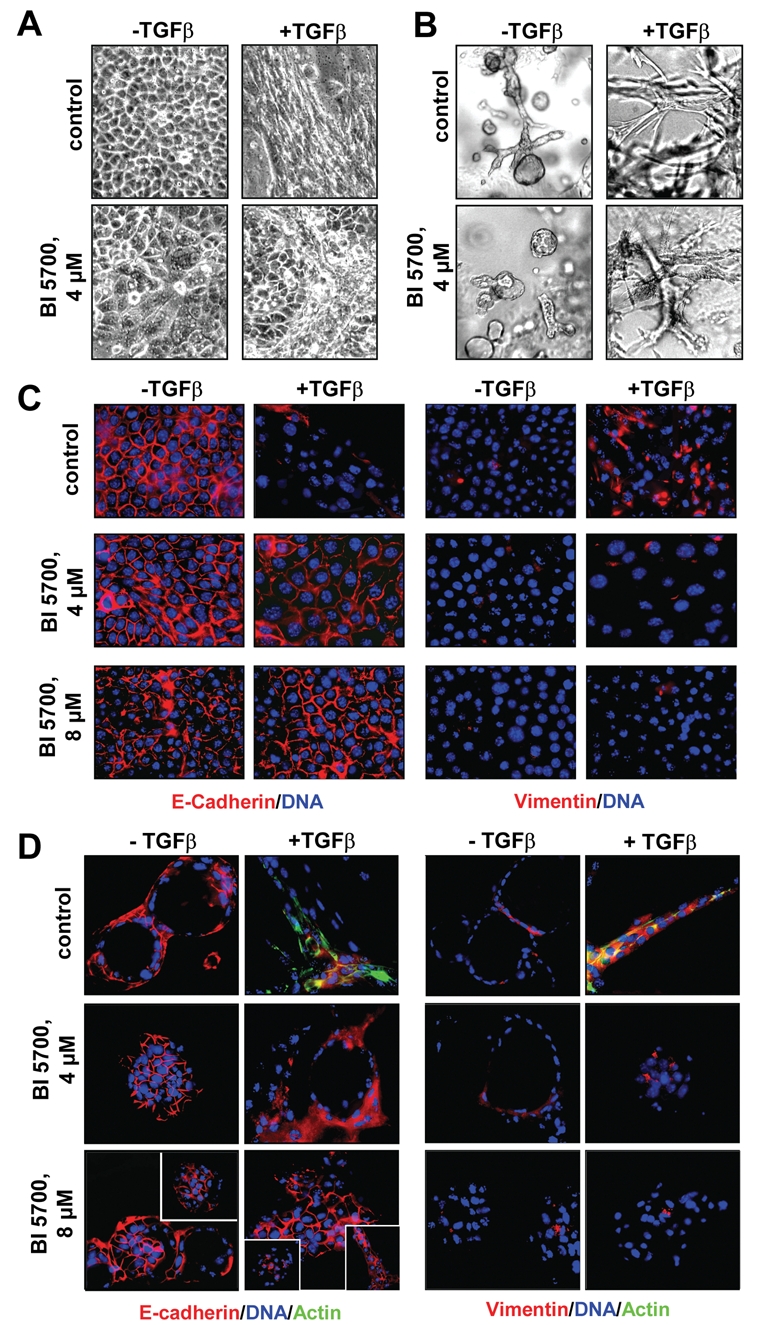
Selective inhibition of IKK2 abrogates EMT in EpRas cells. EpRas cells were seeded on porous supports (filters) or into 3-dimensional collagen gel cultures, exposed (+) or not exposed (–) to TGFβ (5 ng/mL; days 2-7) and treated or not treated with BI 5700 for 7 days at the concentrations indicated. Phase (A) or bright field micrographs (B) were prepared, and cells were then processed for immunofluorescence analysis (C and D). Confocal images of EpRas cells either seeded on porous supports (C) or into collagen gels (D) are shown before or after TGFβ and BI 5700 treatment, immunostained for E-cadherin or vimentin (red) as indicated, with DAPI counterstaining for DNA (blue). In D, cytoplasmic staining of β-actin (green) is also shown in the control + TGFβ panels, while rare structures with persisting EMT are shown in insets of the 8 µM BI 5700 panels. Note that the left panels in Figure 2A show a similar epithelial morphology, the somewhat different appearance in the BI 5700 4 µM panel being caused by a lower cell density. Likewise, the 3-dimensional structures (solid and hollow) in the left panels of Figure 2B are also similar, given the normal variability of the collagen gel structures obtained. Original magnification, 400x.
Upon addition of BI 5700 to EpRas cells exposed to TGFβ on filters and in collagen gels, the cells failed to undergo EMT. Instead, the cells formed epithelial islands sometimes intermingled with more mesenchymal cells on filters and compact but rarely hollow structures in collagen gels (Fig. 2 A and B, bottom panels). EMT was inhibited to a large extent by 4 µM and completely by 8 µM BI 5700, as shown by a full gain and maintenance of plasma membrane E-cadherin, as well as a complete loss of vimentin expression (Fig. 2 C and D, middle and bottom right panels). In conclusion, BI 5700 completely inhibited EMT of EpRas cells at concentrations that block NF-κB activity in EpRas cells as determined in Figure 1.
To further address the specificity of BI 5700-mediated effects, we tested this compound on EpRas cells, in which EMT was not induced by TGFβ but by overexpression of a constitutively active IKK2 mutant (EpRas-CA-IKK2). As a control for side effects of BI 5700 on epithelial polarity, EpRas cells in which EMT was suppressed by transdominant (TD) IκBα15 were employed (EpRas-TD-IκBα cells). As expected, BI 5700 did not affect the maintenance of an epithelial morphology and epithelial marker expression of EpRas-TD-IκBα cells. Even after treatment with TGFβ plus the highest dose of BI 5700 applied (8 µM), these cells fully maintained a cobblestone-like epithelial morphology (not shown), plasma-membrane expression of E-cadherin, and lack of vimentin expression (Supp. Fig. S1A). Thus, BI 5700 caused neither toxicity nor other detectable phenotypic changes on EpRas-TD-IκBα cells regardless of the presence or absence of TGFβ.
In EpRas-CA-IKK2 cells, however, BI 5700 inhibited both the partial EMT caused by CA-IKK2 in the absence of TGFβ and the more complete EMT seen in EpRas-CA-IKK2 cells after exposure to TGFβ.15 This was already detectable at 4 µM BI 5700 but more complete at 8 µM (Supp. Fig. S1B). These results were confirmed by quantitatively evaluating the area of mesenchymal structures in relation to the total area of cells that had grown to confluency on porous supports (Supp. Fig. S1C). In conclusion, BI 5700 even inhibited EMT induced by constitutive, IKK2-mediated NF-κB activation but was completely inactive on EpRas-TD-IκBα cells, in which the activation of NF-κB via IKK2 is already fully blocked by nonphosphorylatable and thus constitutively active IκBα.
BI 5700 Prevents or Reverses EMT in Murine Breast and Colon Carcinoma Cell Lines
To determine if BI 5700 would also interfere with EMT in other murine carcinoma cell lines, we explored its activity in the mammary and colon carcinoma cell lines 4T1 and CT26, respectively. 4T1 cells represent the only fully metastatic member of a series of cell lines that perform increasing numbers of steps of the metastatic process after mammary fat-pad (orthotopic site) injection,26 while little was known about their EMT phenotype. According to published data,37 the 67NR cell line already shows partial but constitutive EMT but lacks enhanced migration and is not metastatic from orthotopic sites, while the 168FARN and 4T07 lines display a complete, constitutive EMT phenotype in culture (A. Csiszar and H.B., unpublished data) (Fig. 3A). 168FARN cells also metastasize after tail-vein injection but not from orthotopic sites.38 Interestingly, 4T1 cells display an epitheloid but highly migratory phenotype,37 but no information was available whether these cells undergo TGFβ-induced EMT.
Figure 3.
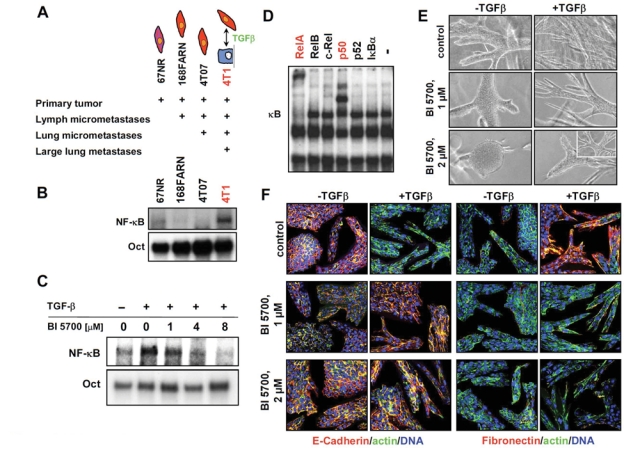
Inhibition of IKK2 prevents TGFβ-induced EMT in 4T1 mouse mammary tumor cells metastasizing from orthotopic tumor sites. (A) Scheme depicting the gross cell culture phenotype of cells from the 4T1 cell series and the in vivo effects of these cells obtained after injection into mice, as revealed by published data37,38 and our own studies. The cell symbols refer to epithelial cells (blue) and to a partial (pink) or complete EMT phenotype (red). (B) Cell extracts from the 4T1 cell series described in A were assayed for NF-κB DNA-binding activity in EMSA assays as described in the legend of Figure 1 (upper panel). (C) 4T1 cells were treated (+) or not treated (–) with TGFβ1 (5 ng/mL) for 2 hours—in the absence or presence of BI 5700 at the concentrations indicated—and analyzed by EMSA (lower panel). (D) Subunit composition of NF-κB in 4T1 cells was assessed by supershift of EMSA-derived NF-κB/DNA complexes with antibodies targeting individual complex members, indicating that the canonical NF-κB pathway (RelA/p50; red) is activated in 4T1 cells. (E) 4T1 cells were seeded into collagen gels for 2 days to start structure formation and then treated (+) or not treated (–) with TGFβ, in the presence or absence of BI 5700 at the concentrations indicated. Note the strong tendency of the inhibitor-treated cells to form more compact structures. (F) 4T1 cells cultivated and treated in collagen gels as described in E were immunostained with antibodies to E-cadherin (red) (left panels) or fibronectin (red) (right panels), both counterstained for β-actin (green). TGFβ induced complete EMT in 4T1 cells (loss of E-cadherin, actin cytoplasmic, or in stress fibers and gain of fibronectin) (top panels), while exposure to 2 µM BI 5700 completely prevented TGFβ-induced EMT in 4T1 cells (E-cadherin and β-actin cortical; no fibronectin) (bottom panels). DAPI-stained nuclei are depicted in blue.
We were especially interested in the 4T1 cell line because it was the only member of the 4T1 cell series (Fig. 3A) that showed highly enhanced NF-κB pathway activity in EMSA assays (Fig. 3B). TGFβ further enhanced this high basal NF-κB activity, which was inhibited by BI 5700 in a dose-dependent fashion (Fig. 3C). Addition of antibodies to RelA and p50 resulted in prominent supershifts, indicating that the canonical NF-κB pathway is activated in 4T1 cells (Fig. 3D). We therefore developed conditions allowing collagen gel cultures of these cells (see Material and Methods) and analyzed the cell phenotype minus and plus TGFβ, before or after addition of BI 5700. The untreated cells formed compact but never hollow structures in collagen gels, which expressed plasma membrane E-cadherin and cortical β-actin but no fibronectin (Fig. 3 E and F, top left). Treatment with BI 5700 further compacted the epitheloid collagen structures obtained in the absence of TGFβ (Fig. 3F, middle and bottom left panels). This effect was also observed with other inhibitors of EMT (not shown) and indicates that BI 5700 induces a more epithelial phenotype in the untreated, epitheloid 4T1 cells. TGFβ treatment, however, caused 4T1 cells to form disorganized strands and chords of fibroblastoid cells, showing marker expression typical for a complete EMT phenotye (no E-cadherin, high vimentin, actin cytoplasmic, or in stress fibers) (Fig. 3 E and F, top panels). BI 5700 inhibitor treatment of 4T1 cells at 2 different concentrations completely prevented TGFβ-induced EMT, as indicated by morphology and marker analysis (Fig. 3 E and F, middle and bottom panels).
To determine whether BI 5700 would also reverse constitutive EMT, we chose the murine colon carcinoma cell line CT26. These cells show complete EMT when grown on plastic but can be reversed to an epitheloid phenotype re-expressing cytoplasmic E-cadherin and showing strongly reduced vimentin by retroviral expression of a dominant-negative (dn) TGFβRII receptor mutant.25 Therefore, this line allows us to determine whether inhibition of IKK2 would induce MET (mesenchymal-epithelial transition). In modified (serum-containing) collagen gels, the untreated cells grew in disorganized clumps of E-cadherin-negative, fibronectin-low cells, while TGFβ treatment further dispersed these clumps and strongly enhanced fibronectin expression (Supp. Fig. S2 A and B, top panels). To our surprise, BI 5700 induced a full MET in CT26 cells treated or not treated with TGFβ, causing the cells to form tight balls with few cells invading the collagen gels, which showed strong cytoplasmic E-cadherin expression and little fibronectin (Supp. Fig. S2 A and B, middle and lower panels), similar to what has been previously observed in dn-TGFβRII-expressing cells on plastic.25 We conclude that BI 5700 can also fully reverse the constitutive EMT occurring in the CT26 model, thus inducing a full MET.
Expression of Transdominant IκBα in 4T1 Cells Decreases EMT In Vivo and Attenuates Lung Metastasis Formation from Orthotopic Tumor Sites
The above results suggested that a relevant mouse model to test BI 5700 for a possible in vivo activity against metastatic tumor progression would be BALB/c mice injected with 4T1 cells. These cells can metastasize from orthotopic tumor sites, while BI 5700 causes a complete block of EMT by inhibiting IKK2 activity in 4T1 cells cultured in vitro (Fig. 3). To establish proof of principle that blocking IKK2 activity could interfere with metastasis from 4T1-induced mammary tumors, we injected BALB/c mice with 4T1 cells expressing TD-IκBα from a retroviral vector using empty vector-expressing cells as controls. As expected, TD-IκBα-4T1 cells showed strong expression of IκBα (Fig. 4A) and exhibited no DNA-binding activity in EMSA assays (Fig. 4B). We then injected 4T1 cells expressing either empty vector or TD-IκBα into mammary gland fat pads of BALB/c mice and checked the mice for occurrence of metastases. Mice were sacrificed when large mammary gland tumors had developed, and lungs were excised to analyze them for metastasis formation. The control mice developed huge metastases that overgrew the entire lung (Fig. 4C, top left), while metastasis formation in mice injected with TD-IκBα cells was much weaker, as indicated by much fewer and smaller metastases in representative lungs (Fig. 4C, top right). Immunohistochemical staining of sections from lung metastases for E-cadherin (Fig. 4C, middle panel) and fibronectin (Fig. 4C, lower panel) showed that the cells in the empty vector controls had undergone EMT (no or delocalized, cytoplasmic E-cadherin, and strong fibronectin staining), while the metastases induced by TD-IκBα cells were characterized by plasma membrane–E-cadherin staining and weak fibronectin staining, indicating an inhibition of EMT resulting from NF-κB blockade in metastatic cancer cells in vivo.
Figure 4.
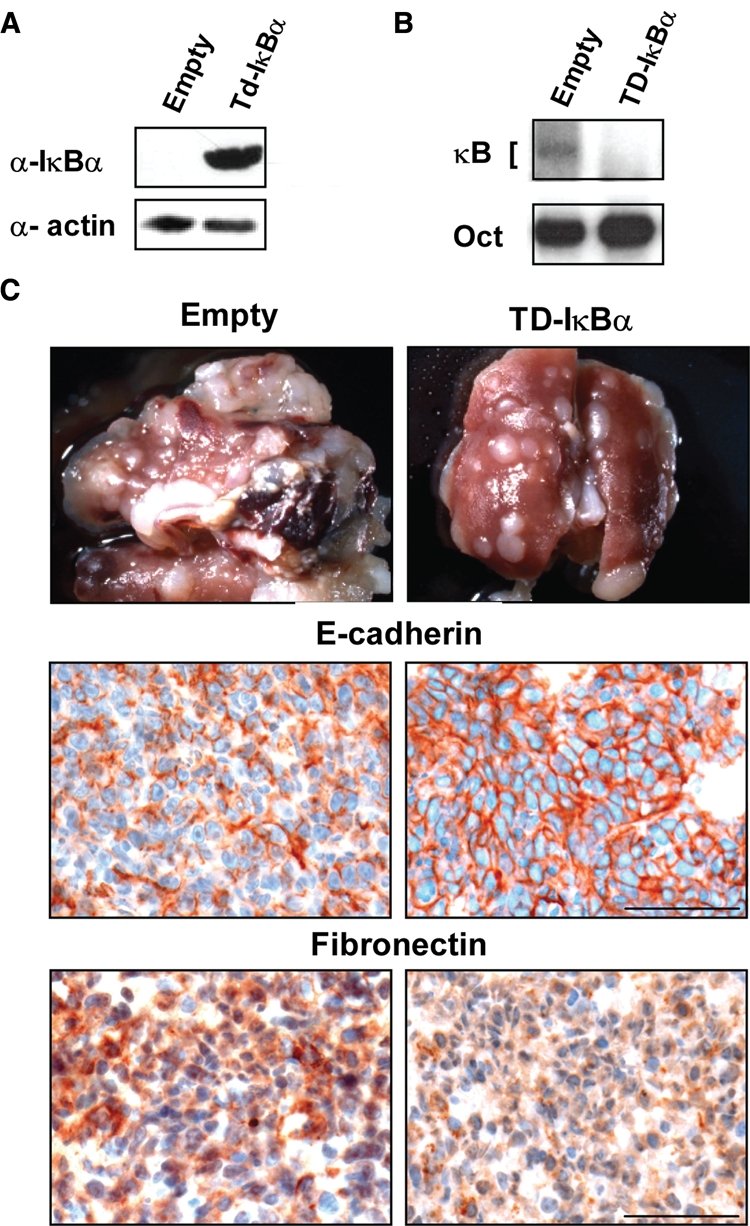
Metastasis formation of 4T1 cells in vivo requires an activated NF-κB pathway. (A) 4T1 cells were stably infected with retroviral vectors containing transdominant (Td) IκBα-cDNA and an empty vector control and assayed for expression of the dominant interfering mutant protein by Western blot analysis of whole-cell extracts, using IκBα- and IKK2-specific antibodies. Equal loading was assessed using an actin-specific antibody (bottom panel). (B) Extracts from the above cells were assayed for NF-κB DNA-binding activity by EMSA assays, showing the expected inhibition of NF-κB DNA binding by Td-IκBα expression. (C) 4T1 cells stably expressing TD-IκBα or empty GFP vector (empty) were injected into the fat pads of nude mice (5 × 105 cells per injection; 4 animals per cell type), and mice were analyzed for the presence of lung metastases. Note the strong reduction in number and size of metastatic lung nodules from mice injected with TD-IκBα cells, as compared to lungs from control cell–injected mice (upper panels). Serial paraffin sections from lung metastases of mice injected either with 4T1/empty or with 4T1-TD-IκBα cells were stained by IHC with antibodies to E-cadherin (middle) and fibronectin (bottom). Note the increased plasma membrane E-cadherin and decreased fibronectin expression in 4T1-TD-IκBα-induced metastases. Bar, 100 µM.
Oral Application of BI 5700 Attenuates Metastatic Capacity of 4T1 Cells in Mice
The above results encouraged us to treat 4T1 cell–injected BALB/c mice with BI 5700 to determine whether a small-molecule inhibitor of IKK2 would show efficacy in reducing metastasis, as has been observed with a transfected IκBα superrepressor. Pharmacokinetic studies after p.o. application to mice (Supp. Fig. S3) revealed a maximal exposure (Cmax) of approximately 18 µM at 6 hours after dosing when applied at a single dose of 150 mg/kg. To enable a continuous plasma concentration above 6 to 8 µM for extended time periods, we settled on a twice daily application of 75 mg/kg, which should produce a BI 5700 concentration enabling at least partial target inhibition for up to 24 hours.
One day after subjecting mice to injection of 4T1 cells into the mammary fat pad, mice were treated with BI 5700 applying the regimen described above, while control mice received the solvent (Natrosol, Ashland Inc., Wilmington, DE) only. After 21 days, all mice had developed macroscopic mammary gland tumors. Determination of tumor sizes yielded a reduction from 375 mm3 in control mice to 261 mm3 in the BI 5700-treated mice (Fig. 5A), a statistically significant but moderate effect. In order to assess the inhibitor effects on metastasis, mice were treated for an additional 7 days, totaling 28 days. This procedure revealed a pronounced effect of BI 5700 on lung metastasis, as determined by an almost 3-fold reduction of additional lung weight due to metastasis (Fig. 5B) and a clear reduction of the number of metastases visible on the lung surface (representative example shown in Fig. 5C). To extend these findings, we also evaluated the size of the lung metastases measured in serial sections through the lung (one section every 3 mm). As depicted in Supplementary Figure S4 A and B, numbers of small metastases had not been significantly reduced, while midsized and large metastases were significantly reduced upon treatment with BI 5700.
Figure 5.
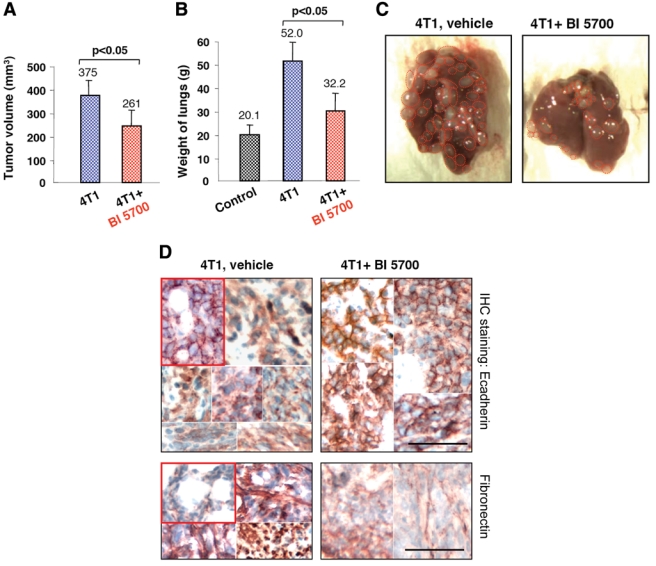
Oral application of the IKK2 inhibitor BI 5700 moderately slows tumor growth and strongly attenuates metastatic potential of 4T1 cells in vivo. (A) In vivo growth properties of 4T1 tumors after injection of 4T1 cells (2 × 105 cells per injection, 2 injection sites per animal) into the mammary glands of Balb-C mice (4 animals per group), treated daily with vehicle (Natrosol, Ashland Inc., Wilmington, DE) or 150 mg/kg BI 5700, administered as a split dose (Δ6 hours) for 21 days. Tumor volumes are shown as mean ± SEM (n = 8), yielding a tumor size reduction by BI 5700 determined as significant by the Student t test (P < 0.05). (B) Lung weights from the same mice as in A, determined 1 day after commencement of treatment with vehicle control or 150 mg/kg BI 5700, as described above and presented as mean ± SEM. BI 5700-treated mice showed a highly significant reduction of lung weights as determined by the Student t test (P < 0.05), approaching lung weights of mice receiving no 4T1 cells. (C) Photographs of representative lungs from a control animal (4T1, vehicle) and a BI 5700-treated animal (4T1 + BI 5700) are shown. Metastases visible at the lung surfaces are marked by dotted, red circles. For more quantitative analyses, see Supplementary Figure S4. (D) Sections from lung metastases from control (vehicle)– and BI 5700-treated mice were stained by immunohistochemistry using antibodies to E-cadherin (top panels) and fibronectin (bottom panels). The insets (red frame) depict representative areas from treated and untreated unaffected lungs as controls, showing E-cadherin localized to the plasma membrane, accompanied by a weak fibronectin staining. Two to 6 typical regions from metastatic lung areas of several mice were assembled into the panels to display the range of phenotypes observed. Note the delocalization of E-cadherin to the cytoplasm and strong fibronectin staining in the metastases from vehicle-treated animals but clear plasma membrane localization of E-cadherin and weak fibronectin staining in the lungs from BI 5700-treated mice. Bar, 100 µM.
We also stained sections from metastatic lungs by immunohistochemistry for E-cadherin and fibronectin (Fig. 4C, middle and lower panels). While many lung metastases from control animals showed strong delocalization of E-cadherin to the cytoplasm and high expression of fibronectin (Fig. 5D, left panels), almost all metastases from the BI 5700-treated mice expressed plasma membrane–located E-cadherin and low levels of fibronectin (Fig. 5D, right panels), comparable to the localization of E-cadherin and fibronectin in normal lung tissue adjacent to the lung metastases (Fig. 5D, insets in left panels). In conclusion, our results indicate that IKK2 is required for EMT and metastasis in the 4T1 model in vivo and that EMT blockade upon treatment with a selective IKK2 kinase inhibitor is paralleled by a clear attenuation of the metastatic potential of 4T1 cells.
Discussion
A number of studies have linked the NF-κB pathway to an enhanced metastatic capability of primary tumors, while the precise mechanisms how this is achieved are only partly understood. In this article, we characterize a novel and highly selective IKK2 small-molecule kinase inhibitor, termed BI 5700, and demonstrate a key role of IKK2 in both the induction and maintenance of EMT in a panel of mouse models of tumor progression both in vitro and in vivo. Our data provide the first evidence that a small-molecule IKK2 inhibitor can reduce the number, size, and dedifferentiation of lung metastases and suggest that a key mechanism for its antimetastatic activity is its effect on blocking epithelial-mesenchymal transitions.
Because of their importance in regulating the expression of proinflammatory cytokines and chemokines, angiogenesis regulators, and prosurvival and proliferation molecules, IKKs and IKK-related kinases have been implicated in the pathogenesis of cancer.17,23,39 For example, IKK2 has been described as a breast cancer oncogene based on its ability to degrade forkhead box O3a (FOXO 3a),40 which is a forkhead transcription factor required both for cell cycle inhibition and reduction of apoptosis. Moreover, the nuclear function of IKK1 has been implicated in prostate cancer metastasis by inhibiting the expression of the tumor suppressor Maspin.41 Also, the IKK-related kinase TBK1 can prevent cancer cell apoptosis via the RalB/Sec5 pathway,42 and IKKε has been identified as a breast cancer oncogene that regulates NF-κB activity, cell proliferation, and invasion.43,44 However, the relative importance of IKK family members in regulating cancer metastasis and EMT has not been previously addressed. Results obtained in this study using BI 5700, which is more than 65-fold more potent on IKK2 as compared to IKK1 and more than 1,000-fold selective versus TBK1 and IKKε, clearly point toward a key role of IKK2 in all models of tumor progression that we have investigated. BI 5700 effectively blocks NF-κB activity in EpRas and 4T1 breast cancer cells and prevents TGFβ-induced EMT while being relatively nontoxic on untreated cells. Importantly, the 4T1 model allows metastasis formation from orthotopic, primary mammary gland carcinomas, thus circumventing the limitation that EpH4/EpRas-based cells form only metastases after tail-vein injection. BI 5700 attenuated spontaneous lung metastasis formation from 4T1-induced, orthotopic mammary tumors with concomitant upregulation and downregulation of epithelial and mesenchymal markers, respectively, indicating that blockade of EMT by BI 5700 is indeed essential for its antimetastatic activity. In contrast, tumor growth is only moderately reduced, indicating that IKK2 is a selective target for antimetastatic therapies.
Based on the complexity of the signaling networks that regulate induction of EMTs5,9 and the plasticity of these transitions, a central question focuses on the most promising approaches toward effective and safe blockade of EMTs, thereby significantly effecting disease outcome. IKK2 appears to be a central player in the regulation of EMTs, integrating signaling inputs from multiple EMT inducers such as TGFβ and TNFα and funneling them via NF-κB to E-cadherin repressors such as Snail. In particular, a key pathway for inflammation-induced metastasis has recently been reported. It has been demonstrated that the inflammatory cytokine TNFα stabilizes the critical EMT transcription factor Snail via the activation of the NF-κB pathway, resulting in loss of E-cadherin expression and the initiation of EMT.18 Thereby, NF-κB can activate Snail both on the transcriptional level45-47 as well as at the posttranslational level.18 It is thus likely that IKK2, which is known to be activated by TNFα, plays an integral role in this pathway and that the IKK2/NF-κB axis regulates the activity of Snail through both transcriptional and posttranslational mechanisms at EMT. Based on the central role of IKK2 in governing EMTs, as well as previous successes using kinase inhibitors in cancer therapy, IKK2 holds promise as a suitable target to block tumor progression.
While the role of IKK2 in tumor progression may be fairly broad and general, our data obtained using BI 5700 indicate that IKK2 contributes to cell proliferation and survival only in a subset of cancer cell lines, that is, those cell lines harboring mutations within the NF-κB pathway (or associated pathways), resulting in enhanced pathway activity. Such cell lines are most sensitive to growth inhibition caused by treatment with BI 5700. For example, our results showing that multiple myeloma cell lines harboring pathway mutations result in enhanced sensitivities toward BI 5700 are in line with a previous study33 that uses the selective IKK2 inhibitor MLN120b. A systematic analysis showed that 20% of patients with multiple myeloma harbor NF-κB-activating mutations, the most common of which was the inactivation of TRAF3.33 Pathway dependence has also been suggested to occur in patients with T-cell acute lymphoblastic leukemia (T-ALL), carrying activating mutations in the NOTCH1 gene.31 In line with the genotype-dependent sensitivities described for T-ALL lines, BI 5700 inhibited proliferation of representative cell lines, whereas it showed limited or no activity on a panel of breast cancer cell lines (Table 1) or melanoma cell lines (not shown) that were thought to harbor constitutive IKK/NF-κB signaling according to studies with less selective IKK2 inhibitors.48 Despite genotype-dependent sensitivities of our cell line panel to BI 5700, the most sensitive cell lines are affected in the low micromolar range, consistent with data obtained by Annunziata et al.,33 using a different selective IKK2 inhibitor. These findings, showing that kinase inhibitory activity of selective IKK2 inhibitors at the nanomolar range is only translated into a single-digit micromolar cellular activity on cancer cell lines, could be explained by a suboptimal cell permeability of these compounds or by partial compensation, for example, by other family members. Interestingly, Lam et al.49 have identified a compensatory role for IKK1 (IKKα) as an IκBα kinase under conditions of IKK2 inhibition in cell lines derived from non-Hodgkin lymphomas. It remains to be studied whether IKK1 can play a role in the models described in this study, either in parallel to IKK2 or as a compensatory mechanism, awaiting suitable tool compounds with sufficient selectivity.
Because of the major role of NF-κB in tumor progression, therapies directed against this pathway are under intense study.23,50 A recent study by Bain et al.29 indicates, however, that results obtained with first-generation IKK2 inhibitors should be interpreted with caution. A comparison of the data in our Supplementary Table S1 and data by Bain et al.29 clearly shows that the 3 IKK2 inhibitors BMS 345.541, PS-1145, and SC514 are not only less potent than BI 5700 but, in contrast to BI 5700, also cross-inhibit important other kinases such as Casein kinase 1/2, PIM1/3, Aurora B, and so on. Furthermore, BI 5700 was also completely inactive on numerous receptor tyrosine kinases (RTKs) (Supp. Table S1), while the 3 earlier IKK inhibitors were not tested on these RTKs.29 The exquisite kinase selectivity of BI 5700 renders it a superior compound to further establish genotype-dependent sensitivities of cancer cell lines and will be useful to further address the role of IKK2 in EMT and metastasis both in vitro and in vivo. It is hoped that a better mechanistic understanding of IKK2 and other IKK family members in tumor initiation, maintenance, and progression, using suitable tool compounds, will ultimately help develop better therapies, leading to improved patient outcome.
Materials and Methods
Chemistry
The thienopyridine BI 5700 was derived from a chemical lead optimization program designed for selective IKK2 inhibitors (issued patent US6974870).
In Vitro Kinase Activity Assays
Kinase assays for human IKK2 were performed as previously described.27 In addition, 59 additional human kinases were profiled for inhibition by BI 5700 by using the Z’LYTE biochemical assay format (SelectScreen profiling service, Invitrogen, Carlsbad, CA) according to the supplier’s instructions. ATP test concentrations amounted to apparent Km values. For Axl, BRAF, MEK1, JNK1, and PDK1, an ATP concentration of 100 µM was used. For IKK1, the ADAPTA assay format with an ATP concentration of 10 µM was applied (SelectScreen, Invitrogen).
Cell Proliferation Assays in Human Cancer Cell Lines
Human tumor cell lines were obtained from the American Type Culture Collection (Manassas, VA) or the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and were cultured according to the supplier’s instructions. Cell proliferation assays were performed by incubation in the presence of various concentrations of BI 5700 or DMSO for 72 hours, and cell growth was assessed by determining alamarBlue dye (AbD Serotec, Kidlington, UK) conversion in a fluorescence spectrophotometer. Effective concentrations at which cellular growth was inhibited by 50% (EC50) were extrapolated from the dose-response curve fit using Graph Pad Software (San Diego, CA).
NF-κB Assays
Western blots, EMSA assays, and semiquantitative RT-PCR analysis of the NF-κB target gene MCP-1 were as described.15 Retroviral infection of 4T1 cells with TD-IκBα and parental vector were done as described.15
EMT Assays in Cell Culture
Origin and culture conditions of EpRas cells were described earlier.20,25 Marker analysis of cells grown on porous support, collagen gel culture, marker analysis, and apoptosis assays were performed as reported earlier15 with the following alterations. A subclone of 4T1 cells26 selected for optimum epithelial morphology in the absence and complete EMT in the presence of TGFβ (ID5) was seeded into collagen gels as described, except that 500 cells were seeded into collagen gels overlaid with DMEM/F12 medium plus all published additions19 plus 0.2% FCS. After 24 hours, this medium was replaced by the same medium containing 5% FCS. Cells were treated with BI 5700 at final concentrations of 200 nM, 400 nM, 1 µM, and 2 µM (from day 2 onward) for 8 days, adding or not adding TGFβ at day 3. Thereafter, gels were fixed and antibody stained as described.15 CT26 cells were tested in the same fashion, seeding 1000 cells per gel and treating the cells with BI 5700 for 5 days.
Tumorigenesis and Metastasis Assays
All animal studies have been approved by the review board of the Institute of Molecular Pathology (IMP), according to IMP-held animal experiment approvals from the Austrian Bundesministerium für Bildung, Wissenschaft und Kultur. For combined tumorigenesis and metastasis assays, 6- to 8-week-old female BALB/c mice were used for mammary gland fat-pad injection (200,000 cells in 100 µL PBS per injection, 2 injection sites). In the lung metastasis assays using fat pad–injected TD-IκBα expressing 4T1 cells, mice were sacrificed when the control mice became moribund, lungs photographed, and then analyzed by IHC staining for E-cadherin and fibronectin as described.28 For tumor and metastasis assays during oral BI 5700 application, mice were sacrificed after 28 days, taking the mice off the drug for 24 hours before termination and analysis. Tumor sizes, lung weights, lung surface metastases, and histology of metastases by H&E staining were analyzed as described.28 Sizes of metastases were determined using an ocular grid with 100 squares covering 45 ± 5 cells per square at the magnification chosen, counting the number of squares occupied by a single metastatic node and calculating cell numbers per section (Supp. Fig. S4B).
Pharmacokinetic Studies
Mice were administered a single oral dose of BI 5700 (150 mg/kg), and blood was collected from the retro-orbital plexus of mice at the indicated time points after dosing (n = 3 mice per time point). Plasma was analyzed using high performance liquid chromatography–mass spectrometry methodology.
Statistical Analysis
We compared tumor volumes, lung weights, and numbers of metastases using a 2-sided Student t test. A P value of less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The authors thank Maria A. Impagnatiello and Katja Flandorfer for discussions and support of in vivo studies. They are grateful to Uta Manfras and Monika Kriz for excellent technical assistance.
Footnotes
Jürgen Braunger, Guido Boehmelt, Jeffrey B. Madwed, Erick R.R. Young, Daniel R. Marshall, and Norbert Kraut are employees of the Boehringer Ingelheim Group of Companies.
Thomas Wirth and Harald J. Maier have received financial support by the German Science Foundation (DFG SFB518).
Supplementary material for this article is available on the Genes & Cancer Web site at http://ganc.sagepub.com/supplemental.
References
- 1. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006;127:679-95 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100: 57-70 [DOI] [PubMed] [Google Scholar]
- 3. Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med 2008;359:2814-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006;12:895-904 [DOI] [PubMed] [Google Scholar]
- 5. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265-73 [DOI] [PubMed] [Google Scholar]
- 6. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial- mesenchymal transitions. Nat Rev Mol Cell Biol 2006;7:131-42 [DOI] [PubMed] [Google Scholar]
- 7. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 2009;28:15-33 [DOI] [PubMed] [Google Scholar]
- 8. Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008;27:6958-69 [DOI] [PubMed] [Google Scholar]
- 9. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005;17:548-58 [DOI] [PubMed] [Google Scholar]
- 10. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cordon-Cardo C, Prives C. At the crossroads of inflammation and tumorigenesis. J Exp Med 1999;190:1367-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cousssens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jackson L, Evers BM. Chronic inflammation and pathogenesis of GI and pancreatic cancers. Cancer Treat Res 2006;130:39-65 [DOI] [PubMed] [Google Scholar]
- 14. Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005;5:749-59 [DOI] [PubMed] [Google Scholar]
- 15. Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, et al. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 2004;114:569-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK β suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007;130:440-55 [DOI] [PubMed] [Google Scholar]
- 17. Naugler WE, Karin M. NF-κB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev 2008;18:19-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009;15:416-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, et al. Ras and TGF-β cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002;156:299-313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 1996;10:2462-77 [DOI] [PubMed] [Google Scholar]
- 21. Shin SR, Sanchez-Velar N, Sherr DH, Sonenshein GE. 7,12-dimethylbenz(a)anthracene treatment of a c-rel mouse mammary tumor cell line induces epithelial to mesenchymal transition via activation of nuclear factor-κB. Cancer Res 2006;66:2570-5 [DOI] [PubMed] [Google Scholar]
- 22. Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol 2007;9:470-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee DF, Hung MC. Advances in targeting IKK and IKK-related kinases for cancer therapy. Clin Cancer Res 2008;14:5656-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-κB and epithelial to mesenchymal transition of cancer. J Cell Biochem 2008;104:733-44 [DOI] [PubMed] [Google Scholar]
- 25. Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 1998;8:1243-52 [DOI] [PubMed] [Google Scholar]
- 26. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117:927-39 [DOI] [PubMed] [Google Scholar]
- 27. Morwick T, Berry A, Brickwood J, Cardozo M, Catron K, DeTuri M, et al. Evolution of the thienopyridine class of inhibitors of IκB kinase-β. Part I: hit-to-lead strategies. J Med Chem 2006;49:2898-908 [DOI] [PubMed] [Google Scholar]
- 28. Waerner T, Alacakaptan M, Tamir I, Oberauer R, Gal A, Brabletz T, et al. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 2006;10:227-39 [DOI] [PubMed] [Google Scholar]
- 29. Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007;408:297-315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med 2007;204:1813-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, et al. Targeting the NF-κB signaling pathway in Notch1-induced T-cell leukemia. Nat Med 2007;13:70-7 [DOI] [PubMed] [Google Scholar]
- 32. Weng AP, Ferrando AA, Lee W, Morris JP, 4th, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269-71 [DOI] [PubMed] [Google Scholar]
- 33. Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007;12:115-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, et al. BMS-345541 is a highly selective inhibitor of I κ B kinase that binds at an allosteric site of the enzyme and blocks NF-κ B-dependent transcription in mice. J Biol Chem 2003;278:1450-6 [DOI] [PubMed] [Google Scholar]
- 35. Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-κB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 2000;96:2537-42 [PubMed] [Google Scholar]
- 36. Gal A, Sjöblom T, Fedorova L, Imreh S, Beug H, Moustakas A. Sustained TGF β exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008;27:1218-30 [DOI] [PubMed] [Google Scholar]
- 37. Lou Y, Preobrazhenska O, auf dem Keller U, Sutcliffe M, Barclay L, McDonald PC, et al. Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev Dyn 2008;237:2755-68 [DOI] [PubMed] [Google Scholar]
- 38. Gumireddy K, Sun F, Klein-Szanto AJ, Gibbins JM, Gimotty PA, Saunders AJ, et al. In vivo selection for metastasis promoting genes in the mouse. Proc Natl Acad Sci USA 2007;104:6696-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol 2007;8:49-62 [DOI] [PubMed] [Google Scholar]
- 40. Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004;117:225-37 [DOI] [PubMed] [Google Scholar]
- 41. Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, et al. Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin. Nature 2007;446:690-4 [DOI] [PubMed] [Google Scholar]
- 42. Chien Y, Kim S, Bumeister R, Korchynskyi O, Zhang M, Gonias SL, et al. RalB GTPase-mediated activation of the IκB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006;127:157-70 [DOI] [PubMed] [Google Scholar]
- 43. Boehm J, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007;129:1065-79 [DOI] [PubMed] [Google Scholar]
- 44. Eddy SF, Guo S, Demicco EG, Romieu-Mourez R, Landesman-Bollag E, Seldin DC, et al. Inducible IκB kinase/IκB kinase ε expression is induced by CK2 and promotes aberrant nuclear factor-κB activation in breast cancer cells. Cancer Res 2005;65:11375-83 [DOI] [PubMed] [Google Scholar]
- 45. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol 2005;168:29-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-κB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007;26:7445-56 [DOI] [PubMed] [Google Scholar]
- 47. Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-κB and snail. Mol Cell Biol 2007;27:3165-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541 targets inhibitor of κB kinase and induces apoptosis in melanoma: involvement of nuclear factor κB and mitochondria pathways. Clin Cancer Res 2006;12:950-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lam LT, Davis RE, Ngo VN, Lenz G, Wright G, Xu W, et al. Compensatory IKKα activation of classical NF-κB signaling during IKKβ inhibition identified by an RNA interference sensitization screen. Proc Natl Acad Sci USA 2008;105:20798-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gilmore TD, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006;25:6887-99 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.