

Abstract
The FLT3 tyrosine kinase receptor is involved in both hematopoiesis and hematological malignancies. The Wnt/β-catenin pathway has been shown to participate in the self-renewal of hematopoietic stem cells and to cooperate with the mutant FLT3 receptors in leukemic transformation. However, the detailed biological impact of such a constitutively activated Wnt pathway remains to be further explored. Here, the authors report that activating mutations of FLT3 constitutively activate β-catenin by inhibition of GSK-3β in a PI3 kinase pathway–dependent manner. Ectopic expression of a dominant negative form of GSK-3β in FLT3-ITD–expressing cells activated β-catenin and blocked the downregulation of the TCF/β-catenin transcriptional activity induced by inhibition of FLT3 kinase. Furthermore, inhibition of cell proliferation and colony formation induced by such suppression of FLT3 kinase activity could be partially reversed by knockdown of GSK-3β and restored by knockdown of either TCF4 or β-catenin. Moreover, exogenous activation of the Wnt pathway also attenuated such inhibitory effect. These findings indicate that the potencies of the inhibitors of FLT3 kinase activity could be modulated by the activity of the Wnt/β-catenin pathway in the cells harboring FLT3-ITD mutations, and FLT3-ITDs signal through GSK-3β to activate β-catenin that this is likely to directly contribute to the leukemic phenotype.
Keywords: mutant FLT3 receptors, Wnt/β-catenin, GSK-3β, drug sensitivity
Introduction
The FLT3 tyrosine kinase receptor plays a critical role in hematopoiesis1 and is also involved in leukemogenesis.2,3 Gain-of-function mutations of FLT3 have been found in approximately 30% to 35% of patients with acute myeloid leukemia (AML), most commonly as in-frame insertions of variable length (internal tandem duplications [ITDs]) in the juxtamembrane domain and the missense point mutations in the kinase domain. Although these mutations have transforming activity,4,5 results from a murine bone marrow transplantation assay6 suggested that FLT3-ITD mutations needed to cooperate with other oncogenes to induce AML.7
The Wingless-type (Wnt) pathway has been shown to participate in self-renewal of hematopoietic stem cells8 and to act synergistically with the FLT3-ITD in leukemic transformation.9-11 Although activation of the Wnt/β-catenin pathway via direct interaction between the FLT3-ITD and β-catenin has been reported,11 it has also been shown that in 32D-FLT3-ITD and 32D-cKit-ITD cells, the ITD mutant kinase activities were associated with GSK-3β and β-catenin activities, although the autocrine mechanism seemed to be ruled out in this system.12 In the canonical Wnt pathway, the central player is β-catenin, whose activity is regulated by a cytosolic destruction complex containing AXIN, adenomatous polyposis coli, and GSK-3β as major components.13,14 Stimulation of the Wnt ligand inhibits the destruction complex and activates β-catenin, which functions as a transcriptional coactivator for lymphoid enhancer–binding factor 1/T cell-specific transcription factor (TCF)15-17 to activate its target genes, such as c-Myc and cyclin D1.18,19
The β-catenin regulations by the destruction complex have been shown to be cell type specific. Phosphorylation of β-catenin by GSK-3β is a critical step in the regulation of the Wnt signaling. Inhibition of GSK-3β by either its dominant negative form or lithium resulted in activation of TCF target transcripts in C57MG fibroblast cells but not in T-lymphocytes.20 Also, in NIH3T3 cells, inhibition of GSK-3β by AKT was not sufficient to induce TCF target genes.21 Thus, regulation of β-catenin needs to be studied carefully in each model system separately.
In this report, we further study the interaction between the FLT3-ITD and the Wnt signaling and specifically investigate β-catenin activation and the resulting biological effects in the context of the gain-of-function mutations of FLT3. Our results demonstrated that FLT3-ITDs signal GSK-3β to activate β-catenin and its downstream targets in a PI3 kinase pathway–dependent manner and that activation of the Wnt/β-catenin pathway in the context of activating mutations of the FLT3 receptor could modulate the potencies of the FLT3 kinase inhibitors.
Results
The Importance of the PI3 Kinase Pathway on the Constitutive Inhibition of GSK-3β and Activation of β-catenin in the Context of Mutant FLT3 Receptors
The activating mutations of FLT3 could render the growth of the murine factor IL3 dependent Ba/F3 and 32D cells factor independence and constitutively activated many downstream signaling pathways, including the PI3K/AKT and p44/42 MAPK pathways.22,23 Such IL3-independent growth biologically distinguishes the Ba/F3 cells expressing FLT3 activating mutations from the parental Ba/F3 cells expressing either empty vector or wild-type FLT3 that could not render the Ba/F3 cells IL3 independence. Because GSK-3β is a potential substrate of the PI3K/AKT pathway,24 we first looked at the activity of GSK-3β in the Ba/F3 cells expressing known activating mutations of the FLT3 receptor, FLT3-ITD and FLT3-N841I,5 as shown in Figure 1A. The phosphorylation of GSK-3β at serine 9, which inhibits the activity of GSK-3β,24-26 was increased in the mutant receptor expressing cells (compare lanes 3 and 5 to lanes 1), suggesting that GSK-3β was inhibited by the mutant FLT3 receptors. More importantly, such reduced GSK-3β activity was restored (lanes 4 and 6 of the pSer9GSK3β blot) when the autophosphorylation of the mutant FLT3 receptors was inhibited by PKC412 (lanes 4 and 6 of pTyr blot), a small-molecule inhibitor of the FLT3 kinase activity,27 indicating a dependence of such reduction on the mutant receptors.
Figure 1.
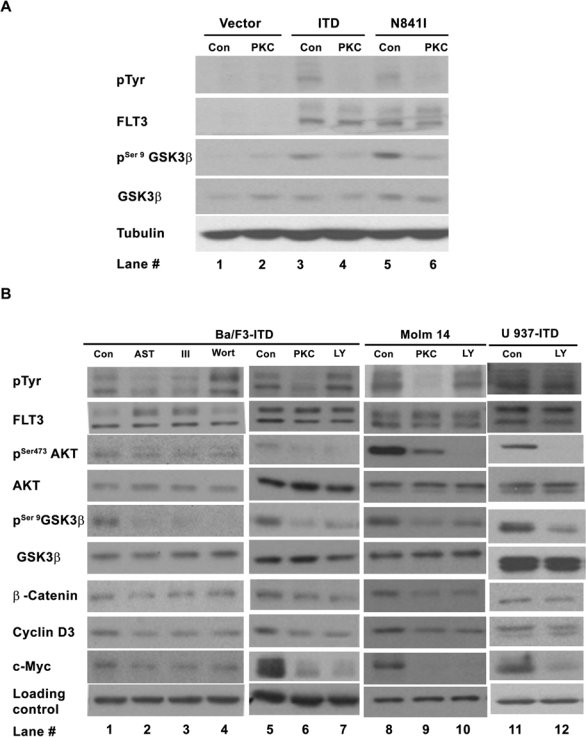
Importance of the PI3K/AKT/GSK-3β on the constitutive activation of β-catenin in mutant FLT3 receptor–expressing cells. (A) Mutant FLT3 receptors constitutively inhibit GSK-3β in a receptor kinase activity–dependent manner. Ba/F3 cells expressing FLT3-ITD, FLT3-N841I, or empty vector (pCIneo) were treated with 0.05 μM PKC412 (PKC) or DMSO (Con) for 30 min. The whole-cell lysis in NP-40 buffer was subjected to immunoblotting analysis using the antibodies indicated. (B) Inhibitions of the FLT3-ITD and PI3K/AKT pathways decrease β-catenin, cyclin D3, and c-Myc in the cells expressing FLT3-ITD. The cells were treated with the inhibitors of FLT3 kinase (0.05 μM PKC412, 0.1 μM AST 487 [AST], and inhibitor III [III]) and the inhibitors of the PI3K/AKT pathway (20 μM Ly294002 [Ly] and 0.2 μM Wortmannin [Wort) or DMSO (Con) for 30 min to 1 h and subjected to Western blotting analysis as described above. Lanes 1 through 10 of the loading level control were probed with antitubulin antibody, and lanes 11 and 12 of the loading level control were probed with anti-GADPH antibody.
GSK-3β activity is controlled by multiple signaling pathways28,29; to determine the importance of PI3 kinase signaling, an inhibitor of the PI3 kinase pathway, LY294002, was used for further study. In the cells either ectopically expressing FLT3-ITD (Ba/F3-ITD and U937-ITD) or derived from an AML patient sample harboring FLT3-ITD (Molm 14; Fig. 1B, lanes 7, 10, and 12), LY294002 reduced the level of phosphorylation at serine 473 of AKT (pSer473AKT) and resulted in decreased levels of phosphorylation at serine 9 of GSK-3β (pSer9GSK-3β), as well as decreased β-catenin, c-Myc, and cyclin D3. These results were further confirmed by a structurally unrelated PI3 kinase inhibitor, Wortmannin, in Ba/F3-ITD cells (Fig. 1B, lane 4). These 2 inhibitors had not much effect on the tyrosine phosphorylation of the mutant FLT3-ITD receptor (pTyr blot of Fig. 1B, lanes 4 and 7; Supp. Fig. S1, lanes 4 and 7). By contrast, an inhibitor of the p44/42 MAPK pathway, U01126, had no effect on the level of β-catenin (data not shown). These data demonstrate that in the absence of IL3, activation of the PI3 kinase pathway results in activation of AKT, inhibition of GSK-3β kinase activity, and increased levels of β-catenin, c-Myc, and cyclin D3 in the FLT3-ITD–expressing cells. Furthermore, 3 FLT3 kinase inhibitors, PKC412, AST 487,30 and inhibitor III,31 reduced the level of tyrosine phosphorylation of the mutant FLT3 receptor (pTyr blot of Fig. 1B, lanes 2, 3, and 6; Supp. Fig. S1, lanes 2, 3, and 6), as well as reduced the levels of pSer473AKT, pSer9GSK-3β, β-catenin, c-Myc, and cyclin D3 (Fig. 1B, lanes 2, 3, 6, and 9), suggesting the dependence of the activations of PI3K/AKT/GSK-3β and β-catenin on FLT3-ITD. Although we do not rule out other potential signaling pathway(s) that may also regulate β-catenin activity, our results strongly indicate that the PI3 kinase pathway, most likely via the AKT/GSK-3β axis, is important for the activation of β-catenin in the context of FLT3-ITD.
FLT3-ITD Constitutively Activates β-catenin via Inhibition of GSK-3β
Although GSK-3β is an important component in the β-catenin destruction box, its regulatory capacity is cell type specific. To determine such capacity in the context of gain-of-function mutations of FLT3 receptor, we investigated the effects of GSK-3β inhibition on the activity of β-catenin.
Pharmacologic Inhibitions of GSK-3β Increase Levels of β-catenin, c-Myc, and Cyclin D3 and Enhance TCF/β-catenin Transcriptional Activity
Two commonly used inhibitors of GSK-3β, lithium chloride32,33 and inhibitor VII, were first tested as shown in Fig. 2A (lanes 2, 3, 5, 6, 8, and 10). Both inhibitors suppressed the kinase activity of GSK-3β, as detected by the increased level of its phosphorylation at serine 9, and had not much effect on the tyrosine phosphorylation on the mutant FLT3-ITD receptor (pTyr blot of Fig. 2A, lanes 2, 3, 5, 6, 8, and 10; Supp. Fig. S1, lanes 10 and 11). The level of β-catenin went up in response to both drugs, indicating that inhibition of GSK-3β alone resulted in accumulation of β-catenin. We then used c-Myc and cyclin D3 to monitor β-catenin activity. In both Ba/F3-ITD and Molm 14 cells, their levels went up upon inhibition of GSK-3β associated with accumulation of β-catenin. In U937-ITD cells, the level of cyclin D3 (lane 10) went up following lithium treatment; the level of c-Myc did not change. It is possible that in U937 cells, expression of FLT3-ITD already resulted in a maximum accumulation of c-Myc (Fig. 2A, compare lane 9 with lane 7). These results indicate that pharmacologic inhibition of GSK-3β in the FLT3-ITD–expressing cells is able to activate β-catenin.
Figure 2.
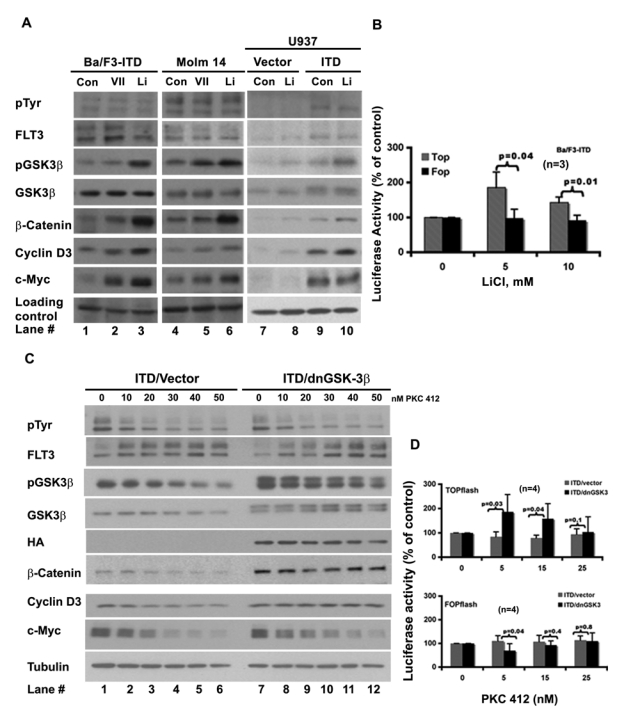
Inhibitions of GSK-3β activate β-catenin and attenuate the regulation of the mutant FLT3 receptor on the β-catenin activity. (A) GSK-3β inhibitions activate β-catenin. GSK-3β inhibitions increase the levels of β-catenin, c-Myc, and cyclin D3. The cells were treated with the inhibitors of GSK-3β (10 mM LiCl, 0.5 μM inhibitor VII) for 30 min. Whole-cell lysates were then subjected to Western blotting analysis using antibodies indicated. Lanes 1 to 6 of the loading level control were probed with antitubulin antibody, and lanes 7 to 10 of the loading level control were probed with anti-GADPH antibody. (B) Lithium increases the TCF/β-catenin–driven transcriptional activity. Ba/F3-ITD cells were coelectroporated with either TCF reporter TOPflash and pRL/thymidine kinase (TK) plasmid or the negative control plasmid of TCF reporter FOPflash and pRL/TK. Twenty to 24 h after electroporation, the cells were treated with lithium chloride for 30 min at the concentrations indicated and analyzed for luciferase activity using the dual-luciferase reporter assay kit. The normalized value of the luciferase activity (TOPflash or FOPflash) in each treatment compared with that of control (100%) was shown. The P values were calculated using an online t-test calculator (http://www.graphpad.com/quickcalcs/ttest1.cfm; the same calculator was used for other P value analyses in this study). (C) Differential responses of β-catenin and its targets to PKC412 inhibition between Ba/F3-FLT3-ITD-vector cells and Ba/F3-FLT3-ITD–dnGSK-3β cells. The Ba/F3-FLT3-ITD cells stably expressing either a dominant negative form of GSK-3β (dnGSK-3β) or the empty vector were treated with increasing concentrations of PKC412 for 3 h and harvested for protein extraction. Whole-cell lysates in NP-40 lysis buffer were then subjected to Western blotting analysis using the antibodies indicated. (D) Differential response of TCF/β-catenin responsible promoter (TOPflash) to PKC412 inhibition. The log phase growing Ba/F3-FLT3-ITD–vector cells and Ba/F3-FLT3-ITD–dnGSK-3β cells were coelectroporated with either TCF reporter, TOPflash, and pRL/TK plasmid or its negative control plasmid, FOPflash, and pRL/TK. Twenty to 24 h after electroporation, the cells were treated with PKC412 at the concentration indicated for 30 min and subjected to the luciferase assay as described in (B).
We then looked at the effect of LiCl on the TCF/ β-catenin driven promoter activity using the TOPflash/FOPflash TCF reporter system. In the TOPflash reporter, a full-length ORF of luciferase is driven by a TK minimal promoter with TCF binding sites, whereas in the FOPflash reporter, the TCF binding sites were mutated. Figure 2B shows that at the tested concentrations, LiCl was able to induce the activity of TOPflash (gray/stripe bar). However, the activity of FOPflash (black bar) remained relevantly constant in response to the treatment. This result further proved that inhibition of GSK-3β could induce the TCF/β-catenin transcriptional activity in the FLT3-ITD–expressing cells.
Expression of a Dominant Negative Form of GSK-3β Attenuated the Response of β-catenin, c-Myc, and Cyclin D3, as Well as TCF/β-catenin Driven Promoter, to the FLT3-ITD Suppression
Additional evidence that GSK-3β regulates β-catenin activity was sought using a dominant negative form of GSK-3β (dnGSK-3β with HA tag)20,32 stably expressed in Ba/F3-FLT3-ITD cells. The FLT3 kinase activity was suppressed by increasing concentrations of PKC412 to examine the response of β-catenin, c-Myc, and cyclin D3, using 3 pairs of ITD/vector and ITD/dnGSK-3β monoclonal lines (Fig. 2C and data not shown). The expression of dnGSK-3β was evidenced by the double bands of GSK-3β blot, as well as by the HA blot (Fig. 2C, lanes 7 to 12). FLT3-ITD autophosphorylation was reduced by increasing concentrations of PKC412 in both vector- and dnGSK-3β–expressing cells, as was phosphorylation at serine 9 of GSK-3β. However, β-catenin, c-Myc, and cyclin D3 showed differential responses. In vector-expressing cells, the levels of β-catenin, c-Myc, and cyclin D3 were reduced by PKC412 in a dose-dependent pattern (Fig. 2C, lanes 1 to 6), whereas in dnGSK-3β–expressing cells, the total levels of β-catenin and cyclin D3 remained relatively constant (Fig. 2C, lanes 7 to 12). The level of c-Myc did go down, but its decline was slower than that in the vector-expressing cells.
To further demonstrate that dnGSK-3β could attenuate the response of β-catenin to the inhibition of the mutant FLT3 receptor, we analyzed the effect of PKC412 on the TCF/β-catenin–driven promoter activity using TOPflash and FOPflash reporter assay as shown in Figure 2D. The top panel shows that in dnGSK-3β–expressing Ba/F3-ITD cells, PKC412 had no inhibitory effect on TOPflash (black bar), whereas in the vector-expressing Ba/F3-ITD cells, the TOPflash activity was inhibited (gray bar). In contrast, the FOPflash reporter showed a similar response pattern to PKC412 treatment in both vector- and dnGSK-3β–expressing Ba/F3-ITD cells, as shown in the bottom panel.
Taken together, these data demonstrate that in the FLT3-ITD–expressing cells, inhibition of GSK-3β induced β-catenin and its target genes c-Myc and cyclin D3 and further rendered these proteins, along with TCF/β-catenin–driven promoter, less sensitive to the inhibition of mutant FLT3 receptor. Our results suggest that inhibition of GSK-3β is sufficient to activate β-catenin in the context of FLT3-ITD, and GSK-3β could serve as a nexus for cross-talk between FLT3-ITD and the Wnt/β-catenin pathways.
Knockdown of GSK-3β Decreases the Potencies of the FLT3 Kinase Inhibitors on Proliferation and Colony Formation in the Ba/F3-FLT3-ITD Cells, and the Potency Could Be Restored via Knockdown of Either TCF4 or β-catenin
The GSK-3β–mediated differential responses of β-catenin to PKC412 inhibition prompted us to further study the potential biological effects induced by GSK-3β inhibition in the context of FLT3-ITD mutation.
Proliferation of Ba/F3-ITD-vector and Ba/F3-ITD–dnGSK-3β cells was assayed. Although the growth rates between these 2 cell lines were similar (data not shown), their proliferation showed differential responses to the FLT3-ITD kinase inhibition (Fig. 3A). At tested concentrations, the growth of Ba/F3-ITD–dnGSK-3β cells had a reduced response to PKC412, with the most significant reduction at 15 nM (Fig. 3A, left). This observation was further confirmed by 2 additional FLT3 kinase inhibitors, AST 48730 and FLT3 inhibitor III31 (middle and right). Such reduced sensitivity to the FLT3-ITD inhibition was further proved by transiently expressing GSK-3β siRNA (Fig. 3B, left). The siRNA suppression of GSK-3β is shown in Figure 3C.
Figure 3.
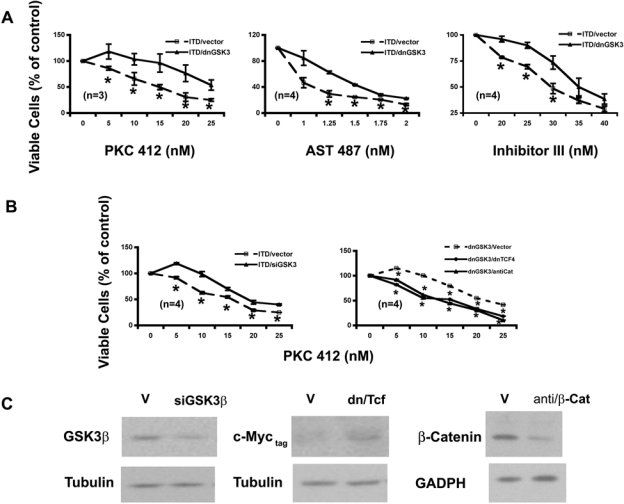
GSK-3β inhibitions attenuate the response of Ba/F3-FLT3-ITD cells to the FLT3 kinase inhibitors on cell proliferation in a Wnt/β-catenin pathway–dependent manner. (A) Stable expression of dnGSK-3β in Ba/F3-FLT3-ITD cells decreases the potencies of the FLT3 kinase inhibitors on proliferation. Ba/F3-FLT3-ITD cells stably expressing either empty vector or dnGSK-3β were treated with FLT3 kinase inhibitors for 68 to 72 h and then stained with trypan blue and counted under microscope, respectively. (B) Transient expression of GSK-3β siRNA in Ba/F3-FLT3-ITD cells also decreases the potency of the FLT3 kinase inhibitor on proliferation. The potency could be restored by transient expression of either the dominant negative form of TCF4 (dnTCF4) or antisense β-catenin (antiCat) in Ba/F3-FLT3-ITD cells stably expressing dnGSK-3β. The cells were electroporated with corresponding plasmid DNAs indicated. Twenty to 24 h after electroporation, cells were analyzed as in (A). (C) Expression levels of GSK-3β, TCF-4 (as detected by c-Myc tag), and β-catenin in transient assays. Cells were collected at the same time as transient assays in (B) and lysed in NP-40 lysis buffer. The whole-cell lysates were then subjected to immunoblotting analysis using antibodies indicated.
We next examined the effect of inhibiting GSK-3β on colony formation using a soft agar assay. More colonies were formed by the cells with knockdown of GSK-3β (Fig. 4C). As in proliferation assay, the potency of PKC412 on colony formation was also reduced by inhibition of GSK-3β via either stably expressing its dominant negative form (Fig. 4A) or transiently expressing its siRNA (Fig. 4B). The most significant reduction was at 5 and 15 nM, respectively. In both assays, the IC50 of PKC412 was increased about 10 nM upon inhibition of GSK-3β. Further analysis using AST 487 and FLT3 inhibitor III confirmed that suppression of GSK-3β did render the FLT3-ITD–expressing cells less sensitive to the inhibition of FLT3-ITD on colony formation (Fig. 4A).
Figure 4.
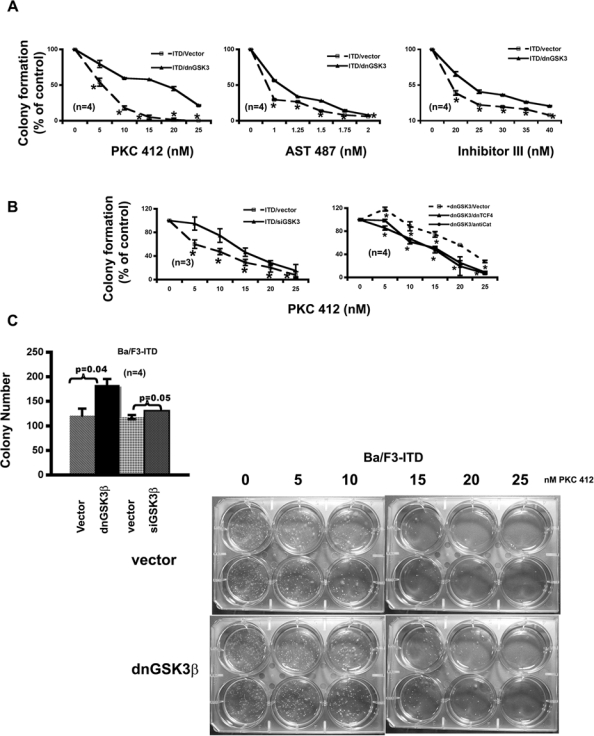
GSK-3β inhibitions attenuate the response of Ba/F3-FLT3-ITD cells to the FLT3 kinase inhibitors on colony formation in a Wnt/β-catenin pathway–dependent manner. (A) Stable expression of dnGSK-3β in Ba/F3-FLT3-ITD cells decreases the potencies of the FLT3 kinase inhibitors on colony formation. Both Ba/F3-FLT3-ITD-vector and Ba/F3-FLT3-ITD–dnGSK-3β cells were seeded at a density of 500 cells/well in 6-well soft agar plates with FLT3 kinase inhibitors at the concentrations indicated. After 2 wk, colonies were counted. (B) Transient expression of GSK-3β siRNA in Ba/F3-FLT3-ITD cells also decreases the potency of the FLT3 kinase inhibitor on colony formation. The potency could be restored by transient expression of either dnTCF4 or antisense β-catenin (AntiCat) in Ba/F3-FLT3-ITD–dnGSK-3β cells. The cells were electroporated with the plasmid DNAs indicated; 20 to 24 h after electroporation, cells were seeded at a density of 500 cells/well in 6-well soft agar plates with FLT3 kinase inhibitor at the concentrations indicated. After 2 wk, colonies were counted. (C) GSK-3β inhibitions in Ba/F3-FLT3-ITD cells enhance colony formation and attenuate the response to PKC412 inhibition. The left panel shows that Ba/F3-FLT3-ITD cells with GSK-3β knockdown formed more colonies. The right panel is the actual soft agar plates showing the differences of colony number formed by Ba/F3-FLT3-ITD–vector cells and Ba/F3-FLT3-ITD–dnGSK-3β cells in response to increasing concentrations of PKC412.
To determine whether such reduced potencies of FLT3 kinase inhibitors by GSK-3β inhibition were Wnt/β-catenin pathway related, a dominant negative form of TCF4 (dnTCF4)17 or antisense β-catenin34 was expressed transiently in the Ba/F3-FLT3-ITD–dnGSK-3β cells. The proliferation and colony formation of the resulted cells with either expression of dnTCF4 or knockdown of β-catenin (Fig. 3C) were assayed in response to increased concentration of PKC412 (Fig. 3B and Fig. 4B, right). The results demonstrated that inhibition of either TCF4 or β-catenin brought the response curves to PKC412 back down to a similar position as that of the parental FLT3-ITD–expressing cells in both assays, suggesting that reduced potencies to the FLT3 inhibitors in the cells with inhibitions of GSK-3β could be due to the activation of the Wnt/β-catenin pathway to a large extent. Together with the biochemical evidence shown in Figures 1 and 2, these data further demonstrated that GSK-3β inhibitions activate β-catenin biologically in the context of FLT3-activating mutations.
Wnt3A Ligand Also Induces Colony Formation and Attenuates the Potencies of the FLT3 Kinase Inhibitors
The finding that endogenous activation of the Wnt/ β-catenin pathway via inhibition of GSK-3β induced colony formation and reduced the potencies of the FLT3 kinase inhibitors led us to hypothesize that exogenous activation of the Wnt pathway should have similar effects. To test this hypothesis, the Wnt3A ligand was applied in FLT3-ITD–expressing cells and then their proliferation and colony formation were assayed in response to 3 FLT3 inhibitors. Wnt3A by itself had little direct effect on proliferation of the FLT3-ITD–expressing cells (data not shown) but did increase the colony formation (Fig. 5C). As expected, Wnt3A stimulation reduced the potencies of these inhibitors on both proliferation (Fig. 6A, B, and C) and colony formation (Fig. 5A and B) in the cells either ectopically expressing FLT3-ITD or derived from AML patient samples harboring FLT3-ITD mutation.
Figure 5.

Exogenous activation of the Wnt/β-catenin pathway by the Wnt3A ligand attenuates the potencies of the FLT3 kinase inhibitors on colony formation. (A) Effects of the FLT3 kinase inhibitors on colony formation of Ba/F3-FLT3-ITD cells with and without the Wnt3A ligand stimulation. Ba/F3-ITD cells were seeded at a density of 500 cells/well in soft agar plates containing either 20% of the Wnt3A conditioned medium (3A) or 20% of the L conditioned medium (Con) with the inhibitors at the concentrations indicated. After 2 wk, colonies were counted. (B) Effects of FLT3 inhibitors on colony formation of the Molm 14 cells. Molm 14 cells were seeded at a density of 1000 cells/well in soft agar plates containing either 20% of 3A conditioned medium (3A) or 20% of L conditioned medium (Con) with the FLT3 inhibitors at the concentrations indicated. After 2 wk, colonies were counted. (C) Wnt3A ligand stimulation enhances colony formation of the FLT3-ITD–expressing cells and attenuates the response of the host cells to PKC412 inhibition on colony formation. (Top) Wnt3A ligand induced colony formation in all of the cells tested. (Bottom) The actual soft agar plates showing the differences in colony number formed by Ba/F3-ITD cells with and without the Wnt3A ligand stimulation in response to increasing concentrations of PKC412. All of the results are representative of at least 2 independent experiments. Points, averages of at least 2 independent experiments; bars, SD; *P ≤ 0.05.
Figure 6.
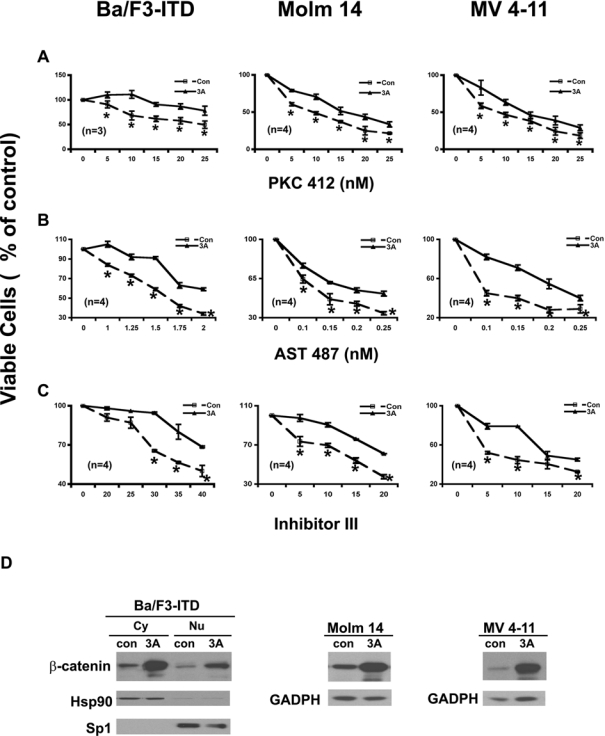
Exogenous activation of the Wnt/β-catenin pathway by the Wnt3A ligand attenuates the potencies of the FLT3 kinase inhibitors on cell proliferation in either Ba/F3 cells ectopically expressing FLT3-ITD or cell lines derived from acute myeloid leukemia (AML) patient samples harboring FLT3-ITD mutations. (A) Effects of PKC412 on proliferation of the FLT3-ITD–expressing cells with and without the Wnt3A ligand stimulation. Ba/F3-FLT3-ITD cells were seeded at a density of 1.5 × 105 cells/mL; Molm 14 and MV 4-11 cells were seeded at a density of 2 × 105 cells/mL in the medium without IL3 but containing either 20% of 3A conditioned medium (3A) or 20% of L conditioned medium (Con) and treated with PKC412 at the concentrations indicated for 68 to 72 h, and then stained with trypan blue and counted under microscope. (B, C) Effects of AST 487 and inhibitor III on the proliferation of FLT3-ITD–expressing cells with and without the Wnt3A ligand stimulation. Analysis was carried out as in (A). (D) Expression levels of β-catenin after Wnt3A ligand stimulation. Ba/F3-FLT3-ITD cells were collected after incubation with either 20% Wnt3A conditioned medium or 20% of L conditioned medium (as control) for 1 h. The proteins of cytoplasmic portion and nuclear portion were separated and subjected to immunoblotting analysis using an antibody against β-catenin. The membrane was stripped and reprobed with Hsp 90 antibody as a marker for the cytoplasmic extraction; the membrane was stripped again and reprobed with Sp1 antibody as a marker for the nuclear extraction. Both Molm 14 and MV4-11 cells were treated with either 20% of the Wnt3A conditioned medium or 20% of the L conditioned medium for 3 h; the whole-cell lysis in NP-40 lysis buffer was then subjected to immunoblotting analysis using an antibody against β-catenin.
The analysis of β-catenin level on the stimulation of the Wnt3A ligand (Fig. 6D) demonstrated that the cells did respond to the Wnt3A stimulation and that the observed biological effects of the Wnt3A ligand stimulation were due to the activation of the Wnt/β-catenin pathway.
Taken together, these results demonstrated that in the FLT3-ITD–expressing cells, activation of the Wnt/β-catenin pathway, both endogenously and exogenously, reduced the potencies of the FLT3 kinase inhibitors and further suggested a critical role of GSK-3β in the cross-talk between these 2 pathways.
Discussion
In this report, we demonstrate the importance of GSK-3β in mediating the induction of β-catenin in response to FLT3-ITD mutations, which culminates in the reduction of the potencies of the FLT3 kinase inhibitors. Activation of the Wnt/β-catenin pathway has been shown to increase self-renewal of both normal hematopoietic stem cells8 and the progenitor cells from patients with chronic myelogenous leukemia35 and other tumors.36-40 Our findings point to an importance of GSK-3β in the pathogenesis of AML.
Although GSK-3β is an important component in the β-catenin destruction complex,13,14 it is reported that both GSK-3β kinase activity and its access to β-catenin21,41 are equally important and that GSK-3β regulation in β-catenin is cell type specific.20,21 Using known targets of the Wnt/ β-catenin pathway, as well as TCF/β-catenin responsible promoter, we demonstrated that inhibitions of GSK-3β resulted in the activation of the Wnt/β-catenin pathway in a mutant FLT3 receptor and PI3 kinase pathway–dependent manner, both biological and biochemical, which suggests that in our system, inhibition of GSK-3β alone is sufficient to activate β-catenin.
An interesting observation in our study is that the potencies of the FLT3 inhibitors were decreased upon activation of the Wnt pathway by either endogenous inhibition of GSK-3β or exogenous stimulation with the Wnt3A ligand. This finding suggests that the cross-talk between the FLT3-ITD and the Wnt pathways may induce partial drug resistance to the inhibitors of this kinase and increase the tolerances of the host cells to cytotoxic agents. A recent study has suggested that interaction of the Wnt pathway with the adhesion-dependent pathway could control the chemosensitivity of AML mediated by GSK-3β and NF-κB.42 Although the mechanism of such increased resistance remained unclear, our findings suggest that the Wnt/β-catenin activation in the context of the FLT3-activating mutations may culminate in genome instability. It would be interesting to find out whether the FLT3-activating mutations could induce a secondary mutation via activation of β-catenin.
Materials and Methods
Reagents, Cell Lines, and Antibodies
GSK-3β inhibitor VII, FLT3 inhibitor III, Ly 294002, Wortmannin, and protease inhibitor cocktail set I were from Calbiochem (San Diego, CA). PKC412 and AST 487 were kindly provided by Dr Johannes Rossel (Novartis Pharma, Switzerland). Lithium chloride was from Sigma (St. Louis, MO). NE-PER nuclear and cytoplasmic extraction reagents were from PIERCE (Rockford, IL). SELECT Agar was from Invitrogen/Gibco (Carlsbad, CA). L cells, L/Wnt3A cells, and U937 cells were from ATCC (Manassas, VA). The following antibodies were used: pTyr(pY99), FLT3(c-20), c-Myc(N-262), c-Myc(9E10), β-catenin(E-5), GSK-3β(H 76), cyclin D3(c-16), TCF4(H125), and Sp1(PEP 2), from Santa Cruz (Santa Cruz, CA); pAKT(Ser 473), AKT, pGSK-3β(ser 9), GADPH, and β-catenin, from Cell Signaling (Danvers, MA); Hsp 90(clone AC 88) from Stressgen (Victoria, BC, Canada); and Tubulin(clone DM 1A) from Sigma.
Cell Culture
Ba/F3-FLT3-ITD, Ba/F3-FLT3-N841I, and 32D-FLT3-ITD cells were cultured in the regular RPMI 1640 medium containing 800 μg/mL G418.5 Ba/F3-pClneo cells were cultured in the same medium but supplemented with 10% of the WEHI3B conditioned medium (as a source of IL3). The Molm 14 and MV4-11 cells were cultured in the regular RPMI 1640 medium. The culture media for U937 cells, L cells, and L/Wnt3A cells were as per the manufacturer’s protocols (ATCC). The cells were maintained in a humidified incubator at 37°C with 5% CO2.
Western Blotting Analysis
Total proteins were isolated using NP-40 buffer following the standard protocol.43 For separation of cytoplasmic and nuclear protein, the cells were lysed in the NE-PER nuclear and cytoplasmic extraction reagents per the manufacturer’s protocol. Protein extracts were then subjected to Western blotting analysis using the antibodies indicated.
Colony Formation Assay
SELECT agar was used for colony formation assay.44 Briefly, cells resuspended in culture medium with 0.4% agar and drug treatments (top agar) were seeded duplicate in a 6-well plate containing 0.5% agar in the culture medium with the same drug treatment as those for the top agar. Colonies were counted on days 10 to 14.
Generation of Stable Cell Lines
Dominant negative GSK-3β (K85A mutant)32 was subcloned into pBABE/puro expression vector45 to facilitate selection. FLT3-ITD46 was subcloned into pCIneo (Promega, Madison, WI) expression vector. pCIneo/FLT3-N841I was generated by site-directed mutagenesis.5 Plasmid DNA was introduced into host cells by electroporation using Gene Pulser Xcell (Bio-Rad, Hercules, CA). Twenty-four to 36 h after electroporation, the cells were grown in the selective medium with appropriate antibiotics (800 μg/mL for neomycin, 2 μg/mL for puromycin) for selection.
Transient Expression Assays
siRNA of GSK-3β was purchased from SABiosciences (Frederick, MD) and manipulated following the manufacturer’s protocol. The cDNAs of dominant negative TCF-417 and antisense–β-catenin34 were used directly without further subcloning. Both Gene Pulser Xcell and the Nucleofector Device (Amaxa Inc., Gaithersburg, MD) were used for introducing target constructs into host cells with 50% to 80% transfection efficiency. The cells were analyzed 20 to 24 h after electroporation.
TOPflash reporter and FOPflash reporter (Upstate/Millipore, Lake Placid, NY) were introduced into host cells by electroporation using Gene Pulser Xcell. Twenty to 24 h after electroporation, the cells were treated and analyzed for luciferase activity using the Dual-Luciferase Reporter Assay Kit (Promega).
Supplementary Material
Acknowledgments
The authors thank Dr Jim Woodgett (University of Toronto, Canada), Dr Bert Vogelstein and Dr Kenneth W. Kinzler (The Johns Hopkins Medical Institutions & Howard Hughes Medical Institute, Baltimore, MD), Dr Jane Trepel (National Institutes of Health/National Cancer Institute, Bethesda, MD), and Dr Ellen Weisberg, Dr Blanca Scheijen, Dr Yuki Yuza, and Dr Matthew Meyerson (Dana-Farber Cancer Institute, Boston, MA) for providing reagents and constructs. They also thank Dr Heidi Greulich and Dr Sarah Walker (Dana-Farber Cancer Institute) for the protocols and suggestions on the colony formation and luciferase assays.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the National Institutes of Health (CA66996, CA36167, and DK50654; J.D.G.) and by a Specialized Center of Research Award from the Leukemia and Lymphoma Society (J.D.G.).
Supplementary material for this article is available on the Genes & Cancer Web site at http://ganc.sagepub.com/supplemental.
References
- 1. Shurin MR, Esche C, Lotze MT. FLT3: receptor and ligand: biology and potential clinical application. Cytokine Growth Factor Rev 1998;9(1):37-48 [DOI] [PubMed] [Google Scholar]
- 2. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 2003;3(9):650-65 [DOI] [PubMed] [Google Scholar]
- 3. Gilliland DG, Griffin JD. Role of FLT3 in leukemia. Curr Opin Hematol 2002;9(4):274-81 [DOI] [PubMed] [Google Scholar]
- 4. Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Muller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000;96(12):3907-14 [PubMed] [Google Scholar]
- 5. Jiang J, Paez JG, Lee JC, Bo R, Stone RM, DeAngelo DJ, et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood 2004;104(6):1855-8 [DOI] [PubMed] [Google Scholar]
- 6. Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 2002;99(1):310-8 [DOI] [PubMed] [Google Scholar]
- 7. Gilliland DG, Jordan CT, Felix CA. The molecular basis of leukemia. Hematology Am Soc Hematol Educ Program 2004:80-97 [DOI] [PubMed] [Google Scholar]
- 8. Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003;423(6938):409-14 [DOI] [PubMed] [Google Scholar]
- 9. Tickenbrock L, Schwable J, Wiedehage M, Steffen B, Sargin B, Choudhary C, et al. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood 2005;105(9):3699-706 [DOI] [PubMed] [Google Scholar]
- 10. Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005;24(14):2410-2420 [DOI] [PubMed] [Google Scholar]
- 11. Kajiguchi T, Chung EJ, Lee S, Stine A, Kiyoi H, Naoe T, et al. FLT3 regulates beta-catenin tyrosine phosphorylation, nuclear localization, and transcriptional activity in acute myeloid leukemia cells. Leukemia 2007;21(12):2476-84 [DOI] [PubMed] [Google Scholar]
- 12. Tickenbrock L, Hehn S, Sargin B, Evers G, Ng PR, Choudhary C, et al. Activation of Wnt signaling in cKit-ITD mediated transformation and imatinib sensitivity in acute myeloid leukemia. Int J Hematol 2008;88(2):174-80 [DOI] [PubMed] [Google Scholar]
- 13. Papkoff J, Rubinfeld B, Schryver B, Polakis P. Wnt-1 regulates free pools of catenins and stabilizes APC-catenin complexes. Mol Cell Biol 1996;16(5):2128-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci U S A 1998;95(6):3020-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996;86(3):391-9 [DOI] [PubMed] [Google Scholar]
- 16. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997;275(5307):1784-7 [DOI] [PubMed] [Google Scholar]
- 17. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275(5307):1787-90 [DOI] [PubMed] [Google Scholar]
- 18. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science 1998;281(5382):1509-12 [DOI] [PubMed] [Google Scholar]
- 19. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 1999;96(10):5522-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Staal FJ, Burgering BM, van de Wetering M, Clevers HC. Tcf-1-mediated transcription in T lymphocytes: differential role for glycogen synthase kinase-3 in fibroblasts and T cells. Int Immunol 1999;11(3):317-23 [DOI] [PubMed] [Google Scholar]
- 21. Yuan H, Mao J, Li L, Wu D. Suppression of glycogen synthase kinase activity is not sufficient for leukemia enhancer factor-1 activation. J Biol Chem 1999;274(43):30419-23 [DOI] [PubMed] [Google Scholar]
- 22. Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000;19(5):624-31 [DOI] [PubMed] [Google Scholar]
- 23. Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwable J, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res 2005;65(21):9643-50 [DOI] [PubMed] [Google Scholar]
- 24. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995;378(6559):785-9 [DOI] [PubMed] [Google Scholar]
- 25. Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J 1994;303(pt 3):701-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, et al. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001;105(6):721-32 [DOI] [PubMed] [Google Scholar]
- 27. Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002;1(5):433-43 [DOI] [PubMed] [Google Scholar]
- 28. Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem 1992;267(24):16878-882 [PubMed] [Google Scholar]
- 29. Eldar-Finkelman H, Seger R, Vandenheede JR, Krebs EG. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J Biol Chem 1995;270(3):987-90 [DOI] [PubMed] [Google Scholar]
- 30. Weisberg E, Roesel J, Bold G, Furet P, Jiang J, Cools J, et al. Antileukemic effects of the novel, mutant FLT3 inhibitor NVP-AST487: effects on PKC412-sensitive and -resistant FLT3-expressing cells. Blood 2008;112(13):5161-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Furet P, Bold G, Meyer T, Roesel J, Guagnano V. Aromatic interactions with phenylalanine 691 and cysteine 828: a concept for FMS-like tyrosine kinase-3 inhibition. Application to the discovery of a new class of potential antileukemia agents. J Med Chem 2006;49(15):4451-4 [DOI] [PubMed] [Google Scholar]
- 32. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 1996;6(12):1664-8 [DOI] [PubMed] [Google Scholar]
- 33. Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol 1997;185(1):82-91 [DOI] [PubMed] [Google Scholar]
- 34. Chung EJ, Hwang SG, Nguyen P, Lee S, Kim JS, Kim JW, et al. Regulation of leukemic cell adhesion, proliferation, and survival by beta-catenin. Blood 2002;100(3):982-90 [DOI] [PubMed] [Google Scholar]
- 35. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004;351(7):657-67 [DOI] [PubMed] [Google Scholar]
- 36. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002;111(2):241-50 [DOI] [PubMed] [Google Scholar]
- 37. Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, et al. Wnt/beta-catenin mediates radiation resistance of Sca1+ progenitors in an immortalized mammary gland cell line. J Cell Sci 2007;120(pt 3):468-77 [DOI] [PubMed] [Google Scholar]
- 38. Khan NI, Bradstock KF, Bendall LJ. Activation of Wnt/beta-catenin pathway mediates growth and survival in B-cell progenitor acute lymphoblastic leukaemia. Br J Haematol 2007;138(3):338-48 [DOI] [PubMed] [Google Scholar]
- 39. de Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci 2007;12:471-91 [DOI] [PubMed] [Google Scholar]
- 40. Gestl SA, Leonard TL, Biddle JL, Debies MT, Gunther EJ. Dormant Wnt-initiated mammary cancer can participate in reconstituting functional mammary glands. Mol Cell Biol 2007;27(1):195-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. Embo J 1998;17(5):1371-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Toni F, Racaud-Sultan C, Chicanne G, Mas VM, Cariven C, Mesange F, et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene 2006;25(22):3113-3122 [DOI] [PubMed] [Google Scholar]
- 43. Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res 2005;65(19):8968-74 [DOI] [PubMed] [Google Scholar]
- 44. Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2005;2(11):e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yuza Y, Glatt KA, Jiang J, Greulich H, Minami Y, Woo MS, et al. Allele-dependent variation in the relative cellular potency of distinct EGFR inhibitors. Cancer Biol Ther 2007;6(5):661-7 [DOI] [PubMed] [Google Scholar]
- 46. Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004;23(19):3338-49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.