

Abstract
Despite the involvement of genetic alterations in neoplastic cell transformation, it is increasingly evident that abnormal epigenetic patterns, such as those affecting DNA methylation and histone posttranslational modifications (PTMs), play an essential role in the early stages of tumor development. This finding, together with the evidence that epigenetic changes are reversible, enabled the development of new antineoplastic therapeutic approaches known as epigenetic therapies. Epigenetic modifications are involved in the control of gene expression, and their aberrant distribution is thought to participate in neoplastic transformation by causing the deregulation of crucial cellular pathways. Epigenetic drugs are able to revert the defective gene expression profile of cancer cells and, consequently, reestablish normal molecular pathways. Considering the emerging interest in epigenetic therapeutics, this review focuses on the approaches affecting DNA methylation, evaluates novel strategies and those already approved for clinical use, and compares their therapeutic potential.
Keywords: DNA demethylating drugs, 5-azacytidine, 5-aza-2′-deoxycytidine, DNMT inhibitors, epigenetic therapy
Introduction
Epigenetics is the study of somatically heritable and reversible modifications of genome functions (gene expression and genomic stability) not involving changes in DNA sequence. DNA methylation, together with histone posttranslational modifications (PTMs), represents the most characterized epigenetic modification and is known to participate in the regulation of gene expression by modifying the chromatin structure and, consequently, the accessibility of transcription factors to regulatory regions of genes. Aberrant epigenetic patterns are involved at early stages of cancer initiation and, altering both global gene expression and genomic stability, strongly contribute to cancer progression.1
DNA methylation involves the covalent addition of a methyl group (-CH3) to the C-5 position of cytosine, mostly in the context of the CpG dinucleotide. This chemical reaction is catalyzed by enzymes belonging to the DNA methyltransferase family (DNMTs). DNMT1 is predominantly responsible for maintaining the preexistent methylation pattern during DNA replication, whereas DNMT3a and DNMT3b are required for de novo DNA methylation.2,3 CpG dinucleotides are clustered in regions, named CpG-islands, present in almost 60% of all gene promoters.4 DNMT activity regulates global gene expression by hypermethylation of promoter CpG-islands and, as recently shown, of regulatory sequences near the promoter (CpG island shores), thus causing gene silencing. Not surprisingly, the DNA methylation pattern is altered in cancer. Paradoxically, tumor cells exhibit global genome hypomethylation and increased levels of DNMT1 expression at the same time. Moreover, both DNA hypo- and hypermethylation occur at specific gene loci, leading to the overexpression of proto-oncogenes and the silencing of tumor suppressor genes, respectively. These phenomena are thought to be promoting factors of cellular transformation.5,6
Evidence regarding the involvement of DNA methylation in tumorigenesis gave rise to the development of new therapeutic strategies that target this modification. In the past few years, many drugs have been proposed. Some have already been approved for clinical use, and many others are still under evaluation. In this review, we focus on antineoplastic strategies affecting DNA methylation, with particular attention to their mechanism of action and the molecular pathways involved. In addition, we compare novel approaches, such as the use of nonnucleoside inhibitors and DNMT silencing with azanucleoside administration, the only demethylating strategy approved for clinical therapy.
The Azanucleosides: Azacytidine and Decitabine
Mechanism of Action
Azanucleoside 5-azacytidine (azacytidine [AZA]) and its derivative 5-aza-2′-deoxycytidine (decitabine [DAC]) were synthesized almost 50 years ago with the original purpose of being used as classical cytostatic drugs.7 Only some years later, Jones and Taylor8 correlated the differentiating effect to the demethylating activity of the 2 compounds. Consequently, a series of clinical studies was aimed to ascertain the role of demethylating agents as “epigenetic” antineoplastic drugs, and AZA and DAC received approval by the US Food and Drug Administration for the treatment of myelodysplastic syndrome (MDS).9,10
The 2 cytidine analogs, once internalized into the cell, are converted into the corresponding active triphosphorylated nucleotides and then incorporated into DNA or RNA as cytosine substitutes. In particular, DAC is exclusively incorporated into DNA, whereas AZA is mainly incorporated into RNA, causing defects in RNA and protein synthesis and tRNA functions. Only about 10% of AZA is incorporated into DNA after its conversion to deoxynucleotide. Under normal conditions, DNMTs establish a covalent bond with the carbon-6 atom of cytosine that is resolved by β-elimination through the carbon-5 atom. The presence of a nitrogen atom at the C-5 position of AZA and DAC prevents DNMT release, causing the covalent trapping of the enzymes to DNA and the subsequent depletion of the DNMT pool.8 Interestingly, DNMT loss seems to be alternatively driven: Ghoshal and colleagues,11 for example, demonstrated that DNMT1 levels also diminish in the absence of DAC genomic incorporation due to its induced degradation through the proteasome pathway.
The peculiarity of DNA methylation patterns in cancerous cells has been widely demonstrated. Generally, global genome hypomethylation, especially at highly and moderately repeated sequences, coexists with hypo- and hypermethylation near and at gene promoter regions. These modifications are responsible, respectively, for the high genomic instability that characterizes cancer cells and the deregulation of genes with crucial roles in differentiation, proliferation, and apoptosis processes.12,13 Although the application of a DNA demethylating strategy in the already demethylated status of cancer cells could be considered paradoxical, the main function of AZA and DAC treatments is in the reexpression of aberrantly hypermethylation-silenced genes, thus restoring important tumor suppressor activities.
Azanucleoside activity has also been linked to the formation of covalent adducts between DNMTs and DNA, which, directly or indirectly, induce DNA double-strand breaks and lead to the activation of both ATM and ATR DNA damage pathways.14,15 Moreover, the DNMT-DNA complex is chemically unstable and can be resolved by the hydrolysis of DAC ring structure. The resulting open ring is the candidate mediator of C:G→G:C transversions frequently observed after DAC administration.16
The prevalence of one mechanism or the other seems to be strictly dependent on the tumor model considered and, most important, the drug dosage. At low doses, indeed, the main effect is the reexpression of methylated genes, possibly associated with reduced proliferation, cell differentiation, apoptosis, and senescence, whereas the DNA damage response, which triggers an apoptotic pathway, is predominant at high doses (Fig. 1).17
Figure 1.
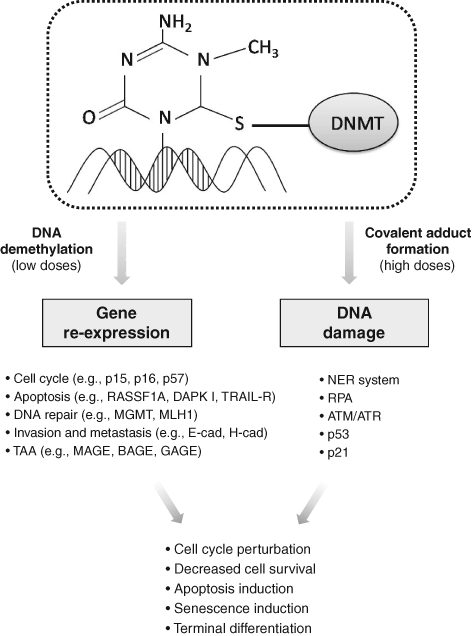
Major effects and cellular responses mediated by azanucleosides.
Another AZA- and DAC-mediated effect that warrants further in-depth examination pertains to organization of the centromeres. Centromeres, which comprise highly repeated sequences named alpha-satellites, are packaged in constitutive heterochromatin through mechanisms involving DNA methylation and posttranslational histone modifications. Such epigenetic properties prevent rearrangements of repetitive sequences and transposable elements, thus contributing to the maintenance of genome integrity and stability.18 AZA and DAC, by direct incorporation into centromeric DNA sequences, could lead to heterochromatin decondensation and altered centromeric structure, resulting in genome destabilization and impaired kinetochore formation. Under these conditions, cells would suffer from altered chromosome alignment and incorrect sister chromatid segregation and, finally, would be irreversibly triggered to die, possibly through mitotic catastrophe.
Azanucleosides can be efficiently administered subcutaneously (SC) or intravenously (IV). Following SC administration, these molecules are rapidly adsorbed and, once in blood circulation, widely distributed. Unfortunately, drug elimination is also rapid because of its low stability. The half-life of AZA, indeed, is 1.5 ± 2.3 h, whereas the half-life of DAC is 20 ± 5 h in aqueous solutions.19,20 The high instability, together with the cell cycle dependency of azanucleoside activity, implies the requirement of prolonged administration schedules. Moreover, due to its prevalent incorporation into DNA and its higher stability, DAC is more effective than AZA in inducing DNA demethylation, and thus it can be used at lower concentrations (100-135 mg/m2/course vs 525 mg/m2/course).9,21-25
Reexpression of Silenced Genes
As mentioned above, low doses of AZA and DAC lead to the reexpression of genes silenced by aberrant DNA methylation. Depending on the function of the reactivated genes, different molecular pathways disrupted in cancer cells, varying from those involved in terminal differentiation and decreased proliferation to those implicated in apoptosis and premature senescence, have the potential to be reestablished. The ability of AZA and DAC to reactivate the expression of genes involved in cell cycle control that have been aberrantly silenced in cancer upon their hypermethylation has been widely demonstrated. Several CDK inhibitors, such as p16, p15, and p57, which regulate G1/S progression, as well as GADD45, involved in G2/M transition, have been found to be reexpressed after DAC exposure in different tumor models.26-30 Loss of CDK inhibitors activity, which contributes to the accumulation of unrepaired DNA damage and the deregulation of cellular proliferation, is strongly involved in tumor development and progression. Restoring their expression and, consequently, their regulatory activity in cell cycle checkpoints impairs the neoplastic potential of cells and limits tumor growth. On the contrary, upregulation of the cell cycle inhibitor p21, although frequently observed following azanucleoside exposure, is independent of the methylation status of the gene and is more likely associated with activation of the DNA damage pathway.31
Azanucleoside treatments are also able to sensitize tumor cells to programmed cell death by restoring the defective expression of genes involved in the apoptotic pathways. Exposure to DAC treatments has been shown to restore the expression of several silenced apoptotic effectors and mediators such as DAPK I, RASSF1A, XAF1, and TRAIL receptor-1, all of which are involved in interferon-induced apoptosis, and APAF1, a downstream element of the p53 apoptotic pathway.32-38 Similarly, the reexpression of the hypermethylated genes HSPA9B, PAWR, PDCD5, NFKBIA, and TNFAIP3, with roles in regulating p53-, Bcl-2-, and NF-κB-related apoptosis, was observed in both osteosarcoma cells and xenografts.30 Recently, it has been shown that DAC is able to induce the reexpression of p300/CBP factor (PCAF), known to be involved in the activation of p53, suggesting a contribution of this phenomenon in increasing the apoptotic responsiveness of cancer cells.39
It has also been shown that DAC can regulate the reexpression and activity of genes involved in DNA repair pathways, such as the MLH1 mismatch repair gene that is reexpressed in cellular and xenograft tumor models.40,41 However, this aspect requires careful attention because, considering their contribution in increasing resistance to DNA-targeting antineoplastic drugs, the reactivation of the DNA repair pathways may not necessarily be considered a therapeutic advantage, as in the case of the MGMT gene in relation to alkylating agents.42,43 The ability of azanucleosides to restore molecular pathways involved in tumor invasiveness and metastatic capacity reduction, through the reexpression of genes such as E-cadherin and H-cadherin, involved in cell-cell adhesion; the antiangiogenic VEGF 189b variant; and thrombospondin-1 also has been shown.44-49
Interestingly, AZA and DAC can augment the expression of molecules specifically recognized by targeted therapies. For example, loss of estrogen receptor (ER) expression in breast cancer induces a more aggressive tumor and results in inefficacy of therapy with selective ER modulators (SERM). Pretreating breast cancer cells with DAC can restore ER expression and function, thereby sensitizing ER-negative cells to endocrine therapy.50 DAC exposure can also induce the reexpression of retinoid acid receptor-β2 (RAR-β2) in human head and neck squamous cell carcinoma cells, sensitizing these tumors to all-trans-retinoic acid (ATRA).51 Comparable effects have been recently observed in prostate cancer cells, in which DAC exposure increased the efficacy of treatment with the growth inhibitor somatostatin by inducing the upregulation of its receptor.52
Similarly, AZA and DAC have been successfully exploited in solid cancers to enhance tumor immunogenicity. Aberrant expression of recognition antigens, one of the main mechanisms used by cancer cells to evade the host immune system, is frequently imputable to promoter hypermethylation of the corresponding genes. Cancer testis antigens (CTAs) constitute a family of tumor-associated antigens (TAA) normally found in testes and ovaries but aberrantly expressed in diverse tumor types. These antigens represent a promising target for immunotherapy because of their specific distribution and capacity of triggering cellular and humoral immune responses. DAC treatment causes a persistent expression of numerous CTAs and normalizes their levels within various populations of neoplastic cells. For example, MAGE, BAGE, and GAGE family members, as well as NY-ESO-1, are induced by DAC both in vitro and in murine melanoma xenografts.53 Moreover, DAC has been shown to upregulate several other molecules that play a central role in immune system regulation, such as human HLA class I antigens and the cell adhesion molecules ICAM-1, found reexpressed in different tumoral models, as well as LFA-3, induced by DAC treatments in melanoma cells.54-58 Due to their “expression-restoring” properties, azanucleosides are able to induce a plethora of effects that contribute to the reversion of the malignant phenotype of cancer cells and, most important, may sensitize tumor cells to other therapeutics as well as to immune system recognition. This plurality of activities likely represents the most interesting feature of these agents.
DNA Damage Response
DNA damage that results from the formation of adducts between DNMTs and DNA is thought to be the other major mechanism of AZA and DAC antitumoral activity. Although the molecular details associated with this response are still under investigation, evidence indicates the involvement of the DNA double-strand break (DSB) repair system. More precisely, the adduct is likely to be first identified by the nucleotide excision repair (NER) system that generates a rupture in one or both DNA strands while removing the adduct.15 The DNA strand breaks are then recognized and coated by the replication protein A (RPA), forming a DNA-RPA complex that recruits the ATR protein and is able to activate p53 through phosphorylation. Activated p53 upregulates p21, leading to inhibition of cell proliferation.59,60 However, in accordance with other studies, our group found that p21 accumulation can also be triggered in a p53-independent manner in malignant pleural mesothelioma (MPM) cells. Moreover, we found that stable silencing of p21 limits the effect of DAC on MPM cells, indicating that p21 is a bona fide effector of DAC antitumoral activity.61 Interestingly, another study demonstrated the involvement of DNMT1, in association with ATM and ATR, in the damage response induced by azanucleosides. However, how DNMT1 contributes to this process is not completely clear, although its role in the regulation of interactions between chromatin and checkpoint kinase 1 (CHK1), an ATR and ATM substrate, has been hypothesized.15
Other Nucleoside Inhibitors
Despite the clinical success in MDS treatment, AZA and DAC administration is limited by serious side effects that are predominantly related to hematological perturbations, as well as by the high chemical instability of the compounds under physiological conditions. For these reasons, at present, special attention is aimed at improving the demethylation strategy through the synthesis of other nucleoside analogs, the identification and synthesis of nonnucleoside inhibitors, and the evaluation of the efficacy of low-dose treatment in combination therapies.
Among the nucleoside derivatives (Table 1), one of the most promising molecules is zebularine (dZTP), a cytidine analog originally developed as a cytidine deaminase inhibitor that was subsequently found to inhibit DNMTs. dZTP shares the same steps of activation and mechanism of action of azanucleosides. Its incorporation into DNA leads to the reactivation of epigenetically silenced genes, such as the CDK inhibitors p15 in acute myelogenous leukemia (AML) cells and thymic T lymphomas in mice; p16 in bladder, colon, and pancreatic cells as well as in a bladder carcinoma xenograft; and p57 in myeloid leukemic cells. In addition, exposure to dZTP leads to reexpression of the cell cycle and apoptosis modulator RASSF1A after demethylation of its promoter in ovarian cancer cells and in a lymphoma mouse model.62-64 However, higher doses of dZTP are required to obtain demethylation and gene reexpression levels comparable to those induced by azanucleosides. This may be because of its lower binding affinity for uridine/cytidine kinase, the enzyme responsible for nucleoside analog activation, or its sequestration by cytidine deaminase. Unlike AZA and DAC, this compound is very stable at both neutral and acidic pH and shows slight cytotoxicity in vitro and in vivo.65,66 Because of its minimal toxicity, dZTP can be used in prolonged treatment protocols, yielding a long-lasting demethylating effect and the prevention of remethylation.67 Furthermore, based on its stability, it was the first DNMT inhibitor to show an in vivo antitumoral activity against T cell lymphoma after oral administration.68
Table 1.
Nucleoside Analogs and Nonnucleoside Molecules
| DNMT Inhibitor | Chemical Structure | Phase of Drug Development | Properties | Reference |
|---|---|---|---|---|
| Nucleosides and derivatives | ||||
| Azacytidine | 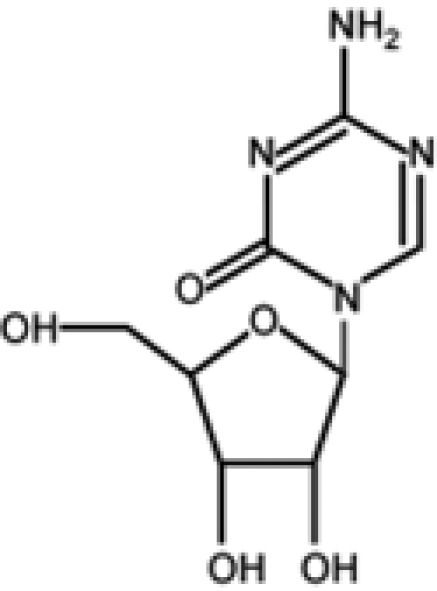 |
Completed | Successfully used for treatment of MDS | 7, 8, 10, 19, 23-25 |
| Decitabine | 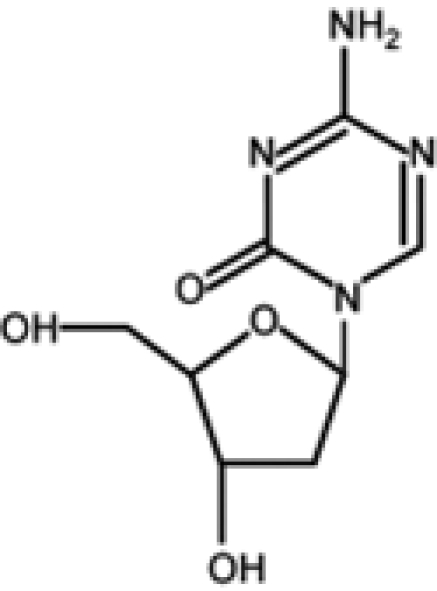 |
Completed |
|
8, 9, 11, 14-17, 20-22, 26-61 |
| Zebularine | 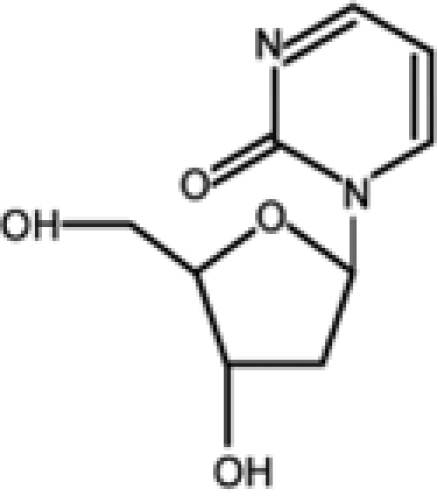 |
Preclinical |
|
62-68 |
| 5-Fluoro-2′-deoxycytidine | 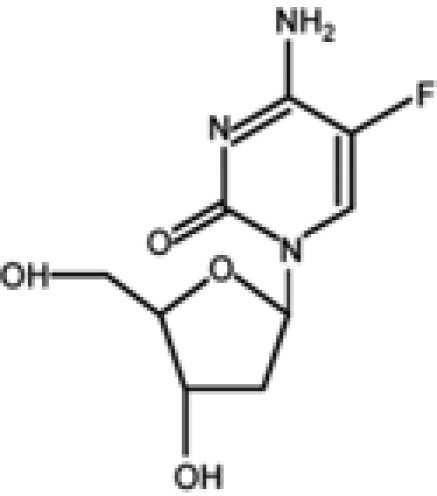 |
Phase I: Breast cancer | Stable | 69-73 |
| Nonnucleosides | ||||
| RG108 | 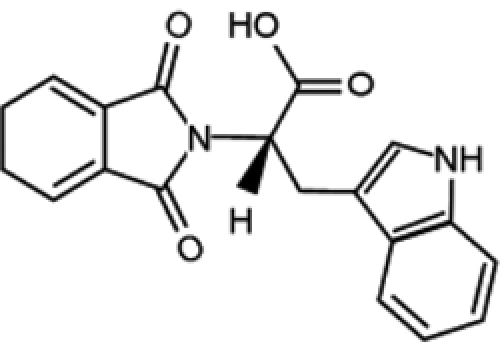 |
Preclinical |
|
76 |
| Procaine | 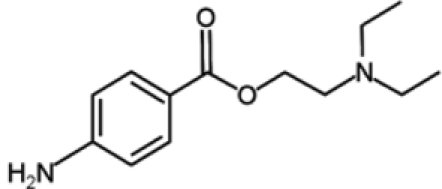 |
Preclinical | Well-known pharmacology in vivo | 77 |
| Procainamide | 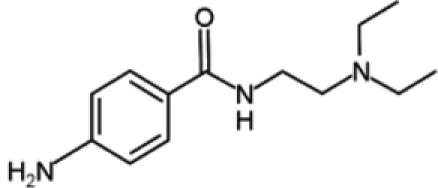 |
Preclinical | Well-known pharmacology in vivo | 78, 79 |
| Hydralazine | 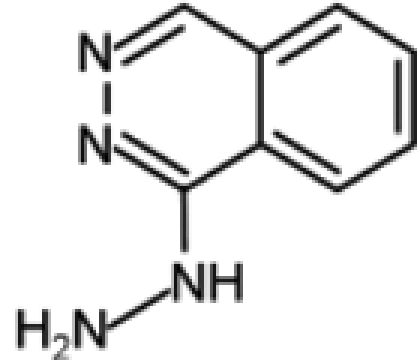 |
|
Well-known pharmacology in vivo | 79-82 |
| Psammaplin A | 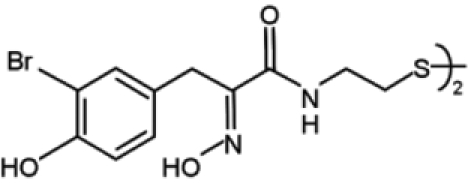 |
Preclinical |
|
83, 84 |
| EGCG | 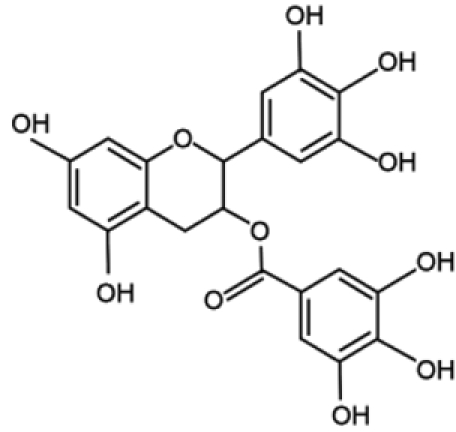 |
Preclinical |
|
85-88 |
DNMT, DNA methyltransferase family; MDS, myelodysplastic syndrome; EGCG, Epigallocatechin-3-Gallate.
Another promising nucleoside analog is 5-fluoro-2′-deoxycytidine (FdCyd), a fluoropyrimidine that is stable in aqueous solutions and inhibits DNA methyltransferases once incorporated into DNA in its triphosphorylated form. Preliminary results indicate that FdCyd causes the demethylation and consequent reexpression of p16 and RASSF1A in bladder carcinoma cells, as well as of the CTA MAGE-1 genes in a melanoma cell line.69-71 Even in these cases, the compound activation and the mechanism of action are the same as azanucleosides. However, the intracellular metabolism of FdCyd involves several different enzymatic pathways, some of which generate active metabolites that do not contribute to DNA demethylation. To diminish the accumulation of collateral metabolites and to promote the incorporation into DNA, FdCyd can be coadministered with tetrahydrouridine (THU), a cytidine deaminase inhibitor. Taking advantage of this combination, FdCyd is under investigation in breast cancer patients in a phase I trial, with encouraging preliminary results.72,73
Because of the limited data from clinical studies, to date, it is impossible to determine whether these novel compounds are more or less efficacious than azanucleosides. Indeed, it has to be considered that improving in vitro efficacy of therapeutic molecules does not necessarily result in an improvement in their antitumoral activity in vivo. This is the case for 5,6-dihydro-5-azacytidine and fazarabine, which reached phase II of clinical trials for several types of solid tumors but were not examined further because of poor outcomes.74,75
Nonnucleoside DNMT Inhibitors
Recently, attention has been focused on the identification of compounds that are able to inhibit DNMT enzymatic activity without trapping them on DNA (Table 1), with the primary aim being to avoid side effects caused by azanucleoside-mediated DNA adduct formation. This is the case for RG108, the first synthetic compound designed to directly inhibit DNMT1 enzymatic activity by targeting its catalytic domain. This molecule was found to induce the reexpression of different hypermethylation-silenced genes, such as p16 and the putative tumor suppressor genes SRFP1 and TIMP-3, in colon cancer cells. However, unlike azanucleosides, this compound does not affect the methylation status of repeated centromeric sequences, suggesting that the lack of effect on chromosomal stability could also contribute to reduced toxicity. Although RG108 does not seem to exert an appreciable antiproliferative effect on cancer cells per se, its possible exploitation as a safe demethylating agent in combination with other antineoplastic therapeutics renders this molecule worthy of particular attention.76
In addition to RG108, several other nonnucleoside inhibitors of DNMTs have been discovered. These molecules are either drugs already used in the clinic for other therapeutic purposes, such as procaine, procainammide, and hydralazine, or natural compounds derived from marine sponges and plants, such as psammaplins and polyphenols.
Procaine and procainammide, both derivatives of 4-amminobenzoic acid, are already employed as local anesthetic and antiarrhythmic drugs, respectively. Recently, their ability to inhibit DNMT activity has been discovered and is associated with direct binding to CpG-rich sequences. More precisely, these molecules act as partial competitive inhibitors of DNMT1, decreasing the affinity of DNMT for its substrates (DNA and S-adenosyl-L-methionine), reducing the processivity of the enzyme and favoring the dissociation of DNMT1 from hemimethylated DNA. These activities result in global genome demethylation of both single-copy genes and centromeric repeats, as well as reactivation of tumor suppressor genes, such as p16, ER, and RAR-β, in bladder and breast cancer cells. In addition, ER expression was shown to be reinduced after hydralazine treatment in breast cancer xenografts.77-79 Although these agents have not been tested in humans as antineoplastic drugs, their long-established and safe employment as pharmaceuticals suggests their possible use for cancer treatment with limited side effects.
The antihypertensive drug hydralazine was tested as a DNMT inhibitor as a consequence of its capability to induce (as a side effect) a lupus-like syndrome known to be related to disorders associated with DNA methylation. Although the details regarding its mechanism of action are still under investigation, some evidence indicates that hydralazine, similar to procaine and procainamide, binds to CpG-rich sequences and interferes with translocation of DNMTs along the DNA strand.79,80 A phase I study has shown that hydralazine is able to induce reexpression of various tumor suppressor genes, including p16 and RAR-β, in cervical cancer patients, even at lower doses than those considered safe for the treatment of cardiovascular disorders.81 Moreover, in a phase II study, hydralazine and magnesium valproate, a histone deacetylase inhibitor, were tested in combination with standard chemotherapeutics in patients with advanced and refractory solid tumors.82 Because treatment was well tolerated and clinical benefits were observed in 80% of cases, a phase III clinical trial, which is still ongoing, was started to further evaluate the chemosensitizing potential of the combined therapy.
Different natural molecules have been tested for their ability to interfere with DNMT activity. Psammaplins, for example, are bisulfide bromotyrosines derived from a marine sponge and are able to inhibit both DNMT1 and histone deacetylase (HDAC) in vitro. Although psammaplin A administration at low doses was found to exert a strong cytotoxic effect in human tumor cell lines and to limit tumor cell growth in a A549 lung xenograft mouse model, DNMT inhibition was not followed by DNA demethylation and reexpression of tumor suppressor genes, suggesting that an intracellular target different from DNMT1 is responsible for the cytotoxic effect of the molecule.83,84
Another natural molecule found to exhibit DNMT inhibitory activities is EGCG (Epigallocatechin-3-Gallate), a polyphenolic component of green tea. EGCG is methylated by the enzyme catechol-O-methyltransferase (COMT), which catalyzes O-methylation of various catecholic compounds. EGCG also inhibits COMT, which shares a common core structure at the active site with DNMT1. As a consequence of this observation, Fang and colleagues85 demonstrated that EGCG is able to inhibit DNMT1 activity by obstructing entry of cytosine in the binding site of the enzyme. Furthermore, this compound was found to induce the demethylation and reactivation of the epigenetically silenced genes p16, RAR-β, MGMT, and MLH1 in esophageal cancer cells, together with inhibition of cellular proliferation. In addition, reversed silencing via promoter demethylation has also been reported for Wnt-inhibitory factor-1 (WIF-1), which is involved in the negative regulation of the Wingless-type (Wnt) oncogenic pathway in 2 different lung cancer cell lines, as well as for tissue factor pathway inhibitor-2 (TFPI-2), a member of the Kunitz-type serine proteinase inhibitor family, in renal cell carcinoma cells.86,87 Interestingly, other dietary catechol-containing polyphenols, such as different tea catechins (catechin, epicatechin) and bioflavonoids (quercetin, fisetin), were also found to inhibit DNMT activity in vitro through mechanisms different from that of EGCG.88 The results obtained from the usage of natural compounds are intriguing, especially considering their ease of use and low costs. However, further investigation is necessary to establish their real efficacy as DNMT inhibitors, as well as to evaluate the toxicity induced by their administration at pharmacological doses.
DNMT Silencing
Excluding some rare exceptions,13 tumor cells generally show increased expression levels of DNMTs.89,90 Although some experimental evidence indicates a role in neoplastic cell transformation, the mechanisms by which DNMTs participate in tumorigenesis are still uncertain and controversial. On one hand, their overexpression may explain promoter hypermethylation of tumor suppressor genes, whereas on the other hand, this is in contrast to the expression of proto-oncogenes and global genome hypomethylation that are frequently observed in cancer cells. Even with the hypotheses that attempt to explain this contradiction, this paradox is far from being resolved. Nevertheless, DNMT silencing has been demonstrated to reactivate the expression of genes that are aberrantly silenced in cancer cells and, most important, to have a strong antineoplastic effect.
Despite its role as maintaining DNMT, many authors reported that DNMT1 silencing simply causes a partial demethylation of aberrantly hypermethylated genes. On the contrary, the simultaneous silencing of DNMT1 and DNMT3b has been shown to induce genomic demethylation and the reactivation of aberrantly silenced genes, such as p16, TIMP-3, and insulin-like growth factor 2 (IGF2) in colorectal cancer cells, as well as RASSF1A and the tumor suppressor gene HIN-1 in ovarian cancer cells, although an antagonistic effect of the silencing of the 2 DNMTs was also reported.91-95 Taken together, these findings cast serious doubt on the direct correlation between DNMT overexpression and promoter hypermethylation in tumor cells and, at the same time, underline the pressing need to further elucidate the mechanism of action of these enzymes to specifically target their activities.96
To date, the majority of the studies conducted have focused attention on the antineoplastic effects exerted by the silencing of DNMT1 and provided interesting results in both cellular and animal neoplastic models. For example, DNMT1 silencing using antisense approaches has been shown to revert the malignant phenotype of tumor cells by inhibiting anchorage-independent growth (tumorigenicity index), inducing the specific demethylation of aberrantly hypermethylated genes such as p16, and inhibiting tumor growth in vivo.97 The apparent contrast between the limited DNA demethylation ability and the robust antitumor effects only can be explained by assuming the presence of other important regulatory abilities exerted by this protein in addition to DNA methylation. Considering this point of view, it is reasonable to hypothesize a DNA methylation-independent property that contributes to the antineoplastic effect caused by the silencing of DNMT1, and this is probably associated with the regulatory functions in which the enzyme is known to be involved.
In the past few years, increasing evidence regarding the efficacy of DNMT silencing has prompted interest in its application in cancer therapy. The most interesting results have been obtained using MG98, an antisense oligonucleotide directed against the 3′ untranslated region of the DNMT1 mRNA. MG98 is able to specifically knock down DNMT1 expression and to induce, at least in part, the demethylation of DNA associated with reexpression of hypermethylated genes such as p16 in bladder and colon cancer cell lines. Treatment of nude mice bearing lung and colon xenografts with MG98 also resulted in tumor growth inhibition.98 Moreover, MG98 acts synergistically in combination with DAC in vitro and in nude mouse models.99 These promising preclinical data provided the rationale for 2 subsequent phase I studies. Despite differences in the therapeutic schedules in terms of mode of administration and dosage, both treatment protocols resulted in significant side effects, such as transaminitis, anemia, weakness, fever, nausea, and anorexia.100,101 The best-tolerated protocol was chosen for phase II studies but, unfortunately, did not show any antitumoral activity in patients with metastatic renal carcinoma.102 In a recent phase I trial, MG98 has been also tested for the treatment of patients with high-risk MDS and AML. Even in this case, this approach failed to result in appreciable effects, and the dose escalation was stopped due to severe toxicity.103 However, the negative results obtained in these trials do not diminish the rationale of DNMT silencing therapy. On the contrary, it can be considered proof in support of the likely theory that targeting DNMT1 alone may be insufficient to cause a relevant antitumoral response. Moreover, the efficacy of this strategy in combination with other therapeutic approaches has never been tested.
Interesting results have come from miR-29b, a microRNA (miRNA) that directly targets DNMT3A and DNMT3B expression. miR-29b induces a decrease in methylation levels and induces the reexpression of hypermethylated tumor suppressor genes FHIT and WWOX in lung cancer cells, as well as of p15 and ER in AML cells. Interestingly, the studies conducted show that miR29b is also able to indirectly inhibit DNMT1 expression in AML cells. Although preliminary, these results support the possibility of miRNA-based approaches.104,105
Nucleosides vs Nonnucleoside Strategies
Because only a few studies have directly analyzed nucleoside and nonnucleoside antitumoral activities using the same model, protocols of treatment, and methods of analysis, it is difficult to compare all of these approaches. Furthermore, diversity of the strategies employed makes the comparison even more difficult. Nevertheless, some observations based on demethylating activity, gene reexpression ability, cellular response, and in vivo applications can be discussed.
To date, only 2 in vitro studies directly compared different hypomethylating agents. In the first study, Chuang and colleagues106 estimated the demethylating effect of DAC and 3 nonnucleoside inhibitors—EGCG, hydralazine, and procainamide—in different cancer cell lines. The results of these studies showed a significant reduction of the methylation levels of Alu and LINE repetitive elements and the reactivation of the silenced genes MAGE-A1, MAGE-B2, RARβ, and p16 only in cells treated with DAC. In another study, Stresemann and colleagues107 performed a comparative analysis in different human cancer cell lines using 3 nucleoside inhibitors (AZA, DAC, and dZTP) and 3 nonnucleoside inhibitors (procaine, EGCG, and RG108). Even in this case, azanucleosides were the strongest demethylanting agents and the most efficient in restoring the expression of the hypermethylated gene TIMP-3. However, RG108 and dZTP exhibited low levels of demethylating activity, whereas ECGC and procaine induced no significant effects on DNA methylation. Nevertheless, with the exception of RG108, all compounds showed the capability to induce, to different degrees, a robust apoptotic response in cancer cells. The results obtained in these studies, although indicative of a limited ability to induce DNA demethylation and tumor-associated gene reexpression, do not exclude the potential of both nonnucleoside and nucleoside inhibitors to be safer alternatives to AZA and DAC.
Despite these results, the data concerning the toxicity of these molecules are limited, and most of these agents have never been tested in humans. Excluding 5,6-dihydro-5-azacytidine and fazarabine, FdCyd and hydralazine are the unique compounds for which a clinical trial has been started.73,81,82 Preliminary preclinical studies have been performed only with dZTP, procainamide, and 2 psammaplins.68,78,84 Although all studies have yielded interesting results, additional studies are necessary to investigate both in vivo efficacy and toxicity, especially in comparison with DAC and AZA.
As previously discussed, DNMT downregulation strategies result in lower demethylating activity when compared to azanucleosides. For example, when evaluating methylation-sensitive restriction pattern data, DNMT1 silencing appears to be less effective than DAC in producing extensive DNA demethylation. Interestingly, DAC-treated cells show demethylation of satellite 2, 3, and alphoid sequences whereas only methylation of satellite 2 and 3 is affected in DNMT1−/− cells. As expected, the different demethylating ability corresponds to a different capability to restore silenced-gene expression. In fact, when comparing results obtained from microarray-based gene expression analysis, DNMT knockdown seems to be less effective in inducing reexpression of hypermethylated genes than DAC.93
In contrast to these results, Jung and colleagues94 found that DNMT1 downregulation by siRNAs is sufficient to restore expression of aberrantly hypermethylated genes, despite the fact that its demethylating effect is weaker than that induced by DAC treatment. Moreover, these authors demonstrated that siRNAs seem to inhibit cancer cell proliferation and induce lower levels of DNA damage compared to DAC. In accordance with these results, our group has recently demonstrated that DAC and DNMT1 silencing exerts their antitumoral activity by regulating different cellular responses, although both strategies drastically decrease the survival of malignant pleural mesothelioma cells.61 However, the data regarding the efficacy of DNMT1 silencing in vivo are still controversial. Most notably, this strategy does not seem able to improve the efficacy of DAC and AZA treatments, mainly because of the problems associated with the administration of siRNAs and antisense oligonucleotides to humans; this includes toxicity, instability, risk of nonspecific effects, and complexity in developing a suitable delivery system.108
The finding that DNMT1 silencing induces a different response when compared to DAC is likely linked to the absence of DNA damaging activity and suggests that DNMT silencing strategies are worthy of further investigation. Moreover, to date, DNMT1 has been the main target of such studies, although evidence indicates that this strategy might be improved by targeting other enzymes in addition to DNMT1, such as de novo DNMT3a and DNMT3b.
Combination Therapy
Recent studies have demonstrated that treatment with demethylating agents such as DAC is able to sensitize cells and overcome the resistance of tumor cells to traditional antineoplastic therapies (Table 2). Yang and colleagues109 have shown that pretreatment with DAC induces the reexpression of MLH1, which is involved in the mismatch repair system (MMR), sensitizing ovarian and colon tumor xenografts to cisplatin, carboplatin, temozolimide, and epirubicin. Subsequently, a phase I trial showed that it is possible to safely combine low doses of DAC and carboplatin in different solid tumors (colon carcinoma, breast cancer, melanoma, sarcoma, gallbladder, pleural mesothelioma), achieving epigenetic changes equivalent to or even greater than those observed in tumor xenografts.21 In another study, treatment of colon adenocarcinoma with a combination of DAC and the topoisomerase-I inhibitor irinotecan (CPT-11) was found to induce a marked suppression of tumor growth compared to DAC or CPT-11 alone, both in vitro and in vivo, and restored the expression of tumor suppressor genes such as p14 and p16.110
Table 2.
Recent Studies on the Efficacy of Combination Therapy with Decitabine and Other Therapeutic Agents
| Drug Name | Main Activity | Tumors Tested | Type of Study | Reference |
|---|---|---|---|---|
| Carboplatin | Alkylating agent | Different types of solid tumors | Phase I trial | 21 |
| IL-2 | Cytokine | Metastatic melanoma | Phase I trial | 111 |
| Irinotecan | Topoisomerase-I inhibitor | Colon adenocarcinoma | In vivo | 110 |
| Zebularine | CR deaminase inhibitor | Lymphoid leukemia | In vivo | 63 |
| Valproic acid | HDAC inhibitor | T cell leukemia and acute myeloid leukemia | In vitro | 109 |
Interesting results were also obtained from the combination of DAC with nonchemotherapeutic agents. Cotreatment of leukemia cells with DAC and valproic acid (a histone deacetylase inhibitor) results in a significant synergistic effect on cell viability reduction and apoptosis induction associated with the upregulation of p21 and p57.109 More interestingly, the ability of DAC low-dose administration to enhance IL-2 efficacy against metastatic melanoma was also demonstrated in a phase I trial.111
DAC has also been shown to enhance the sensitivity of gastric cancer cells to radiotherapy. This result is not surprising considering that DAC exposure arrests tumor cells in G2-M, a phase of the cell cycle known to be the most sensitive to irradiation. Moreover, this effect was specific for tumor cells, probably as a consequence of the sensitivity of rapidly dividing cells to the drug. These findings suggest that DAC might be advantageous as it will avoid side effects on the surrounding organs caused by X-ray irradiation.112
Not surprisingly, cancer cells can acquire resistance also to DAC. In fact, increased activity of cytidine (CR) deaminase, a key enzyme in the metabolism of nucleoside analogs, may reduce viability and sensitivity to DAC, leading to insensitivity of tumor cells to the drug.113 Cotreatment with CR deaminase inhibitors, such as dZTP, was demonstrated to overcome this resistance, improving the efficacy of DAC against resistant tumor cells.63,114
Conclusion and Future Perspectives
DNA demethylating agents have recently arisen as a promising therapeutic tool for the treatment of cancer. In the past few years, the severe toxicities related to the administration of azanucleosides have prompted research into new DNA demethylating compounds and strategies. In this field, a plethora of drugs, even if they have different mechanisms of action that are not fully understood, have demonstrated interesting anticancer properties. Unfortunately, although most of the new compounds exhibit favorable toxicity profiles, their capability to act as DNA demethylating agents with antitumor properties that are comparable to azanucleosides has not yet been achieved. The use of DNMT-silencing strategies has also been problematic with respect to the in vivo delivery of siRNAs and antisense oligonucleosides. Nevertheless, research in this field is at a relative early stage: many compounds have not been sufficiently researched in vivo and, in many cases, have never been tested in humans.
With our current level of knowledge regarding their functions, DNA demethylating agents, in our opinion, are unlikely to be used as single agents in cancer therapy. However, this does not diminish their importance in the fight against cancer for different reasons: first, as shown for MDS, these agents are potent and effective therapeutics; second, at low doses, these compounds can induce the reexpression of aberrantly silenced tumor suppressor genes, allowing tumor cells to revert to a normal phenotype (terminal differentiation) and/or reacquire cellular pathways crucial for cell cycle regulation and apoptosis induction. Based on their ability to induce the reexpression of tumor suppressor genes, DNA demethylating drugs can be used as “biosensitizers” by increasing the sensitivity of cancer cells to standard therapeutics (e.g., cisplatin, carboplatin, irinotecan, IL-2) or therapeutic procedures (e.g., radiotherapy). This aspect is even more important when considering the frequency with which drug resistance, whether intrinsic or acquired after the first cycles of treatment, negatively influences patients’ outcomes. Another aspect underlining the importance of the gene expression restoring activity of these agents pertains to their ability to potentiate the innate immune response against tumors by inducing cancer cells to reacquire the expression of silenced TAAs and improve the efficacy of targeted therapies by increasing the expression of their target molecules.
Finally, it has to be considered that long-term, chronic disease has recently arisen as a somewhat viable option for the treatment of seemingly incurable patients. DNA demethylating approaches could have a central role in this context, especially considering their known ability to induce a premature cellular senescent response that is similar to that caused by low-dose treatments with cytotoxic drugs.115 Hematological malignancies have provided a good starting point for the application of DNA demethylating agents, but interesting results have recently been obtained with different solid tumors. However, further studies are necessary to evaluate the efficacy and toxicity of known compounds as well as new agents. Despite the number of problems to be solved, combination therapy using DNA demethylating drugs and conventional therapeutics has a reasonably good chance to be used in the clinic in the near future.
Acknowledgments
We have tried to cite as many works to the best of our knowledge but apologize to those omitted because of space limitations.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The authors did not receive grants or outside funding of any kind in support of their research for the preparation of this manuscript.
References
- 1. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007;4:683-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet 2000;9:2395-402 [DOI] [PubMed] [Google Scholar]
- 3. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999;99:247-57 [DOI] [PubMed] [Google Scholar]
- 4. Antequera F. Structure, function and evolution of CpG island promoters. Cell Mol Life Sci 2003;60:1647-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. Genome-wide methylation analysis of human colon cancer reveals similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009;41:178-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehrlich M. DNA methylation and cancer: too much, but also too little. Oncogene 2002;21:5400-13 [DOI] [PubMed] [Google Scholar]
- 7. Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964;20:202-3 [DOI] [PubMed] [Google Scholar]
- 8. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980;20:85-93 [DOI] [PubMed] [Google Scholar]
- 9. Kantarjian H, Issa JPJ, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006;106:1794-803 [DOI] [PubMed] [Google Scholar]
- 10. Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, et al. Approval summary: azacytidine for the treatment of myelodysplastic syndrome subtypes. Clin Cancer Res 2005;11:3604-8 [DOI] [PubMed] [Google Scholar]
- 11. Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol 2005;25:4727-41 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005;6:597-610 [DOI] [PubMed] [Google Scholar]
- 13. Fanelli M, Caprodossi S, Ricci-Vitiani L, Porcellini A, Tomassoni-Ardori F, Amatori S, et al. Loss of pericentromeric DNA methylation pattern in human glioblastoma is associated with altered DNA methyltransferases expression and involves the stem cell compartment. Oncogene 2008;27:358-65 [DOI] [PubMed] [Google Scholar]
- 14. Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci USA 1994;91:11797-801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol 2008;28:752-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc Natl Acad Sci USA 1997;94:4681-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferguson AT, Vertino PV, Spitzneri JR, Baylin SB, Muller MT, Davidson NE. Role of estrogen receptor gene demethylation and DNA methyltransferase-DNA adduct formation in 5-Aza-2′-deoxycytidine-induced cytotoxicity in human breast cancer cells. J Bio Chem 1997;272:32260-6 [DOI] [PubMed] [Google Scholar]
- 18. Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature 1998;395:89-93 [DOI] [PubMed] [Google Scholar]
- 19. Rudek MA, Zhao M, He P, Hartke C, Gilbert J, Gore SD, et al. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J Clin Oncol 2005;123:3906-11 [DOI] [PubMed] [Google Scholar]
- 20. Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin Hematol 2005;42:S9-S16 [DOI] [PubMed] [Google Scholar]
- 21. Appleton K, Mackay HJ, Judson I, Plumb JA, McCormick C, Strathdee G, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol 2007;25:4603-9 [DOI] [PubMed] [Google Scholar]
- 22. Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and cronic myelomonocytic leukemia. Blood 2007;109:52-7 [DOI] [PubMed] [Google Scholar]
- 23. Kornblith AB, Herndon JE, Silverman LR, Demakos EP, Odchimar-Reissig R, Holland JF, et al. Impact of azacytidine in the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a cancer and leukaemia group B study. J Clin Oncol 2002;20:2441-52 [DOI] [PubMed] [Google Scholar]
- 24. Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacytidine in patients with the myelodysplastic syndrome: a study of the cancer and leukaemia group B. J Clin Oncol 2002;20:2429-40 [DOI] [PubMed] [Google Scholar]
- 25. Silverman LR, McKenzie DR, Peterson BL, Holland JF, Backstrom JT, Beach CL, et al. Further analysis of trial with azacytidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the cancer and leukemia group B. J Clin Oncol 2006;24:3895-903 [DOI] [PubMed] [Google Scholar]
- 26. Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res 1998;58:95-101 [PubMed] [Google Scholar]
- 27. Herman JG, Jen J, Merlo A, Baylin SB. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B. Cancer Res 1996;56:722-7 [PubMed] [Google Scholar]
- 28. Shin JY, Kim HS, Park J, Park JB, Lee JY. Mechanism for inactivation of the KIP family cyclin-dependent kinase inhibitor genes in gastric cancer cells. Cancer Res 2000;60:262-5 [PubMed] [Google Scholar]
- 29. Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol 2007;212:330-44 [DOI] [PubMed] [Google Scholar]
- 30. Al-Romaih K, Somers GR, Bayani J, Hughes S, Prasad M, Cutz JC, et al. Modulation by decitabine of gene expression and growth of osteosarcoma U2OS cells in vitro and in xenografts: identification of apoptotic genes as targets for demethylation. Cancer Cell Int 2007;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiemjit A, Fandy TE, Carraway H, Bailey KA, Baylin S, Herman JG, et al. p21WAF1/CIP1 induction by 5-azacytosine nucleosides requires DNA damage. Oncogene 27:3615-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kissil JL, Feinstein E, Cohen O, Jones PA, Tsai YC, Knowles MA, et al. DAP-kinase loss of expression in various carcinoma and B-cell lymphoma cell lines: possible implications for role as tumor suppressor gene. Oncogene 1997;15:403-7 [DOI] [PubMed] [Google Scholar]
- 33. Katzenellenbogen RA, Baylin SB, Herma JG. Hypermethylation of the DAP-kinase CpG island is a common alteration in B-cell malignancies. Blood 1999;93:4347-53 [PubMed] [Google Scholar]
- 34. Tang X, Wu W, Sun SY, Wistuba, Hong WK, Mao L. Hypermethylation of the death associated protein kinase promoter attenuates the sensitivity to TRAIL-induced apoptosis in human non-small cell lung cancer cell. Mol Cancer Res 2004;2:685-91 [PubMed] [Google Scholar]
- 35. Reu FJ, Leaman DW, Maitra RR, Bae SI, Cherkassky L, Fox MW, et al. Expression of RASSF1A, an epigenetically silenced tumor suppressor, overcomes resistance to apoptosis induction by interferons. Cancer Res 2006;66:2785-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reu FJ, Bae SI, Cherkassky L, Leaman DW, Lindner D, Beaulieu N, et al. Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J Clin Oncol 2006;24:3771-9 [DOI] [PubMed] [Google Scholar]
- 37. Eramo A, Pallini R, Lotti F, Sette G, Patti M, Bartucci M, et al. Inhibition of DNA methylation sensitizes glioblastoma for tumor necrosis factor-related apoptosis-inducing ligand mediated destruction. Cancer Res 2005;65:11469-77 [DOI] [PubMed] [Google Scholar]
- 38. Soengas MS, Capodieci P, Polski D, Mora J, Esteller M, Opitz-Araya X, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001;409:207-11 [DOI] [PubMed] [Google Scholar]
- 39. Zhang C, Li K, Wei L, Li Z, Yu P, Teng L, et al. p300 expression repression by hypermethylation associated with tumour invasion and metastasis in oesophageal squamous cell carcinoma. J Clin Pathol 2007;60:1249-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998;95:6870-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999;21:103-7 [DOI] [PubMed] [Google Scholar]
- 42. Bae SI, Lee HS, Kim SH, Kim WH. Inactivation of O6-methylguanine-DNA methyltransferase by promoter CpG island hypermethylation in gastric cancers. Br J Cancer 2002;86:1888-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Danam RP, Howell SR, Brent TP, Harris LC. Epigenetic regulation of O6-methylguanine-DNA methyltransferase gene expression by histone acetylation and methyl-CpG binding proteins. Mol Cancer Ther 2005;4:61-9 [PubMed] [Google Scholar]
- 44. Miller-Kasprzak E, Jagodzinski PP. 5-Aza-2′-deoxycytidine increases the expression of anti-angiogenic vascular endothelial growth factor 189b variant in human lung microvascular endothelial cells. Biomed Pharmacother 2008;62:158-63 [DOI] [PubMed] [Google Scholar]
- 45. Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995;55:5195-9 [PubMed] [Google Scholar]
- 46. Corn PG, Smith BD, Ruckdeschel ES, Douglas D, Baylin SB, Herman JG. E-cadherin expression is silenced by 5′ CpG island methylation in acute leukemia. Clin Cancer Res 2000;6:4243-8 [PubMed] [Google Scholar]
- 47. Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res 2001;61:4556-60 [PubMed] [Google Scholar]
- 48. Nam JS, Ino Y, Kanai Y, Sakamoto M, Hirohashi S. 5-Aza-2′-deoxycytidine restores the E-cadherin system in E-cadherin-silenced cancer cells and reduces cancer metastasis. Clin Exp Metastasis 2004;21:49-56 [DOI] [PubMed] [Google Scholar]
- 49. Li Q, Ahuja N, Burger PC, Issa JP. Methylation and silencing of the Thrombospondin-1 promoter in human cancer. Oncogene 1999;18:3284-9 [DOI] [PubMed] [Google Scholar]
- 50. Ferguson AT, Lapidus RG, Baylin SB, Davidson NE. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res 1995;55:2279-83 [PubMed] [Google Scholar]
- 51. Youssef EM, Lotan D, Issa JP, Wakasa K, Fan YH, Mao L, et al. Hypermethylation of the retinoic acid receptor-β2 gene in head and neck carcinogenesis. Clin Cancer Res 2004;10:1733-42 [DOI] [PubMed] [Google Scholar]
- 52. Liu Z, Marquez M, Nilsson S, Holmberg AR. Incubation with somatostatin, 5-aza decitabine and trichostatin up-regulates somatostatin receptor expression in prostate cancer cells. Oncol Rep 2008;20:151-4 [PubMed] [Google Scholar]
- 53. Coral S, Sigalotti L, Colizzi F, Spessotto A, Nardi G, Cortini E, et al. Phenotypic and functional changes of human melanoma xenografts induced by DNA hypomethylation: immunotherapeutic implications. J Cell Physiol 2006;207:58-66 [DOI] [PubMed] [Google Scholar]
- 54. Coral S, Sigalotti L, Gasparollo A, Cattarossi I, Visintin A, Cattelan A, et al. Prolonged upregulation of the expression of HLA class I antigens and costimulatory molecules on melanoma cells treated with 5-aza-2′-deoxycytidine (5-AZA-CdR). J Immunother 1999;22:16-24 [DOI] [PubMed] [Google Scholar]
- 55. Arnold JM, Cummings M, Purdie D, Chenevix-Trench G. Reduced expression of intercellular adhesion molecule-1 in ovarian adenocarcinomas. Br J Cancer 2001;85:1351-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Calabrò L, Fonsatti E, Altomonte M, Pezzani L, Colizzi F, Nanni P, et al. Methylation-regulated expression of cancer testis antigens in primary effusion lymphoma: immunotherapeutic implications. J Cell Physiol 2005;202:474-7 [DOI] [PubMed] [Google Scholar]
- 57. Sigalotti L, Coral S, Fratta E, Lamaj E, Danielli R, Di Giacomo AM, et al. Epigenetic modulation of solid tumors as a novel approach for cancer immunotherapy. Semin Oncol 2005;32:473-8 [DOI] [PubMed] [Google Scholar]
- 58. Lettini AA, Guidoboni M, Fonsatti E, Anzalone L, Cortini E, Maio M. Epigenetic remodelling of DNA in cancer. Histol Histopathol 2007;22:1413-24 [DOI] [PubMed] [Google Scholar]
- 59. Wang H, Zhao Y, Li L, McNutt MA, Wu L, Lu S, et al. An ATM- and Rad3-related (ATR) signaling pathway and a phosphorylation-acetylation cascade are involved in activation of p53/p21Waf1/Cip1 in response to 5-Aza-2′-deoxycytidine treatment. J Bio Chem 2008;283:2564-74 [DOI] [PubMed] [Google Scholar]
- 60. Zhu WG, Hileman T, Ke Y, Wang P, Lu S, Duan W, et al. 5-Aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J Bio Chem 2004;279:1516-6 [DOI] [PubMed] [Google Scholar]
- 61. Amatori S, Papalini F, Lazzarini R, Donati B, Bagaloni I, Rippo MR, et al. Decitabine, differently from DNMT1 silencing, exerts its antiproliferative activity through p21 upregulation in malignant pleural mesothelioma (MPM) cells. Lung Cancer 2009;66:184-90 [DOI] [PubMed] [Google Scholar]
- 62. Scott SA, Lakshimikuttysamma A, Sheridan DP, Sanche SE, Geyer CR, DeCoteau GF. Zebularine inhibits human acute myeloid leukaemia cell growth in vitro in association with p15INK4B demethylation and reexpression. Exp Hematol 2007;35:263-73 [DOI] [PubMed] [Google Scholar]
- 63. Lemaire M, Momparler LF, Bernstein ML, Marquez VE, Momparler RL. Enhancement of antineoplastic action of 5-aza-2′-deoxycytidine by zebularine on L1210 leukemia. Anti-Cancer Drug 2005;16:301-8 [DOI] [PubMed] [Google Scholar]
- 64. Balch C, Yan P, Craft T, Young S, Skalnik DG, Huang THM, et al. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol Cancer Ther 2005;4:1505-14 [DOI] [PubMed] [Google Scholar]
- 65. Cheng JC, Matsen CB, Gonzalez FA, Ye W, Greer S, Marquez VE, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst 2003;95:399-409 [DOI] [PubMed] [Google Scholar]
- 66. Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, et al. Preferential response of cancer cells to zebularine. Cancer Cell 2004;6:151-8 [DOI] [PubMed] [Google Scholar]
- 67. Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG, et al. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol 2004;24:1270-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Herranz M, Martin-Caballero J, Fraga MF, Ruiz-Cabello J, Flores JM, Desco M, et al. The novel DNA methylation inhibitor zebularine is effective against the development of murine T-cell lymphoma. Blood 2006;107:1174-7 [DOI] [PubMed] [Google Scholar]
- 69. Li C, Villacorte D, Newman EM. Analyzing p16 DNA methylation and expression in EJ6 cells after treatment with the DNA (cytosine-5-)-methyltransferase inhibitor 5-fluoro-2′-deoxycytidine by bisulfite genomic sequencing and real-time RT PCR. P Am Assoc Cancer Res 2004;45:1605 [Google Scholar]
- 70. Li C, Villacorte D, Newman EM. The DNA (cytosine-5-)-methyltransferase inhibitor 5-fluoro-2′-deoxycytidine induces RASSF1A expression in EJ6 human bladder carcinoma cells. P Am Assoc Cancer Res 2005;45:4114 [Google Scholar]
- 71. Li C, Villacorte D, Newman EM. 5-fluoro-2′-deoxycytidine induces re-expression of hypermethylation-silenced genes in the human breast cancer cell line MDA-MB 231. P Am Assoc Cancer Res 2006;45:1613 [Google Scholar]
- 72. Beumer JH, Eiseman JL, Parise RA, Joseph E, Holleran JL, Covey JM, et al. Pharmacokinetics, metabolism, and oral bioavailability of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine in mice. Clin Cancer Res 2006;12:7483-9 [DOI] [PubMed] [Google Scholar]
- 73. Beumer JH, Parise RA, Newman EM, Doroshow JH, Synold TW, Lenz HJ, et al. Concentrations of the DNA methyltransferase inhibitor 5-Fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother Pharmacol 2008;62:363-8 [DOI] [PubMed] [Google Scholar]
- 74. Goffin J, Eisenhauer E. DNA methyltransferase inhibitors: state of the art. Ann Oncol 2002;13:1699-716 [DOI] [PubMed] [Google Scholar]
- 75. Samuels BL, Herndon JE, Harmon DC, Carey R, Aisner J, Corson JM, et al. Dihydro-5-azacytidine and cisplatin in the treatment of malignant mesothelioma: a phase II study by the Cancer and Leukemia Group B. Cancer 1998;82:1578-84 [PubMed] [Google Scholar]
- 76. Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res 2005;65:6305-10 [DOI] [PubMed] [Google Scholar]
- 77. Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res 2003;63:4984-9 [PubMed] [Google Scholar]
- 78. Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem 2005;280:40749-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res 2003;9:1596-603 [PubMed] [Google Scholar]
- 80. Singh N, DueÇas-Gonzalez A, Lyko F, Medina-Franco JL. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. Chem Med Chem 2009;4:792-9 [DOI] [PubMed] [Google Scholar]
- 81. Zambrano P, Segura-Pacheco B, Perez-Cardenas E, Cetina L, Revilla-Vazquez A, Taja-Chayeb L, et al. A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer 2005;5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Candelaria M, Gallardo-Rincón D, Arce C, Cetina L, Aguilar-Ponce JL, Arrieta O, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol 2007;18:1529-38 [DOI] [PubMed] [Google Scholar]
- 83. Godert AM, Angelino N, Woloszynska-Read A. An improved synthesis of psammaplin A. Bioorg Med Chem Lett 2006;16:3330-3 [DOI] [PubMed] [Google Scholar]
- 84. Pina IC, Gautschi JT, Wang G. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J Org Chem 2003;68:3866-73 [DOI] [PubMed] [Google Scholar]
- 85. Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (–)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 2003;63:7563-70 [PubMed] [Google Scholar]
- 86. Gao Z, Xu Z, Hung MS, Lin YC, Wang T, Gong M, et al. Promoter demethylation of WIF-1 by epigallocatechin-3-gallate in lung cancer cells. Anticancer Res 2009;29:2025-30 [PubMed] [Google Scholar]
- 87. Gu B, Ding Q, Xia G, Fang Z. EGCG inhibits growth and induces apoptosis in renal cell carcinoma through TFPI-2 overexpression. Oncol Rep 2009;21:635-40 [PubMed] [Google Scholar]
- 88. Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol 2005;68:1018-30 [DOI] [PubMed] [Google Scholar]
- 89. Szyf M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry 2005;7:533-49 [DOI] [PubMed] [Google Scholar]
- 90. MacLeod AR, Szyf M. Expression of antisense to DNA methyltransferase mRNA induces DNA demethylation and inhibits tumorigenesis. J Biol Chem 1995;270:8037-43 [DOI] [PubMed] [Google Scholar]
- 91. Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002;416:552-6 [DOI] [PubMed] [Google Scholar]
- 92. Leu YW, Rahmatpanah F, Shi H, Wei SH, Liu JC, Yan PS, et al. Double RNA interference of DNMT3b and DNMT1 enhances DNA demethylation and gene reactivation. Cancer Res 2003;63:6110-5 [PubMed] [Google Scholar]
- 93. Gius D, Cui H, Bradbury CM, Cook J, Smart DK, Zhao S, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 2004;6:361-71 [DOI] [PubMed] [Google Scholar]
- 94. Jung Y, Park J, Kim TY, Park JH, Jong HS, Im SA, et al. Potential advantages of DNA methyltransferase 1 (DNMT1)-targeted inhibition for cancer therapy. J Mol Med 2007;85:1137-48 [DOI] [PubMed] [Google Scholar]
- 95. Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000;404:1003-7 [DOI] [PubMed] [Google Scholar]
- 96. Szyf M, Knox DJ, Milutinovic S, Slack AD, Araujo FD. How does DNA methyltransferase cause oncogenic transformation? Ann NY Acad Sci 2000;910:156-74 [DOI] [PubMed] [Google Scholar]
- 97. Ramchandani S, MacLeod AR, Pinard M, von Hofe E, Szyf M. Inhibition of tumorigenesis by a cytosine-DNA, methyltransferase, antisense oligodeoxynucleotide. Proc Natl Acad Sci USA 1997;94:684-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Beaulieu N, Fournel M, Macleod A. Antitumor activity of MG98, an antisense oligonucleotide targeting DNA methyltransferase-1 (DNMT1). Clin Cancer Res 2001;7(suppl):3800S [Google Scholar]
- 99. Beaulieu N, Fournel M, Siders B, Macleod A. Synergistic antitumor activity of MG98 (antisense oligonucleotide targeting DNMT1) in combination with 5-aza-deoxycytosine. Clin Cancer Res 2001;7(suppl):3800S-01S [Google Scholar]
- 100. Davis AJ, Gelmon KA, Siu LL, Moore MJ, Britten CD, Mistry N, et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as 21-day continuous infusion every 4 weeks. Invest New Drugs 2003;21:85-97 [DOI] [PubMed] [Google Scholar]
- 101. Stewart DJ, Donehower RC, Eisenhaue EA, Wainman N, Shah AK, Bonfils C, et al. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann Oncol 2003;14:766-74 [DOI] [PubMed] [Google Scholar]
- 102. Winquist E, Knox J, Ayoub JP, Wood L, Wainman N, Reid GK, et al. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest New Drugs 2006;24:159-67 [DOI] [PubMed] [Google Scholar]
- 103. Klisovic RB, Stock W, Cataland S, Klisovic MI, Liu S, Blum W, et al. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin Cancer Res 2008;12:2444-49 [DOI] [PubMed] [Google Scholar]
- 104. Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009;113:6411-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 2007;104:15805-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chuang JC, Yoo CB, Kwan JM, Li TW, Liang G, Yang AS, et al. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2′-deoxycytidine. Mol Cancer Ther 2005;4:1515-20 [DOI] [PubMed] [Google Scholar]
- 107. Stresemann C, Brueckner B, Musch T, Stopper H, Lyko F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines methylation inhibitors. Cancer Res 2006;66:2794-800 [DOI] [PubMed] [Google Scholar]
- 108. Jansen B, Zangemeister-Wittke U. Antisense therapy for cancer—the time of truth. Lancet Oncol 2002;3:672-83 [DOI] [PubMed] [Google Scholar]
- 109. Yang H, Hoshino K, Sanchez-Gonzales B, Kantarjian H, Garcia-Manero G. Antileukemia activity of the combination of 5-aza-2′-deoxycytidine with valproic acid. Leukemia Res 2005;29:739-48 [DOI] [PubMed] [Google Scholar]
- 110. Ishiguro M, Iida S, Uetake H, Morita S, Makino H, Kato K, et al. Effect of combined therapy with low-dose 5-aza-2-deoxycytidine and irinotecan on colon cancer cell line HCT-15. Ann Surgical Oncol 2006;14:1752-62 [DOI] [PubMed] [Google Scholar]
- 111. Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M, Moran K, et al. Phase I trial of sequential low-dose 5′-aza-2′deoxycitydine plus high-dose intravenous bolus interleukin-2 in patients with melanoma or renal cell carcinoma. Clin Cancer Res 2006;12:4619-27 [DOI] [PubMed] [Google Scholar]
- 112. Qiu H, Yashiro M, Shinto O, Matsuzaki T, Hirakawa K. DNA methyltransferase inhibitor 5-aza-CdR enhances the radiosensitivity of gastric cancer cells. Cancer Sci 2009;100:181-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Eliopoulos N, Cournoyer D, Momparler RL. Drug resistance to 5-aza-2-deoxycytidine, 2,2-difluorodeoxycytidine and cytosine arabinoside conferred by retroviral-mediated transfer of human cytidine deaminase cDNA into murine cells. Cancer Chemother Pharmacol 1998;42:373-8 [DOI] [PubMed] [Google Scholar]
- 114. Lemaire M, Momparler LF, Raynal NJM, Bernstein ML, Momparler RL. Inhibition of cytidine deaminase by zebularine enhances the antineoplastic action of 5-aza-2-deoxycytidine. Cancer Chemother Pharmacol 2009;63:411-16 [DOI] [PubMed] [Google Scholar]
- 115. Timmermann S, Hinds PW, Münger K. Re-expression of endogenous p16ink4a in oral squamous cell carcinoma lines by 5-aza-2′-deoxycytidine treatment induced a senescence-like state. Oncogene 1998;17:3445-53 [DOI] [PubMed] [Google Scholar]