

Abstract
Cancer is an evolutionary process that arises due to mutations and expands through the selection of clones with higher reproductive success that will outcompete their peers. Most tumors require many mutations to explain the cancer phenotype, making it difficult to identify the gene(s) that confer the reproductive fitness to the clone. Moreover, the impact of any oncogene is context dependent: it can increase the fitness of particular stages of cell differentiation but not other stages. In addition, the fitness advantage of an oncogene is not irreversible: sometimes it can be reversed with targeted therapy, for example. The understanding of these dynamical processes and their consequences may be greatly simplified when addressed from an evolutionary perspective. Using the dynamics of chronic myeloid leukemia—perhaps the best understood human neoplasm—as an example, we show how three fundamental evolutionary behaviors provide insights into the dynamics of this disease: (1) BCR-ABL does not affect the reproductive success of any cell within the stem cell pool (resulting therefore in neutral drift), (2) BCR-ABL expression gives a fitness (selective) advantage to progenitor cells, and (3) imatinib therapy reduces the fitness of progenitor cells expressing the oncogene (selective disadvantage) and consequently leads to significant reductions in disease burden. These three different evolutionary dynamics scenarios based on the interpretation of mutation and gene expression as potentially leading to a fitness imbalance of cell populations clearly explain the course of the disease, providing as such a better grasp of cancer dynamics and the role of related therapies.
Keywords: oncogenes, mutant fitness, clonal expansion, selection, small-molecule inhibitors
Introduction
Multicellularity
The emergence of multicellularity was one of the major transitions in evolution and occurred independently several times in the history of life on Earth.1-3 Multicellularity brought with it several advantages to organisms, including (1) resistance to predation, (2) functional specialization and division of labor between cells, and (3) more efficient energy utilization.4 However, multicellularity required that the majority of cells within the organism (the soma) “sacrificed” their reproductive potential1,4-6 such that only the germ cells are responsible for reproduction of the organism. This not withstanding, essentially all tissues in the body undergo turnover, and in order to maintain tissue homeostasis, somatic cells still require a limited reproductive potential.
Because cells can acquire mutations either due to errors during DNA replication7 or as a consequence of exposure to genotoxic agents, most tissues have an architecture that limits the probability that mutant cells survive for long periods of time.8 Every epithelium in the body as well as hematopoiesis are organized in a hierarchical manner: at the root, one finds stem cells that divide at a relatively slow pace, being able to self-renew and give rise to more committed progenitor cells. Progenitor cells replicate faster, and their daughter cells differentiate even further until mature cells are produced that cannot divide and generally live for a short period of time.8-13
Cancer is one of the consequences of multicellularity. The acquisition of mutations is a stochastic process, and as a result, the probability that a given specific mutation occurs depends on the number of cells at risk, the mutation rate, and the life expectancy of the host.14 Of course, whether a mutation leads to a given phenotype depends on the host cell where it occurs15 and on the mutation type. It is easy to see that in any “average”-sized human being with ~1014 cells, there will be many cells with a mutation in any given gene because the normal mutation rate has been estimated to be ~10−7 per gene per replication.16 Cancer is not more frequent because (1) many mutations, being context and cell specific, do not occur in cells with the potential to cause cancer15; (2) most mutations are deleterious to the cell and, as a consequence, will lead to cell death; (3) one specific mutation is normally not enough to lead to cancer,17,18 and hence the cell will have to live for a long enough time to acquire the additional mutations necessary for transformation19; and (4) immune surveillance may eliminate mutant cells.20,21 Genomic instability may enhance the probability that mutations occur,22 but it is not essential for the development of cancer.23-25 Recently, it has been shown that many tumors have a hierarchical cellular organization similar to normal tissues.26 The bulk of the tumor population is composed of relatively short-lived cells, with their population being maintained by cancer stem cells (CSC). Initially described in acute myeloid leukemia,27 CSC now appear to be present in most tumors,26,28 although some skeptics claim that their existence is an artifact of xenotransplantation in immunodeficient mice. One of the questions that arises relates to the cellular origin of these CSC. There is evidence that normal stem cells (e.g., hematopoietic stem cells, HSC) can become CSC due to acquired mutations,26 but it is also possible that more differentiated cells reacquire stem cell–like properties due to mutations.29,30
Evolution
Evolution is a natural consequence of reproduction, mutation, and selection within populations.5 Given the large number of cells in most multicellular organisms and the inevitable occurrence of mutations, aberrant clones are developing in such organisms (e.g., in the human body) all the time, at par with normal cells. The natural history of such clones depends on the location of the initiating mutant cell within the hierarchical organization of the specific tissue and the reproductive advantage (fitness) that the mutation(s) confer(s) to the cell that harbors it. Cells with a higher relative fitness on average will have more progeny cells, enabling the mutant cell population to expand and outcompete its neighbors, whereas cells harboring a deleterious mutation tend to have a reduced average reproductive fitness that normally would result in elimination of the clone. Many mutations do not alter the fitness of the cell (neutral fitness), and the number of cells harboring such a mutation can only change (increase or decrease) stochastically by neutral drift.31 In such a scenario, the outcome depends sensitively on the size of the populations that are competing with each other. Large populations are protected from significant invasions of neutral mutations, but they are at increased risk from mutant clones with a higher fitness.14 Finally, it is also likely that many mutations present in cancer cells have no impact on the tumor phenotype, and these have been labeled “passenger mutations.”32,33
Mutations causing cancer typically increase the fitness of cells either by enhancing their reproductive potential or by generally prolonging their survival.8,12,23-25,34,35 Furthermore, enhanced reproductive fitness need not derive from the replication rate of a single cell, being instead observed at a cell population level36: a different probability of self-renewal of mutant cells compared to their competitors may be enough to enable one clone to eventually outgrow another.37
Although these concepts are generally accepted, it has been difficult to apply them specifically to any tumor. The difficulty with this approach lies at multiple levels, including (1) many tumors arise due to the cumulative effect of multiple mutations, making it difficult to infer the specific impact of a given gene on reproductive fitness; (2) fitness is a phenotypic property that is defined by the environment where the mutant cell exists, and it is difficult to reproduce this environment in vitro; and (3) drugs that specifically alter the fitness of cancer cells are uncommon.
We believe that chronic myeloid leukemia (CML) is a paradigm-setting neoplasm that illustrates all aspects of tumor evolution from neutral drift to cells with higher fitness due to oncogene expression and reduced fitness as a result of targeted therapy. In the following, we discuss these evolutionary aspects of cancer using CML as a prototypic example.
Chronic Myeloid Leukemia
CML is a classic myeloproliferative neoplasm.38,39 The disease is characterized by the presence of the Philadelphia chromosome,40 that is, the outcome of a balanced translocation between chromosomes 9 and 22 [t(9;22)]. As a result, the c-abl proto-oncogene, present on chromosome 9, is translocated to the major breakpoint cluster region (bcr) on chromosome 22 with the formation of the BCR-ABL fusion gene and the aberrant expression of the BCR-ABL oncogene.41 This oncoprotein is the target of imatinib mesylate, the first of a series of reversible abl-kinase inhibitors42-44 that gives high response rates in patients with this disease45—that is, it leads to a significant reduction of disease burden in the chronic phase of the disease. It is generally accepted that BCR-ABL expression alone is enough for the development of the chronic phase of CML.46 This is supported by animal models where aberrant expression of BCR-ABL in hematopoietic stem cells (more precisely, CD34+ hematopoietic cells) leads to a condition similar to the chronic phase of the disease.47,48 Moreover, the age-specific incidence of CML is also compatible with a single mutation (“one-hit”) event.49 Because BCR-ABL is found in both myeloid and lymphoid cells, including a small fraction of T and NK cells,50 one concludes that CML is a true HSC disorder. A characteristic feature of CML is an expansion of hematopoietic cell output, with an increase in the circulating number of granulocytes and their precursors, as well as extramedullary hematopoiesis leading to splenomegaly. Although normal marrow output in an adult is ~3.5 × 1011 cells/day, hematopoietic output in patients with CML often exceeds 1012 cells/day.46
Neutral Evolution: CML Stem Cells
Several studies have now shown that patients with CML have a population of primitive cells (that can be CD34+ or CD34−) that harbor the Philadelphia chromosome but do not express BCR-ABL.51-54 Clearly, these cells are independent of BCR-ABL and would not be expected to be sensitive to tyrosine kinase inhibitor therapy. In addition, it has been argued that BCR-ABL expression in HSC does not enhance their self-renewal properties.55-59 Consequently, it is not surprising that there is also (indirect) independent evidence suggesting that the HSC pool may not expand in this disease.29,36,60 Experimental and theoretical estimates show that the pool of active HSC in humans is small and of the order of 400, and these cells replicate slowly—on average, each cell replicates once per year.61-63 The appearance of the first HSC with the t(9;22) is a rare event because the probability of any chromosomal translocation in a cell is of the order of 10−5 (per cell per replication), and as discussed above, the number of active HSC is small and cells replicate slowly. Because primitive HSC may not express BCR-ABL, they will not be influenced by it, and so the time development of the HSC population that harbors the t(9;22) abnormality will be determined by neutral drift. A convenient framework in which to describe such a process is via the well-known Moran process from population genetics. We have estimated that, in most patients, the number of active CSC is probably less than 8.64 Perhaps this small number of CSC may be a surprise to the reader because it appears “easy” to induce a disease like CML by injecting human CML progenitor cells in immunosuppressed mice. Although these cells engraft, if the mice are observed long enough, the clone disappears,65 which supports the idea that cells injected in mice are not CSC but early progenitor cells. On the other hand, this small number of CSC would be expected, given that their dynamics is governed by neutral drift, as BCR-ABL does not seem to affect stem cells dynamics.
Neutral drift has implications for the fate of the clone. After the appearance of the first CSC, the mutant cell can be selected to divide. If the cell divides, the clone will expand, increasing the probability that the patient will develop disease. On the other hand, the CSC may be selected for export from the active stem cell pool. The export of such a cell to the progenitor pool will start the process leading to disease66 (see below). Under neutral drift, both selection processes occur with the same probability, and hence it is possible that all CSC are selected for export, disappearing from the HSC population. Indeed, it is possible to show that in a substantial fraction of virtual patients with simulated CML, the stem cell(s) in which the disease originated are no longer contributing to hematopoiesis when increased marrow output leads to the diagnosis of CML, the disease being maintained by the inertia of progenitor cells that derived from the differentiation of the leukemic stem cell(s).66
Fitness Advantage: CML Progenitor Cells
It has been shown that progenitor cells that express BCR-ABL produce IL-3 and G-CSF that may act in an autocrine fashion to enhance the self-renewal of these leukemic progenitors leading to clonal expansion.67,68 In the language of evolution, leukemic progenitors acquire a relative fitness advantage compared to normal progenitors, unlike CSC and HSC, which undergo neutral evolution. Such a fitness advantage leads to the expansion of the leukemic progenitor population, which enables CML progenitors to take over hematopoiesis and effectively drive the disease. Therefore, while CML arises in the HSC, it is driven by the expanded progenitor cell clone.
Combining mathematical modeling of hematopoiesis and CML (see below) with serial BCR-ABL data from patients with CML who were being treated with imatinib64 allowed us to determine the level of enhancement of limited self-renewal of progenitor cells.69,70 A major difference between HSC and progenitor self-renewal is that HSC can self-renew for a much longer time than progenitor cells. Hence, HSC contribute to hematopoiesis for years while committed progenitor cells may contribute to hematopoiesis for just a few weeks to months.9,26
A mathematical model of hematopoiesis
Under normal conditions, hematopoiesis can be metaphorically represented by a multicompartmental model in dynamic equilibrium in which cells transfer from one compartment to the next, in a conveyor belt fashion, as they become increasingly differentiated (see Fig. 1).9 As stated before, in a healthy adult, approximately 400 HSC, each replicating on average once per year,61,63 are responsible for a daily marrow output of ~3.5 × 1011 cells. In any compartment i downstream of the stem cell pool, a cell division leads to two daughter cells that are either (1) transferred to the next downstream compartment (i + 1, compatible with differentiation) or (2) stay in the same compartment, as the daughter cells retain the properties of their parent (self-renewal).9 Under stationary conditions, the size of each compartment remains constant on average. The probability of differentiation (ε) is for simplicity considered to be constant across hematopoiesis while the corresponding probability of self-renewal is given by 1 − ε. The normal (physiologic) value of ε was determined as ε0 = 0.85. In the case of normal hematopoiesis, our estimate of the total number of replications (K) that link HSC with the circulating blood cell compartment is 31, whereas the exponential increase in replication rate that occurs between compartments is r ≈ 1.26.9 Our estimate for K (~31) is similar to prior predictions,71-73 and the estimated values of both ε and r are essentially independent of the number of HSC contributing to hematopoiesis, strongly suggesting that they constitute characteristic features of hematopoiesis.
Figure 1.
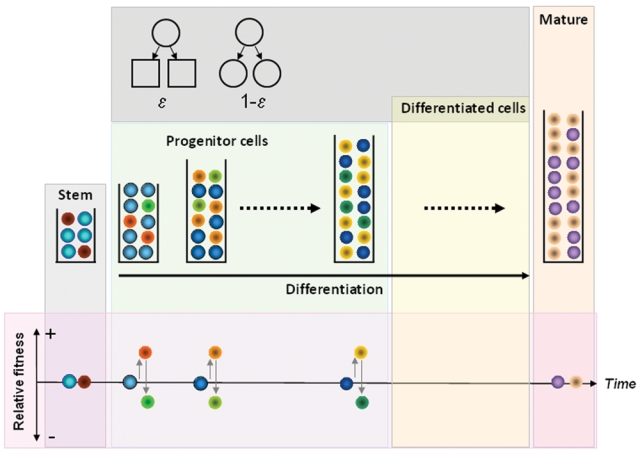
Dynamics of normal and chronic myeloid leukemia (CML) cells in hematopoiesis. Normal (blue) and leukemic (red) stem cells may coexist under neutral drift at the level of the stem cell pool. Downstream of the stem cell pool, cells that are selected to replicate differentiate with probability ε or self-renew with probability 1 − ε (top inset). Differentiation leads to migration of cells to the next downstream compartment (on the right, see time arrow) while self-renewal keeps the daughter cells in the same compartment as their parent. Progenitor cells produced from stem cell replication populate downstream hematopoietic compartments. BCR-ABL expression increases the relative fitness of CML cells (different shades of red and yellow), enabling them to gradually take over hematopoiesis (normal progenitors depicted in different shades of blue). As a result, the clone expands and is increasingly represented in downstream hematopoietic compartments. BCR-ABL expression has no impact on mature cells. The impact of BCR-ABL on the more mature cells is unclear at present. Imatinib therapy affects a fraction of CML cells (in different shades of green) and reduces their relative fitness compared to normal cells. Imatinib has no impact on stem cells or mature cells.
The same model can be employed to understand serial BCR-ABL data from patients with CML under therapy. In accord with what was discussed before, the phenotypic effect of BCR-ABL expression in CML cells is to enhance their self-renewal capability, which corresponds, in the model, to a change of ε from ε0 = 0.85 to εCML = 0.72,64 compatible with what Marley et al.74,75 have reported. Such a change in ε has consequences in what concerns the fitness of CML cells. The existence of two cell populations with different properties leads the hematopoietic system to evolve toward a new stationary regime in which the two cell populations coexist. Because εCML = 0.72 compared to ε0 = 0.85, CML cell numbers grow faster with compartment number than normal cell numbers. As a result, one observes that the number of normal cells contributing to hematopoiesis decreases in CML compared to normal (CML-free) conditions. We may recast this information in an evolutionary perspective by computing the ratio between the probability of self-renewal and that of differentiation for these different cell types, which is independent of their number and is given by . Hence, one can define the relative reproductive fitness of the CML cell type with respect to normal cells as ; replacing the respective values of ε gives fCML = 2.2, a significant relative fitness advantage in evolutionary terms.76 Given this value for the relative fitness advantage of CML progenitors, the takeover of hematopoiesis by the CML clone is not surprising, even though it is known that normal HSC persist in the bone marrow of patients with this disease.57,59,77 In addition, because it appears that differentiation in hematopoiesis is linked to replication, the enhanced self-renewal of leukemic progenitors leads to more cell divisions on the path to circulating blood and consequently to an “output” of cells with shorter telomeres compared to normal cells.78 Therefore, CML progenitors take over hematopoiesis by increasing their self-renewal compared to normal progenitors, a process that takes considerable time but yields a robust exponential growth of the CML cell population. Indeed, the model is compatible with the observation that it takes approximately 5 years from the initial insult to the diagnosis of disease.64,66,79
Lower Fitness: BCR-ABL and Imatinib Therapy
The same combination of mathematical modeling with serial data enables an estimate of the impact of imatinib therapy on the behavior of CML progenitors. Interestingly, it is found that imatinib reduces the self-renewal probability of CML progenitor cells from εCML = 0.72 to εIMAT = 0.89 − 0.90 (Fig. 1).64 This model prediction turns out to be compatible with experimental observations of the growth kinetics of CFU-GM isolated from patients with CML and exposed to pharmacologically achievable concentrations of imatinib in vitro.42,75 Such values of εIMAT imply that imatinib-treated cells are now evolutionarily disadvantageous. Using the same reasoning as in the previous section, the relative fitness disadvantage of CML cells in the presence of imatinib can be calculated as fIMAT = 0.70.76 Therefore, from an evolutionary standpoint, imatinib therapy reduces the fitness advantage of CML progenitors compared to normal progenitors, enabling the latter to regain control of hematopoiesis. Under imatinib therapy, CML cells transit through hematopoiesis faster but are unable to amplify their population. As a consequence, CML cells exiting hematopoiesis for the circulation have longer telomeres compared to untreated controls.80 This reduced fitness in the presence of a drug may explain why imatinib therapy is so effective in reducing the disease burden in CML.
Conclusions
We have presented an evolutionary dynamic view of CML, perhaps the best understood human neoplasm. The disease illustrates many aspects that are at the core of evolutionary biology: (1) neutral fitness of cells (CML stem cells) that impart a significant stochastic component to disease progression and can have major implications as a consequence of their offspring; (2) high fitness of CML progenitors that take over hematopoiesis, leading to disease diagnosis; and (3) conditional reduced fitness in the presence of specific drug therapy that suppresses the impact of an oncogene on the cell that expresses it. Clearly, the phenotype is context dependent, and the impact of a gene depends on not only the cell where it is expressed but also the environment that may select for or against it. Viewed in this way, the appearance of an HSC with BCR-ABL is similar to the proverbial butterfly that can lead to a hurricane by fluttering its wings. Neutral evolution need not be benign81: the downstream consequences of a neutral mutation can be highly significant and even deadly. This evolutionary perspective on cancer dynamics can help us to understand how the disease unfolds in time and its response to therapy and also explains phenomena that are difficult to grasp from a more traditional clinical point of view.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
DD is supported by Mayo Foundation, AT is supported by DFG and DAAD (Project 0813008), TL acknowledges support from FNRS, and JMP is supported by FCT Portugal and DAAD.
References
- 1. Maynard Smith J, Szathmary E. The major transitions in evolution. Oxford, UK: Freeman; 1995 [Google Scholar]
- 2. Bonner JT. The origins of multicellularity. Integr Biol 1998;1:28-36 [Google Scholar]
- 3. Carroll SB. Chance and necessity: the evolution of morphological complexity and diversity. Nature 2001;409:1102-9 [DOI] [PubMed] [Google Scholar]
- 4. Grosberg RK, Strathmann RR. The evolution of multicellularity: a minor major transition? Annu Rev Ecol Evol Syst 2007;38:621-54 [Google Scholar]
- 5. Nowak MA. Evolutionary dynamics. Cambridge, MA: Belknap; 2006 [Google Scholar]
- 6. King N. The unicellular ancestry of animal development. Dev Cell 2004;7:313-25 [DOI] [PubMed] [Google Scholar]
- 7. Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem 2000;69:497-529 [DOI] [PubMed] [Google Scholar]
- 8. Nowak MA, Michor F, Iwasa Y. The linear process of somatic evolution. Proc Natl Acad Sci USA 2003;100:14966-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dingli D, Traulsen A, Pacheco JM. Compartmental architecture and dynamics of hematopoiesis. PLoS ONE 2007;2:e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnston MD, Edwards CM, Bodmer WF, Maini PK, Chapman SJ. Mathematical modeling of cell population dynamics in the colonic crypt and in colorectal cancer. Proc Natl Acad Sci USA 2007;104:4008-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnston MD, Edwards CM, Bodmer WF, Maini PK, Chapman SJ. Examples of mathematical modeling: tales from the crypt. Cell Cycle 2007;6:2106-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nat Rev Cancer 2004;4:197-205 [DOI] [PubMed] [Google Scholar]
- 13. Michor F, Iwasa Y, Rajagopalan H, Lengauer C, Nowak MA. Linear model of colon cancer initiation. Cell Cycle 2004;3:358-62 [PubMed] [Google Scholar]
- 14. Lopes JV, Pacheco JM, Dingli D. Acquired hematopoietic stem-cell disorders and mammalian size. Blood 2007;110:4120-2 [DOI] [PubMed] [Google Scholar]
- 15. Pierce A, Owen-Lynch PJ, Spooncer E, Dexter TM, Whetton AD. p210 Bcr-Abl expression in a primitive multipotent haematopoietic cell line models the development of chronic myeloid leukaemia. Oncogene 1998;17:667-72 [DOI] [PubMed] [Google Scholar]
- 16. Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, et al. A quantitative measurement of the human somatic mutation rate. Cancer Res 2005;65:8111-7 [DOI] [PubMed] [Google Scholar]
- 17. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10:789-99 [DOI] [PubMed] [Google Scholar]
- 18. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009;458:719-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, et al. Genetic progression and the waiting time to cancer. PLoS Comput Biol 2007;3:e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenberg AH, Greene M. Non-adaptive rejection of small tumour inocula as a model of immune surveillance. Nature 1976;264:356-9 [DOI] [PubMed] [Google Scholar]
- 21. Penninger JM, Wen T, Timms E, Potter J, Wallace VA, Matsuyama T, et al. Spontaneous resistance to acute T-cell leukaemias in TCRV gamma 1.1J gamma 4C gamma 4 transgenic mice. Nature 1995;375:241-4 [DOI] [PubMed] [Google Scholar]
- 22. Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci USA 2006;103:18238-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tomlinson I, Bodmer W. Selection, the mutation rate and cancer: ensuring that the tail does not wag the dog. Nat Med 1999;5:11-12 [DOI] [PubMed] [Google Scholar]
- 24. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70 [DOI] [PubMed] [Google Scholar]
- 25. Bodmer W, Bielas JH, Beckman RA. Genetic instability is not a requirement for tumor development. Cancer Res 2008;68:3558-60; discussion 3560-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105-11 [DOI] [PubMed] [Google Scholar]
- 27. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730-7 [DOI] [PubMed] [Google Scholar]
- 28. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009;15:1010-2 [DOI] [PubMed] [Google Scholar]
- 29. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004;351:657-67 [DOI] [PubMed] [Google Scholar]
- 30. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006;442:818-22 [DOI] [PubMed] [Google Scholar]
- 31. Kimura M. DNA and the neutral theory. Philos Trans R Soc Lond B Biol Sci 1986;312:343-54 [DOI] [PubMed] [Google Scholar]
- 32. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463:191-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cairns J. Mutation selection and the natural history of cancer. Nature 1975;255:197-200 [DOI] [PubMed] [Google Scholar]
- 35. Dingli D, Traulsen A, Pacheco JM. Stochastic dynamics of hematopoietic tumor stem cells. Cell Cycle 2007;6:461-6 [DOI] [PubMed] [Google Scholar]
- 36. Buckle AM, Mottram R, Pierce A, Lucas GS, Russell N, Miyan JA, et al. The effect of Bcr-Abl protein tyrosine kinase on maturation and proliferation of primitive haematopoietic cells. Mol Med 2000;6:892-902 [PMC free article] [PubMed] [Google Scholar]
- 37. Dingli D, Traulsen A, Michor F. (A)Symmetric stem cell replication and cancer. PLoS Comput Biol 2007;3:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fialkow PJ, Jacobson RJ, Papayannopoulou T. Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am J Med 1977;63:125-30 [DOI] [PubMed] [Google Scholar]
- 39. Goldman JM. Chronic myeloid leukemia—still a few questions. Exp Hematol 2004;32:2-10 [DOI] [PubMed] [Google Scholar]
- 40. Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973;243:290-3 [DOI] [PubMed] [Google Scholar]
- 41. Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984;36:93-9 [DOI] [PubMed] [Google Scholar]
- 42. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561-6 [DOI] [PubMed] [Google Scholar]
- 43. Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 2006;354:2542-51 [DOI] [PubMed] [Google Scholar]
- 44. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006;354:2531-41 [DOI] [PubMed] [Google Scholar]
- 45. Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355:2408-17 [DOI] [PubMed] [Google Scholar]
- 46. Holyoake TL, Jiang X, Drummond MW, Eaves AC, Eaves CJ. Elucidating critical mechanisms of deregulated stem cell turnover in the chronic phase of chronic myeloid leukemia. Leukemia 2002;16:549-58 [DOI] [PubMed] [Google Scholar]
- 47. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990;247:824-30 [DOI] [PubMed] [Google Scholar]
- 48. Zhao RC, Jiang Y, Verfaillie CM. A model of human p210(bcr/ABL)-mediated chronic myelogenous leukemia by transduction of primary normal human CD34(+) cells with a BCR/ABL-containing retroviral vector. Blood 2001;97:2406-12 [DOI] [PubMed] [Google Scholar]
- 49. Michor F, Iwasa Y, Nowak MA. The age incidence of chronic myeloid leukemia can be explained by a one-mutation model. Proc Natl Acad Sci USA 2006;103:14931-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin PJ, Najfeld V, Hansen JA, Penfold GK, Jacobson RJ, Fialkow PJ. Involvement of the B-lymphoid system in chronic myelogenous leukaemia. Nature 1980;287:49-50 [DOI] [PubMed] [Google Scholar]
- 51. Keating A, Wang XH, Laraya P. Variable transcription of BCR-ABL by Ph+ cells arising from hematopoietic progenitors in chronic myeloid leukemia. Blood 1994;83:1744-9 [PubMed] [Google Scholar]
- 52. Schulze E, Krahl R, Thalmeier K, Helbig W. Detection of bcr-abl mRNA in single progenitor colonies from patients with chronic myeloid leukemia by PCR: comparison with cytogenetics and PCR from uncultured cells. Exp Hematol 1995;23:1649-54 [PubMed] [Google Scholar]
- 53. Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002;99:319-25 [DOI] [PubMed] [Google Scholar]
- 54. Lemoli RM, Salvestrini V, Bianchi E, Bertolini F, Fogli M, Amabile M, et al. Molecular and functional analysis of the stem cell compartment of chronic myelogenous leukemia reveals the presence of a CD34– cell population with intrinsic resistance to imatinib. Blood 2009;114:5191-200 [DOI] [PubMed] [Google Scholar]
- 55. Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004;6:587-96 [DOI] [PubMed] [Google Scholar]
- 56. Dazzi F, Capelli D, Hasserjian R, Cotter F, Corbo M, Poletti A, et al. The kinetics and extent of engraftment of chronic myelogenous leukemia cells in non-obese diabetic/severe combined immunodeficiency mice reflect the phase of the donor’s disease: an in vivo model of chronic myelogenous leukemia biology. Blood 1998;92:1390-6 [PubMed] [Google Scholar]
- 57. Dazzi F, Hasserjian R, Gordon MY, Boecklin F, Cotter F, Corbo M, et al. Normal and chronic phase CML hematopoietic cells repopulate NOD/SCID bone marrow with different kinetics and cell lineage representation. Hematol J 2000;1:307-15 [DOI] [PubMed] [Google Scholar]
- 58. Eisterer W, Jiang X, Christ O, Glimm H, Lee KH, Pang E, et al. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia 2005;19:435-41 [DOI] [PubMed] [Google Scholar]
- 59. Petzer AL, Eaves CJ, Barnett MJ, Eaves AC. Selective expansion of primitive normal hematopoietic cells in cytokine-supplemented cultures of purified cells from patients with chronic myeloid leukemia. Blood 1997;90:64-9 [PubMed] [Google Scholar]
- 60. Udomsakdi C, Eaves CJ, Swolin B, Reid DS, Barnett MJ, Eaves AC. Rapid decline of chronic myeloid leukemic cells in long-term culture due to a defect at the leukemic stem cell level. Proc Natl Acad Sci USA 1992;89:6192-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Buescher ES, Alling DW, Gallin JI. Use of an X-linked human neutrophil marker to estimate timing of lyonization and size of the dividing stem cell pool. J Clin Invest 1985;76:1581-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rufer N, Brummendorf TH, Kolvraa S, Bischoff C, Christensen K, Wadsworth L, et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J Exp Med 1999;190:157-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dingli D, Pacheco JM. Allometric scaling of the active hematopoietic stem cell pool across mammals. PLoS ONE 2006;1:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dingli D, Traulsen A, Pacheco JM. Chronic myeloid leukemia: origin, development, response to therapy, and relapse. Clin Leukemia 2008;2:133-9 [Google Scholar]
- 65. Hoyle CF, Negrin RS. Engraftment of chronic myeloid leukemia in SCID mice. Hematol Oncol 1998;16:87-100 [DOI] [PubMed] [Google Scholar]
- 66. Lenaerts T, Pacheco JM, Traulsen A, Dingli D. Tyrosine kinase inhibitor therapy can cure chronic myeloid leukemia without hitting leukemic stem cells. Haematologica 2009. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang X, Lopez A, Holyoake T, Eaves A, Eaves C. Autocrine production and action of IL-3 and granulocyte colony-stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci USA 1999;96:12804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Marley SB, Lewis JL, Gordon MY. Progenitor cells divide symmetrically to generate new colony-forming cells and clonal heterogeneity. Br J Haematol 2003;121:643-8 [DOI] [PubMed] [Google Scholar]
- 69. Marley SB, Gordon MY. Chronic myeloid leukaemia: stem cell derived but progenitor cell driven. Clin Sci (Lond) 2005;109:13-25 [DOI] [PubMed] [Google Scholar]
- 70. Gordon MY, Marley SB, Lewis JL, Davidson RJ, Nguyen DX, Grand FH, et al. Treatment with interferon-alpha preferentially reduces the capacity for amplification of granulocyte-macrophage progenitors (CFU-GM) from patients with chronic myeloid leukemia but spares normal CFU-GM. J Clin Invest 1998;102:710-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Lansdorp PM. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci USA 1994;91:9857-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. MacKey MC. Cell kinetic status of haematopoietic stem cells. Cell Prolif 2001;34:71-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shochat E, Stemmer SM, Segel L. Human haematopoiesis in steady state and following intense perturbations. Bull Math Biol 2002;64:861-86 [DOI] [PubMed] [Google Scholar]
- 74. Marley SB, Davidson RJ, Goldman JM, Gordon MY. Effects of combinations of therapeutic agents on the proliferation of progenitor cells in chronic myeloid leukaemia. Br J Haematol 2002;116:162-5 [DOI] [PubMed] [Google Scholar]
- 75. Marley SB, Deininger MW, Davidson RJ, Goldman JM, Gordon MY. The tyrosine kinase inhibitor STI571, like interferon-alpha, preferentially reduces the capacity for amplification of granulocyte-macrophage progenitors from patients with chronic myeloid leukemia. Exp Hematol 2000;28:551-7 [DOI] [PubMed] [Google Scholar]
- 76. Traulsen A, Pacheco JM, Dingli D. Reproductive fitness advantage of BCR-ABL expressing leukemia cells. Cancer Lett 2010. February 12 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 77. Petzer AL, Eaves CJ, Lansdorp PM, Ponchio L, Barnett MJ, Eaves AC. Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood 1996;88:2162-71 [PubMed] [Google Scholar]
- 78. Brummendorf TH, Rufer N, Holyoake TL, Maciejewski J, Barnett MJ, Eaves CJ, et al. Telomere length dynamics in normal individuals and in patients with hematopoietic stem cell-associated disorders. Ann N Y Acad Sci 2001;938:293-303; discussion 303-4 [DOI] [PubMed] [Google Scholar]
- 79. Ichimaru M, Ishimaru T, Mikami M, Yamada Y, Ohkita T. Incidence of leukemia in a fixed cohort of atomic bomb survivors and controls, Hiroshima and Nagasaki October 1950–December 1978. In: Technical Report RERF TR 13-81. Hiroshima, Japan: Radiation Effects Research Foundation; 1981 [Google Scholar]
- 80. Brummendorf TH, Ersoz I, Hartmann U, Balabanov S, Wolke H, Paschka P, et al. Normalization of previously shortened telomere length under treatment with imatinib argues against a preexisting telomere length deficit in normal hematopoietic stem cells from patients with chronic myeloid leukemia. Ann N Y Acad Sci 2003;996:26-38 [DOI] [PubMed] [Google Scholar]
- 81. Dingli D, Luzzatto L, Pacheco JM. Neutral evolution in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA 2008;105:18496-500 [DOI] [PMC free article] [PubMed] [Google Scholar]