

Abstract
The (c-)Myc oncoprotein and its cousins, the N-Myc and L-Myc proteins, show all hallmarks of transcriptional activator proteins: Myc carries a carboxy-terminal DNA binding domain, which mediates sequence-specific binding to DNA. At its amino-terminus, Myc carries a transcriptional regulatory domain that strongly activates transcription when fused to an ectopic DNA binding domain; moreover, the strength of activation of different members of the Myc family correlates with their ability to transform rodent cells. Furthermore, activation of conditional alleles of Myc, either tetracycline or estrogen inducible, upregulates expression of a large number of genes, both in tissue culture and in transgenic animals. Indeed, many of these genes have essential roles in cell proliferation, cell growth, and metabolism; two of them, odc, encoding ornithine decarboxylase, a rate-limiting enzyme of polyamine biosynthesis, and rpl24, encoding a constituent of the large ribosomal subunit, are haploinsufficient for Myc-induced lymphomagenesis but not for normal development, arguing very strongly that upregulation of both genes is critical for Myc-dependent tumor formation. Undoubtedly, therefore, Myc exerts part of its biological activities via transcriptional upregulation of a large number of target genes. One of the key issues in the field is whether there are additional biochemical activities of the Myc protein and, if so, whether and how they contribute to Myc biology. This review summarizes evidence demonstrating that Myc has the ability to repress transcription and that this may be an important function during oncogenic transformation.
Keywords: Miz1, Sp1, p15Ink4b, p21Cip1, integrin beta1, Arf
Gene Repression by Myc
Ectopic expression of Myc leads to downregulation of both specific mRNAs and of specific microRNAs.1 The first identified target of Myc-mediated repression was c-myc itself, arguing for negative feedback regulation of c-myc.2 Further experiments documented that Myc autosuppression acts at the level of the c-myc promoter and that it is disrupted in multiple transformed cells. Although the mechanism of this direct autosuppression remains unresolved, more recent studies also revealed that two micro-RNAs (miR-17-5p and miR-20a), which are induced by Myc, target E2F1.3 As E2F1 in turn can activate the c-myc promoter, these findings strongly support the notion that negative feedback is a major mechanism that maintains low levels of c-myc expression in untransformed cells.
Not surprisingly, multiple genes that are repressed by Myc encode negative regulators of cell proliferation. Historically, c/EBPα, a transcription factor that promotes the differentiation of adipocytes and exerts a very potent cell cycle arrest in culture, was the first target identified among this group of proteins4; the related c/EBPδ protein is also repressed by Myc.5 In addition, several inhibitors of the cell cycle, notably cdkn2b (encoding p15Ink4b), cdkn2c (p18Ink4c), cdkn1a (p21Cip1), cdkn1b (p27Kip1), and cdkn1c (p57Kip2), are targets for repression by Myc.6-12 A number of other genes that are repressed by Myc are also negative regulators of cell proliferation, yet their mechanism of action is not completely clear: notable examples are the ndrg genes (N-Myc downregulated gene), which are growth-suppressive genes that are consistently downregulated upon expression of N-Myc.13
Collectively, these observations suggest that repression of cell cycle inhibitors is a major pathway via which Myc proteins promote cell proliferation both during normal development and during oncogenesis. Several observations support this hypothesis: for example, co-deletion of cdkn2c and cdkn1b partly alleviates the defects in cell proliferation caused by N-Myc deficiency in the cerebellum.14 Similarly, deletion of c-myc strongly attenuates tumor development in a chemical model of skin carcinogenesis and leads to upregulation of cdkn1a; co-deletion of cdkn1a restores rapid tumor development in c-myc −/− epidermis.15 In tissue culture, depletion of cdkn1a alleviates the arrest of several tumor cells in the G1 phase of the cell cycle upon depletion of Myc.16 Similarly, colon carcinoma cells express high levels of c-myc due to mutations in the APC pathway, and c-myc represses both cdkn1a and terminal differentiation of these cells; surprisingly, ectopic expression of cdkn1a restores not only cell cycle arrest but also differentiation in this model.17 It is less certain whether repression of other cell cycle inhibitors contributes to Myc-dependent cellular phenotypes; potentially, this is due to their redundancy. For example, loss of cdkn2b has little phenotype in wild-type animals but strongly enhances tumorigenesis of mice that are deficient in cdkn2a (encoding p16Ink4a and p19Arf), arguing that the functions of these cell cycle regulators in tumor suppression are redundant.18 If so, repression of cdkn2b may be a critical oncogenic function of Myc in tumors that have sustained deletions of the cdkn2a locus.
Another large group of genes that is repressed by Myc and N-Myc encodes proteins involved in cell adhesion.19 Potentially, the most intensely studied example of this class of genes is itgb1, which encodes integrin β1, a subunit of multiple heterodimeric integrin complexes.20-22 Due to binding of multiple ligands such as collagen, fibronectin, or laminin, these complexes mediate cell-cell interactions as well as contact to the extracellular matrix.
Mechanisms of Gene Repression by Myc
In all cases that have been analyzed to date, Myc binds to the core promoter of the genes it represses directly. Although this is often true also for genes that are activated by Myc, there are many examples where Myc activates transcription from distant sites, suggesting that the mechanism of gene repression, but not necessarily activation, by Myc is intimately linked to the events that occur at the transcription start site. Early experiments had suggested a specific interaction of Myc with proteins that recognize the “initiator” element of transcription; however, the relevance of these interactions remains uncertain.23
Myc interacts with two zinc finger transcription factors, Sp1 and Miz1, that bind to core promoters.24,25 Both factors stimulate transcription when bound to DNA in the absence of Myc, and binding of Myc interferes with transcriptional activation by both factors. Both proteins are present at the promoter of many Myc-repressed genes: for Miz1, this has been demonstrated in multiple cases by chromatin immunoprecipitation; for Sp1, the presence of the cognate DNA binding site strongly suggests that the protein is present, although this has often not formally been shown. Miz1 transactivates the promoter of many of the genes that are repressed by Myc and enhances gene expression both in tissue culture and in vivo (Fig. 1A); for example, expression of cdkn2b is attenuated in the skin of mice that express a nonfunctional Miz1 protein.26 A point mutant of Myc, MycV394D, that binds to Max and activates transcription but has lost the ability to bind to Miz1 fails to repress many target genes that are repressed by Myc in tissue culture.10,27 In a transgenic model of lymphomagenesis, MycV394D fails to repress cdkn2b and cdkn1c, demonstrating that the interaction of Myc with Miz1 is critical for repression of both target genes in vivo.28 In contrast, the mutant is not impaired in repression of cdkn1a in the same model; therefore, other interactions, potentially with Sp1, are sufficient for mediating repression of Myc at the cdkn1a promoter.28 Notably, Miz1 mediates repression not only by Myc but also by other repressor proteins (Fig. 1B). For example, the Bcl-6 oncoprotein contains at its amino-terminus, like Miz1, a POZ/BTB domain and uses this domain to heterodimerize with Miz1 and repress cdkn1a expression via Miz1; the same is true for the POZ domain transcription factor Zbtb4.29-31 Apparently, these complexes form independently of Myc. In contrast, the Gfi-1 repressor forms a ternary complex with Myc and Miz1 at the cdkn2b, cdkn1a, and cdkn1b promoter to repress transcription.32,33
Figure 1.
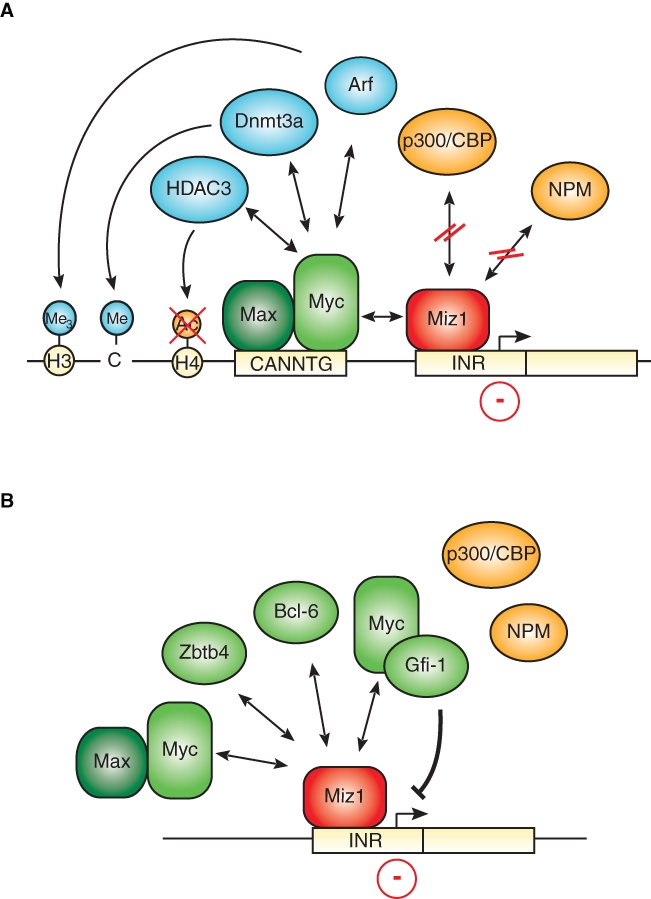
Transcriptional repression mediated by Miz1. (A) Myc blocks the transcription of Miz1-dependent gene expression. Whereas free Miz that is bound to the core promoter in the promoter region of its target genes activates transcription via recruitment of coactivators, including p300 and nucleophosmin, Myc represses Miz1-dependent transcriptional activation through disruption of the interaction between Miz1 and these cofactors. Furthermore, Myc recruits the histone acetylase HDAC3 as well as the DNA methylase Dnmt3a. In addition, formation of a heterotrimeric Myc/Miz1/Arf complex was shown to increase trimethylation of histone H3 at lysine 9, leading to transcriptional repression. Recruitment of Myc itself might involve its binding to noncanonical E-boxes (CANNTG) close to the transcriptional start site in a complex with its partner protein Max. (B) Miz1 might act as a platform for recruitment of several repressor complexes. Besides Myc, Miz1 associates with other repressor proteins such as Bcl-6, Zbtb4, or Gfi-1 in the promoter regions of its target genes, which results in the repression of gene expression.
Whether interactions of Myc with Sp1 or Miz1 are required for recruitment of Myc to repressed promoters or whether these interactions are required for the establishment of a repressed promoter state is an open question. Binding of Myc to the cdkn2b promoter, for example, depends on heterodimerization with Max, arguing that binding of Myc to DNA may contribute to targeting Myc to repressed promoters.34 Indeed, E-box elements are found in the core promoter of repressed genes, and some, like in the c/EBPα promoter, confirm to the consensus sequence CACGTG, suggesting that interactions with Sp1 or Miz1 may not necessarily be critical for recruitment of Myc to sites of repression. Our own unpublished experiments show that altering the nonconsensus sequence CAGCTG in the core of the human cdkn2b promoter to a consensus sequence renders the promoter more sensitive to inhibition by low amounts of Myc but does not alter the direction of regulation by Myc (J.F. Naud and M.E., unpublished observation). If this finding extends to the regulation of endogenous genes, it argues that the sequence of Myc binding sites at the core promoter does not predict the mode of how a gene is regulated by Myc.
Reporter assays show that Myc represses transcription by Miz1 and potentially also by Sp1 by displacing coactivators, including the p300 histone acetyl transferase and nucleophosmin.7,35 A similar mechanism has been proposed for the interference of Myc with transactivation by c/EBPα.36 The knowledge about other effector mechanisms via which Myc may inhibit transcription is limited. Myc-dependent histone deacetylation has been suggested to account for repression of the Id2 and Gadd153 genes because Myc recruits HDAC3 to these genes37 (Fig. 1A). In the presence of the Arf tumor suppressor protein, the Myc/Miz1 complex can induce the formation of heterochromatin on its target sites, which is indicated by the accumulation of H3K9-trimethylated histones; whether formation of heterochromatin is a general mechanism of transcriptional repression by Myc remains to be determined.38 Similarly, the Myc/Miz1 complex has been shown to recruit the DNA methyl transferase Dmnt3a to the cdkn1a promoter, leading to the cytosine methylation of the DNA and silencing of gene expression.39 Strikingly, CpG islands in the cdkn2b promoter are heavily methylated in leukemias, which express very high levels of Myc, suggesting that recruitment of Dmnt3a by Myc/Miz1 may facilitate DNA methylation at that site.40 However, it is less certain whether repression by Myc and Miz1 requires Dmnt3a and DNA methylation under physiological, not pathophysiological, conditions. Currently, therefore, it is not clear whether covalent modifications of either histones or DNA are required for Myc-mediated repression.
Alternatively, transcriptional repression is mechanistically related to the mechanism of transcriptional activation of Myc. Here, a number of analyses of individual genes or groups of genes had demonstrated that Myc has little or no effect on the recruitment of RNA polymerase II to its target promoters but rather promotes polymerase clearance and transcriptional elongation, thereby establishing a model of how Myc cooperates with other transcription factors in gene activation.41-44 These findings have been recently confirmed by a global analysis of Myc function in embryonic stem cells; conversely, therefore, Myc/Miz1 complexes may retain RNA polymerase at the promoter and prevent elongation.45
Pathways of Repression
The localization of the Myc/Miz1 binding sites close to the start site of transcription suggests a model in which the target promoters of this complex are under dual control: one level of control is exerted by the upstream transcription factors that regulate promoter and enhancer activity, a second by the presence of either free Miz1 or of Miz1/Myc complexes at the core promoter. One pathway for which this model holds is the cellular response to the antiproliferative cytokine, Tgfβ. Addition of Tgfβ activates a multitude of genes in keratinocytes, and a subset of these genes is sensitive to Miz1-dependent repression by Myc (Fig. 2A).27 Myc therefore does not generally block the cellular responses to Tgfβ or other antimitogenic stimuli but alters the response profile by blocking induction of a subset of target genes—and also by cooperating with the Tgfβ-regulated Smad proteins during induction of other target genes of Tgfβ such as Snail. As a result, Myc blocks the cytostatic response to Tgfβ but facilitates Tgfβ-induced epithelial-mesenchymal transition.46
Figure 2.
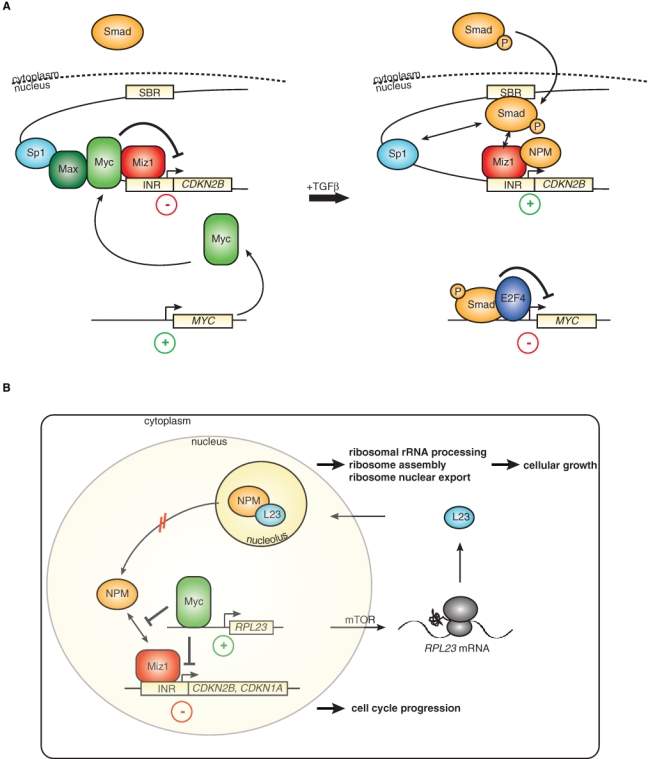
Repression via the Myc/Miz1 complex in different cellular settings. (A) Dual-input model of Tgfβ-mediated cell cycle arrest (modified from Seoane et al.6). In the absence of Tgfβ signaling, Myc represses the expression of cdkn2b in a complex with Miz1. Increased levels of the cytokine Tgfβ lead to the phosphorylation of Smad proteins and their subsequent translocation to the nucleus, where they bind to Smad binding regions (SBR) and cooperate with Miz1 in inducing gene expression. In parallel, activated Smads block the expression of the myc gene in a complex with E2f4. (B) The Myc/Miz1 complex links cell cycle arrest to cell growth via a potential feedback loop, including the ribosomal protein Rpl23. In addition to disrupting the interaction of Miz1 with its coactivator nucleophosmin (NPM), Myc induces the transcription of Rpl23. Nucleophosmin and L23 are mainly localized to the nucleolus, where they promote ribosomal biogenesis and cell growth. Rpl23 negatively regulates the transcriptional activity of Miz1 by retaining NPM in the nucleolus. Therefore, this regulatory circuit provides a potential mechanism for the ability of Myc to coordinate cell cycle progression and cell growth.
According to this model, the cdkn2b promoter could be regulated in two possible manners. First, Tgfβ regulates both the Smad and the Myc/Miz1 “arm” of this circuit to establish a particular robust activation of cdkn2b expression. Alternatively, there might be a separate set of upstream regulators of the Myc/Miz1 complex, such that the cdkn2b promoter integrates two separate inputs. Evidence exists for both models because addition of Tgfβ leads to the formation of an E2f4/Smad complex that represses transcription of the c-myc promoter and thereby relieves repression of cdkn2b by Myc/Miz1 complexes in addition to activating Smad proteins.47 On the other hand, transcriptional activation by Miz1 requires nucleophosmin as a coactivator, and nucleophosmin is largely sequestered in growing cells in the cell nucleolus and therefore not available for transcriptional activation.35
Several stress signals such as DNA damage can release nucleophosmin from the nucleolus, arguing that transactivation by Miz1 is regulated by specific upstream signals. One key example is provided by the ribosomal protein Rpl23 that retains nucleophosmin in the nucleolus of unstressed cells, and levels of Rpl23 therefore indirectly affect transcriptional activation by Miz1 (Fig. 2B).35 rpl23 is a direct target gene that is transactivated by Myc, and translation of the rpl23 mRNA is under the control of the TOR pathway. This model suggests that Miz1 may link cell cycle progression to cell growth because Miz1-dependent cell cycle arrest is regulated by factors that promote ribosome biogenesis and cell growth. Strikingly, this is a specific function of Rpl23 because other ribosomal proteins cannot substitute for Rpl23 in regulating Miz1-dependent transactivation; this suggests that mechanisms exist that measure the levels of Rpl23 in a growing cell.
Cell Adhesion
Repression of genes encoding proteins involved in cell adhesion has been observed in a multitude of cells types, ranging from neuroblastoma cells to hematopoietic stem cells, arguing that it is a central function of Myc.21,22,48 For example, Myc represses itgb1 and other integrins in murine keratinocytes and therefore impairs adhesion and spreading; many of these genes are regulated by the Myc/Miz1 complex.22,27 The repression of itgb1 and other cell adhesion genes by ectopic Myc in murine skin also reduces the adhesive interactions of stem cells with their niche (Fig. 3). As a consequence, expression of Myc in stem cells results in the entry into the transit-amplifying compartment and subsequently in premature differentiation in culture, which are reflected by defects in wound healing in vivo.20,22 Similarly, Myc regulates adhesion of hematopoietic stem cells with the niche via multiple integrin receptors. This regulation may be part of the physiological processes that link exit from the stem cell niche to the enhanced proliferation of transit-amplifying cells.49 However, repression of integrins may also provide a critical failsafe mechanism that eliminates stem cells, which have acquired oncogenic Myc levels. One key argument to indicate that this may be the case is the observation that the tumor suppressor protein p19Arf, which is induced by supraphysiological levels of Myc, not only blocks transactivation by Myc but also promotes the assembly of the Myc/Miz1 complex and enhances the Myc-mediated repression of adhesion genes.38,50 This is consistent with findings that p19Arf acts as a p53-independent tumor suppressor in skin, which has been demonstrated in a Ras-dependent tumor model.51
Figure 3.
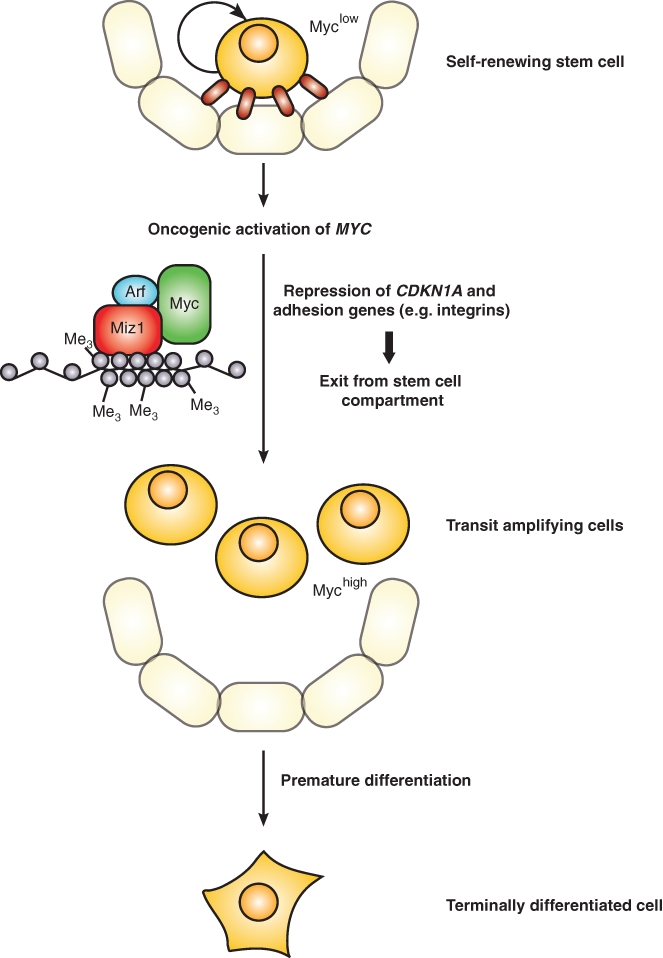
Regulation of cell adhesion by Myc as a potential tumor-protective process. Whereas stem cells are often in a quiescent, nonproliferating state, myc expression results in the proliferation of these cells. Sustained or dramatically increased levels of Myc due to oncogenic activation lead to the repression of cdkn1a and cell adhesion genes. The loss of adhesion promotes the exit of these proliferating stem cells from the niche and subsequent entry in the transit-amplifying compartment, thereby initiating terminal differentiation. As the tumor suppressor Arf and Myc jointly regulate adhesion genes, this mechanism might serve to prevent uncontrolled cell proliferation in a p53-independent manner.
Outlook
The current state of the field raises a number of central questions: for example, we do not know how many sites in the genome are jointly regulated by Myc and Miz1 or Sp1, and therefore the repressive Myc complexes potentially have a much broader spectrum of biological functions than those reported here. Potentially most important is how transcriptional repression by endogenous Myc protein contributes to normal development and to the many oncogenic functions of Myc that are discussed in this issue.
Acknowledgments
We thank our colleagues in the laboratory and in other laboratories for many stimulating discussions.
Footnotes
The work in the author’s laboratory has been supported by grants from the Deutsche Forschungsgemeinschaft and from the European Union.
The authors declare no conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1. Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Penn LJZ, Brooks MW, Laufer EM, Land H. Negative autoregulation of c-myc transcription. EMBO J. 1990;9:113-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839-43 [DOI] [PubMed] [Google Scholar]
- 4. Yang B-S, Gilbert JD, Freytag SO. Overexpression of Myc suppresses CCAAT transcription factor/nuclear factor 1-dependent promoters in vivo. Mol Cell Biol. 1993;13:3093-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Si J, Yu X, Zhang Y, DeWille JW. Myc interacts with Max and Miz1 to repress C/EBPdelta promoter activity and gene expression. Mol Cancer. 2010;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400-8 [DOI] [PubMed] [Google Scholar]
- 7. Staller P, Peukert K, Kiermaier A, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3:392-9 [DOI] [PubMed] [Google Scholar]
- 8. Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699-712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729-34 [DOI] [PubMed] [Google Scholar]
- 10. Herold S, Wanzel M, Beuger V, et al. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell. 2002;10:509-21 [DOI] [PubMed] [Google Scholar]
- 11. Yang W, Shen J, Wu M, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20:1688-702 [DOI] [PubMed] [Google Scholar]
- 12. Adhikary S, Peukert K, Karsunky H, et al. Miz1 is required for early embryonic development during gastrulation. Mol Cell Biol. 2003;23:7648-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimono A, Okuda T, Kondoh H. N-myc-dependent repression of ndr1, a gene identified by direct subtraction of whole mouse embryo cDNAs between wild type and N-myc mutant. Mech Dev. 1999;83:39-52 [DOI] [PubMed] [Google Scholar]
- 14. Zindy F, Knoepfler PS, Xie S, Sherr CJ, Eisenman RN, Roussel MF. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proc Natl Acad Sci U S A. 2006;103:11579-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oskarsson T, Essers MA, Dubois N, et al. Skin epidermis lacking the c-Myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21Cip1 gene. Genes Dev. 2006;20:2024-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H, Mannava S, Grachtchouk V, et al. c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene. 2008;27:1905-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van de Wetering M, Sancho E, Verweij C, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241-50 [DOI] [PubMed] [Google Scholar]
- 18. Krimpenfort P, Ijpenberg A, Song JY, et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448:943-6 [DOI] [PubMed] [Google Scholar]
- 19. Inghirami G, Grignani F, Sternas L, Lombardi L, Knowles DM, Dalla Favera R. Down-regulation of LFA-1 adhesion receptors by C-myc oncogene in human B lymphoblastoid cells. Science. 1990;250:682-6 [DOI] [PubMed] [Google Scholar]
- 20. Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet. 2001;28:165-8 [DOI] [PubMed] [Google Scholar]
- 21. Judware R, Culp LA. Over-expression of transfected N-myc oncogene in human SKNSH neuroblastoma cells down-regulates expression of beta 1 integrin subunit. Oncogene. 1995;11:2599-607 [PubMed] [Google Scholar]
- 22. Gandarillas A, Watt FM. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997;11:2869-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roy AL, Carruthers C, Gutjahr T, Roeder RG. Direct role for Myc in transcription initiation mediated by interactions with TFII-I. Nature. 1993;365:359-61 [DOI] [PubMed] [Google Scholar]
- 24. Gartel AL, Ye X, Goufman E, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A. 2001;98:4510-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M. An alternative pathway for gene regulation by Myc. EMBO J. 1997;16:5672-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gebhardt A, Kosan C, Herkert B, et al. Miz1 is required for hair follicle structure and hair morphogenesis. J Cell Sci. 2007;120:2586-93 [DOI] [PubMed] [Google Scholar]
- 27. Gebhardt A, Frye M, Herold S, et al. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J Cell Biol. 2006;172:139-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Riggelen J, Muller J, Otto T, et al. The interaction between Myc and Miz1 is required to antagonize TGFbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 2010;24:1281-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054-60 [DOI] [PubMed] [Google Scholar]
- 30. Saito M, Novak U, Piovan E, et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:11294-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weber A, Marquardt J, Elzi D, et al. Zbtb4 represses transcription of P21CIP1 and controls the cellular response to p53 activation. EMBO J. 2008;27:1563-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Q, Basu S, Qiu Y, Tang F, Dong F. A role of Miz-1 in Gfi-1-mediated transcriptional repression of CDKN1A. Oncogene. 2010;29:2843-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Basu S, Liu Q, Qiu Y, Dong F. Gfi-1 represses CDKN2B encoding p15INK4B through interaction with Miz-1. Proc Natl Acad Sci U S A. 2009;106:1433-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mao DY, Watson JD, Yan PS, et al. Analysis of Myc bound loci identified by CpG island arrays shows that max is essential for Myc-dependent repression. Curr Biol. 2003;13:882-6 [DOI] [PubMed] [Google Scholar]
- 35. Wanzel M, Russ AC, Kleine-Kohlbrecher D, Colombo E, Pelicci PG, Eilers M. A ribosomal protein L23-nucleophosmin circuit coordinates Mizl function with cell growth. Nat Cell Biol. 2008;10:1051-61 [DOI] [PubMed] [Google Scholar]
- 36. Steinmann S, Schulte K, Beck K, Chachra S, Bujnicki T, Klempnauer KH. v-Myc inhibits C/EBPbeta activity by preventing C/EBPbeta-induced phosphorylation of the co-activator p300. Oncogene. 2009;28:2446-55 [DOI] [PubMed] [Google Scholar]
- 37. Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008;68:3624-9 [DOI] [PubMed] [Google Scholar]
- 38. Herkert B, Dwertmann A, Herold S, et al. The Arf tumor suppressor protein inhibits Miz1 to suppress cell adhesion and induce apoptosis. J Cell Biol. 2010;188:905-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brenner C, Deplus R, Didelot C, et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005;24:336-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quesnel B, Guillerm G, Vereecque R, et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood. 1998;91:2985-90 [PubMed] [Google Scholar]
- 41. Eberhardy SR, Farnham PJ. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem. 2001;276:48562-71 [DOI] [PubMed] [Google Scholar]
- 42. Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem. 2002;277:40156-62 [DOI] [PubMed] [Google Scholar]
- 43. Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004;23:2830-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martinato F, Cesaroni M, Amati B, Guccione E. Analysis of Myc-induced histone modifications on target chromatin. PLoS One. 2008;3:e3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith AP, Verrecchia A, Faga G, et al. A positive role for Myc in TGFbeta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene. 2009;28:422-30 [DOI] [PubMed] [Google Scholar]
- 47. Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19-32 [DOI] [PubMed] [Google Scholar]
- 48. Wilson A, Murphy MJ, Oskarsson T, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development. 2003;130:2793-808 [DOI] [PubMed] [Google Scholar]
- 50. Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431:712-7 [DOI] [PubMed] [Google Scholar]
- 51. Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS Biol. 2004;2:E242. [DOI] [PMC free article] [PubMed] [Google Scholar]