

Abstract
Over the past few years, high-throughput analyses have provided important novel insights into molecular pathways that play a crucial role in the progression from early to advanced melanoma, and at the same time, they have led to the identification of genes that as part of melanoma progression are upregulated in advanced melanoma. In the present study, we provide evidence that Aurora kinases A and B, 2 key regulators of M phase progression, are upregulated to high levels with progression from melanoma in situ to primary and metastatic melanoma and that inhibiting the expression of these 2 genes by RNA interference or blocking their function with an Aurora kinase–specific small-molecule inhibitor severely impairs melanoma cell proliferation and cell cycle progression and induces melanoma cell apoptosis. In addition, we present the results of systemic treatment of human melanoma xenografts with an Aurora kinase small-molecule inhibitor as well as Aurora kinase targeting vectors.
Keywords: melanoma, Aurora kinases, molecular targeting, xenograft studies
Introduction
Aneuploidy due to chromosome instability is one of the hallmarks of cancer, and advanced melanoma ranks high on the list of solid malignancies that exhibit a large number of changes in chromosome number and structure.1 One of the causes, underlying genome instability, is molecular errors that occur during mitosis. However, regarding melanoma, little information is yet available concerning molecular processes and individual genes/proteins that may be dysregulated as primary and metastatic melanomas progress through M phase.
In the context of a previously conducted whole-genome microarray analysis of tissue samples ranging from normal human skin to melanoma-infiltrated lymph nodes,2 we obtained a first indication that changes in M phase regulation are associated with progression from melanoma in situ to primary melanoma in the vertical growth phase (VGP melanoma) and melanoma in the metastatic growth phase (MGP melanoma). Specifically, these data revealed that 2 distinct molecular profiles govern melanoma progression—one that is specific for and encompasses normal skin, benign and atypical nevi, and melanoma in situ, and the other that includes VGP and MGP melanomas and melanoma-infiltrated lymph nodes. Furthermore, and equally important, we found that this switch from one genetic profile to the other occurs precisely with the transition from melanoma in situ to VGP melanoma and that the leading gene ontology (GO) group, most prominently associated with this switch, is the mitotic cell cycle. Subsequently, another whole-genome expression profiling study of laser microdissected primary and metastatic melanoma tissues3 showed that genes associated with the GO terms cell cycle, mitotic cell cycle, M phase of mitotic cell cycle, mitosis, and chromosome condensation were significantly enriched on the list of genes that were upregulated in melanoma metastases.
Mechanisms that segregate chromosomes in mitosis and divide the cell in cytokinesis—the process that leads to 2 identical daughter cells—have been an important research topic for more than 2 decades. However, the scientific aspect that over the past several years has drawn particular attention to the complex network of molecular events that controls and assures accuracy of spindle formation, chromosome segregation, and cytokinesis is the finding that the Aurora kinases A and B are upregulated to high levels in the advanced stages of a significant number of solid and hematological malignancies.4-8
Because, to date, little is known regarding molecular events associated with cell cycle progression that may be dysregulated in advanced melanoma, we pursued the study summarized herein. Specifically, we provide data, which demonstrate that in VGP and MGP melanomas but not in benign or atypical nevi, or melanomas in situ, Aurora kinases A and B are expressed at high levels and that inhibiting the expression and likewise the function of these 2 Aurora kinases severely interferes with melanoma cell proliferation and the cells’ progression through G2/M and that it causes melanoma cells to undergo apoptosis. In addition, we present the results of a preclinical study that focused upon systemic treatment of human melanoma xenografts with an Aurora kinase small-molecule inhibitor, which when administered alone and even more effectively when given in combination with the chemotherapeutic agent paclitaxel impaired the growth of these tumors.
Results
Status of Aurora kinase A and Aurora kinase B expression in nevus and melanoma tissues and melanoma cell lines
Probe sets from a whole-genome microarray analysis, which we previously performed,2 of cryopreserved normal skin, benign nevi, atypical nevi, which are the precursors and risk markers of melanoma, and melanomas in situ, which although noninvasive, are the first stage of melanoma development, VGP and MGP melanomas, and melanoma-infiltrated lymph nodes, provided a first indication that the Aurora kinases A and B are upregulated with progression from early to advanced melanoma (Fig. 1). This observation prompted us to probe 1) cryopreserved tissue specimens, ranging from normal skin all the way to melanoma-infiltrated lymph nodes; 2) a nevus > melanoma progression tissue microarray (TMA), comprised of more than 180 tissue cores; and 3) tissue sections from randomly selected formalin-fixed, paraffin-embedded (FFPE) melanoma specimens with an antibody to Aurora kinase A and, likewise, an antibody to Aurora kinase B. With the exception of some epidermal keratinocytes and/or dermal fibroblasts in normal skin, benign and atypical nevi, and melanoma in situ that stained positive for Aurora kinase B, the cryopreserved tissues exhibited little expression of Aurora kinase B (Fig. 2A) or Aurora kinase A (data not shown). In contrast, Aurora kinase B and likewise Aurora kinase A (data not shown) were strongly expressed in cryopreserved tissue samples representing VGP and MGP melanomas and melanoma-infiltrated lymph nodes (LN) (Fig. 2A). Scored on a signal-intensity scale of 0 > 3, the nevus > melanoma progression TMA analysis yielded very similar results (data not shown). In addition, the TMA data revealed that the number of VGP, MGP, and LN melanoma tissue cores that demonstrated expression of Aurora kinase B was 5-fold higher than the number of Aurora kinase A–positive melanoma tissue cores (data not shown). Depicted in Figure 2B are examples of an MGP melanoma TMA core and 2 adjacent tissue sections of a randomly selected FFPE MGP melanoma specimen, probed with Aurora kinase A, and likewise Aurora kinase B antibody.
Figure 1.
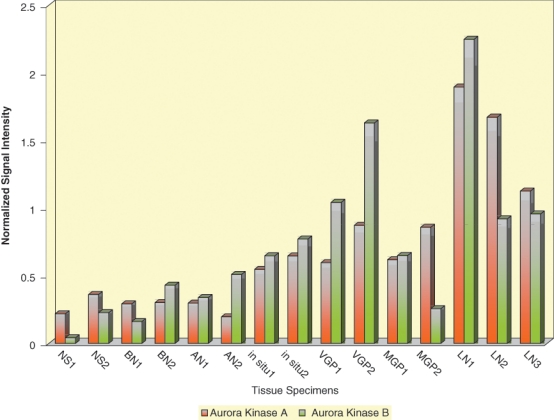
Expression of Aurora kinases A and B in normal skin and nevus and melanoma tissue specimens subjected to whole-genome microarray analysis. Levels of Aurora kinase A (orange-colored bars) and Aurora kinase B (green-colored bars) expression in cryopreserved tissue samples representing normal human skin (NS1, NS2), benign nevi (BN1, BN2), atypical nevi (AN1, AN2), melanomas in situ (in situ1, in situ2), VGP melanomas (VGP1, VGP2), MGP melanomas (MGP1, MGP2), and melanoma-infiltrated lymph nodes (LN1, LN2, LN3).
Figure 2.
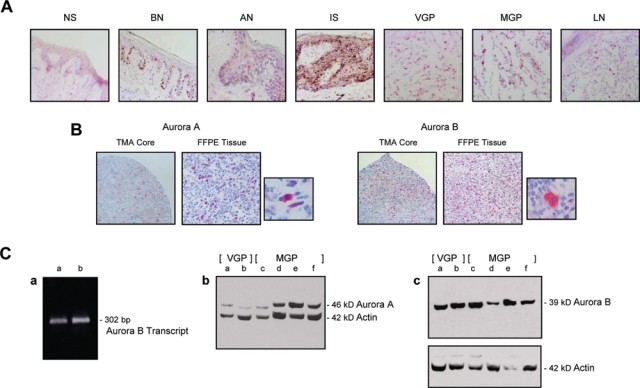
Aurora kinase A and Aurora kinase B expression in cryopreserved and archival nevus and melanoma tissues and VGP and MGP melanoma cell lines. (A) Cryopreserved tissue sections, prepared from normal human skin (NS), a benign (BN) and an atypical nevus (AN), a melanoma in situ (IS), a VGP melanoma (VGP), an MGP melanoma (MGP), and a melanoma-infiltrated lymph node (LN), were probed with an antibody to Aurora kinase B. (B) Images of 2 representative tissue cores of a melanoma tissue microarray (TMA), probed with an antibody to Aurora kinase A and likewise an antibody to Aurora kinase B. Depicted in addition are 2 adjacent tissue sections, prepared from an FFPE MGP melanoma, that were stained by standard immunohistochemistry with an antibody to Aurora kinase A and likewise an antibody to Aurora kinase B. Next to each of the 2 tissue sections is an image, captured at higher magnification, which shows individual cells in the Aurora kinase antibody-probed FFPE MGP melanoma tissue. All tissue sections depicted (A and B) were counterstained with hematoxylin. (C) (a) RT-PCR analysis of Aurora kinase B mRNA expression in WM1158 (lane a) and WM983-B (lane b) MGP melanoma cell lines. (b) Immunoblot analysis of Aurora kinase A and (c) Aurora kinase B protein expression in VGP melanoma cell lines WM983-A (lane a) and WM98-2 (lane b) and in MGP melanoma cell lines WM373 (lane c), WM852 (lane d), WM983-B (lane e), and WM1158 (lane f). The immunoblots were probed with an antibody to β-actin serving as loading control.
In addition to these tissues, we also analyzed VGP and MGP melanoma cell lines for the status of Aurora kinase A and Aurora kinase B expression. RT-PCR analysis of 2 MGP melanoma cell lines (WM1158 and WM983-B) with a human Aurora kinase B–specific set of primers led to the amplification of a single 302-bp Aurora kinase B transcript (Fig. 2C, panel a), and immunoblot analysis of 2 VGP (WM983-A and WM98-2) and 4 MGP melanoma cell lines (WM373, WM852, WM983-B, and WM1158) demonstrated the presence of Aurora kinase A (Fig. 2C, panel b) and Aurora kinase B protein (Fig. 2C, panel c) in every one of these cell lines.
Downregulating the expression of Aurora kinase A or B leads to inhibition of melanoma cell proliferation
Using a pool, comprised of 4 Aurora kinase A and likewise 4 Aurora kinase B–specific siRNAs, we transfected WM1158 MGP melanoma cells, which as determined by immunoblot analysis led to downregulation of Aurora kinase A (Fig. 3A, panel a) and, similarly, Aurora kinase B expression (Fig. 3A, panel b) at 24, 48, and 72 hours following transfection. In addition, phosphorylation of the Aurora kinase B substrate, Ser10 on histone 3 (pHisH3), was reduced starting at 48 hours following transfection with the Aurora kinase B–specific siRNAs (Fig. 3A, panel c). Furthermore, starting at 48 hours, and becoming more apparent thereafter, the proliferation of the Aurora kinase A and similarly, albeit less pronounced, the Aurora kinase B siRNA-transfected WM1158 MGP melanoma cells was inhibited compared with the proliferation of WM1158 cells that, serving as controls, had received only the siRNA delivery vehicle, Lipofectamine, or were transfected with a pool of 4 nontargeting siRNAs (Fig. 3C and 3B).
Figure 3.
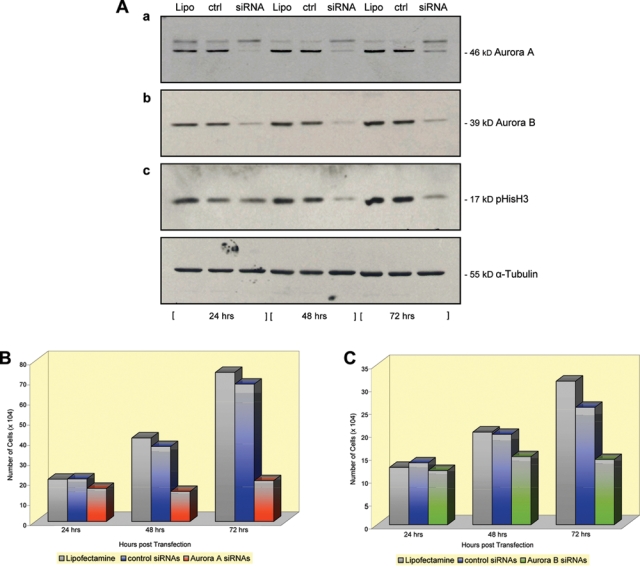
Immunoblot and cell proliferation analysis of Aurora kinase A and Aurora kinase B siRNA-transfected MGP melanoma cells. (A) Total cell lysates of WM1158 MGP melanoma cells, transfected with (a) 150 nM Aurora kinase A siRNAs (siRNA) or (b and c) 150 nM Aurora kinase B siRNAs (siRNA), were probed with antibody to (a) Aurora kinase A, (b) Aurora kinase B, or (c) pHisH3 at 24, 48, and 72 hours following transfection. WM1158 MGP melanoma cells transfected with only Lipofectamine (a-c; Lipo) or 150 nM control siRNAs (a-c; crtl) served as controls. The immunoblots were probed with an antibody to α-tubulin for loading control. (B and C) Proliferation of WM1158 MGP melanoma cells at 24, 48, and 72 hours following their transfection with (B) 150 nM of Aurora kinase A or (C) 150 nM of Aurora kinase B siRNAs. Controls were WM1158 MGP melanomas that received only Lipofectamine or were transfected with control siRNAs.
Treatment of melanoma cells with an Aurora kinase small-molecule inhibitor leads to overt changes in melanoma cell morphology and cell division
To determine whether in addition to inhibiting expression of the Aurora kinases A and B, blocking the function of these 2 molecules would interfere with the biological features of advanced melanoma, we obtained the Aurora kinase small-molecule inhibitor, PF-03814735, whose IC50 value (nmol/L) for Aurora kinase A is 5 ± 3 and for Aurora kinase B is 0.8 ± 0.6.9 Using as a first step the concentrations of 1 nM and 10 nM as well as 0.1 µM, 1 µM, and 10 µM, we found that starting at 1 µM and becoming most pronounced at 10 µM, VGP (WM983-A) and several MGP melanoma cells, including the WM1158 MGP melanoma cells (Fig. 4A, panel c), rapidly severed their cell-cell contacts, in some cases formed long dendrites, a process indicative of onset of terminal differentiation, and starting at about 72 hours following addition of the Aurora kinase small-molecule inhibitor, massively dislodged into the growth medium. Furthermore, cells that had detached from the surface of the Petri dish and dislodged into the growth medium did not reattach to a tissue culture dish after they had been rinsed several times with complete growth medium not containing the inhibitor.
Figure 4.
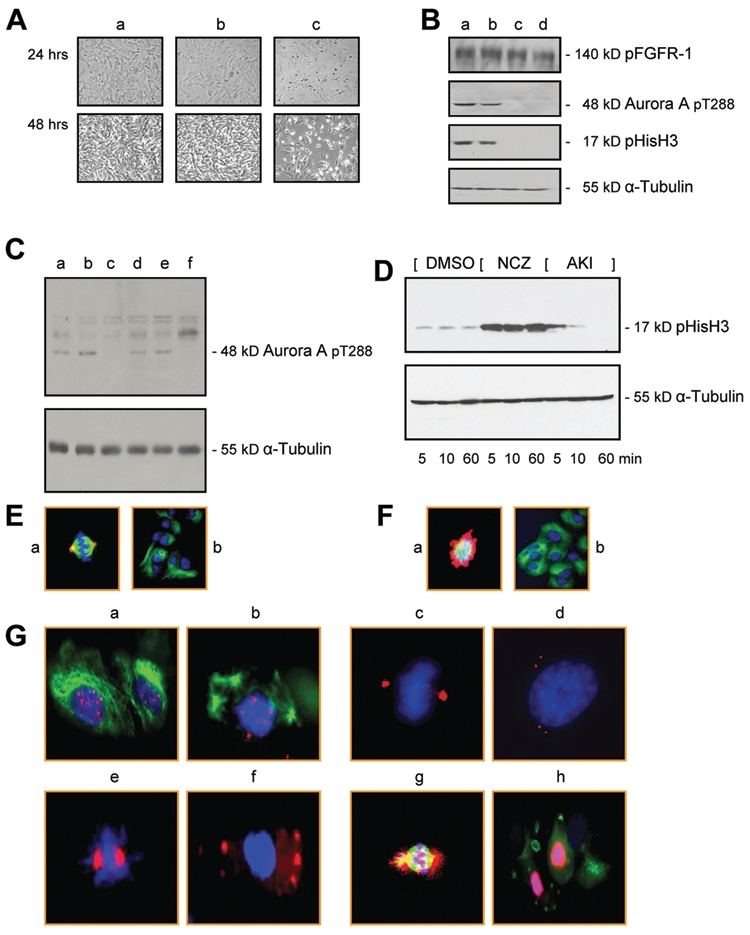
Aurora kinase inhibitor treatment of MGP melanoma cells. (A) Morphology of MGP melanoma cells (WM1158) not treated (panel a), that received only DMSO (panel b), or were treated with 10 µM of Aurora kinase inhibitor (panel c) for 24 or 48 hours. (B) Immunoblot analysis of WM1158 MGP melanoma cells, treated for 1 hour with Aurora kinase inhibitor, PF-03814735, at a dose of 10 nM (lane a), 100 nM (lane b), 1 µM (lane c), or 10 µM (lane d), that were probed with antibody to pFGFR-1, Aurora kinase A pT288, or pHisH3, and α-tubulin for loading control. (C) Immunoblot analysis of WM1158 MGP melanoma cells, treated with 10 µM of Aurora kinase inhibitor for 24 hours (lane c) or 48 hours (lane f) and probed with antibody to Aurora A pT288 or α-tubulin for loading control. WM1158 MGP melanoma cells not treated (lanes a and d) or treated with only DMSO (lanes b and e) served as controls. (D) Immunoblot analysis of WM1158 MGP melanoma cells incubated in the presence of 50 ng/mL of nocodazole (NCZ) for 20 hours, followed by addition of 10 µM of Aurora kinase inhibitor (AKI) for 5, 10, or 60 minutes (lane i), that were probed with an antibody to pHisH3. WM1158 MGP melanoma cells that received only DMSO (DMSO) for 5, 10, or 60 minutes or only 50 ng/mL of nocodazole for 5, 10, or 60 minutes served as controls. (E) Immunofluorescence analysis of WM1158 MGP melanoma cells not treated (panel a) or treated (panel b) with 10 µM of Aurora kinase inhibitor for 2 hours that were stained with an antibody to Aurora kinase A pT288 (pseudocolored red) and α-tubulin (pseudocolored green) and counterstained with fluorescent DAPI (pseudocolored blue). (F) Immunofluorescence analysis of WM1158 MGP melanoma cells incubated in the presence of nocodazole (20 ng/mL) for 20 hours (panel a) or incubated in the presence of nocodazole (20 ng/mL) for 20 hours, followed by treatment with the Aurora kinase inhibitor (10 µM) for 2 hours (panel b), that were stained with antibody to pHisH3 (pseudocolored red) and α-tubulin (pseudocolored green) and counterstained with fluorescent DAPI (pseudocolored blue). (G) Immunofluorescence analysis of WM1158 MGP melanoma cells incubated in the presence of 20 ng/mL of nocodazole for 20 hours (panels a, c, e, g) or incubated in the presence of 20 ng/mL of nocodazole for 20 hours, followed by treatment with 10 µM of Aurora kinase inhibitor for 2 hours (panels b, d, f, h), that were stained with antibody to CREST (pseudocolored red, panels a and b) and α-tubulin (pseudocolored green, panels a and b), y-tubulin (pseudocolored red, panels c and d), Aurora kinase A (pseudocolored red, panels e and f), and Aurora kinase B (pseudocolored red, panels g and h) and α-tubulin (pseudocolored green, panels g and h). Nuclei were counterstained with fluorescent DAPI (pseudocolored blue).
To determine to which extent this small-molecule inhibitor when added to melanoma cells blocked primarily the function of the 2 Aurora kinases, we pursued a series of immunoblot and optical imaging studies. Like in the case of almost all small-molecule inhibitors, PF-03814735 has been reported to inhibit, in addition to Aurora kinase A and B, other molecules including Flt1, FAK, TrkA, Met, and FGFR-1, albeit with significantly lower affinity.9 However, we did not obtain experimental evidence that, for example, FGFR-1, which correlating with melanocytic progression is upregulated to high levels in advanced melanoma,10,11 was not or no longer phosphorylated in melanoma cells treated with the PF-03814735 inhibitor (Fig. 4B). In contrast, treatment of melanoma cells for 1 hour with 1 µM or 10 µM of the inhibitor revealed that the kinase activity of Aurora kinase A and phosphorylation of Ser10 on histone 3 were impaired (Fig. 4B, lanes c and d). Similarly, Aurora kinase A was no longer phosphorylated when the cells were treated with 10 µM of the inhibitor for 24 hours (Fig. 4C, lane c) or 48 hours (Fig. 4C, lane f). In addition, immunoblot analysis of WM1158 MGP melanoma cells incubated in the presence of nocodazole for 20 hours, followed by addition of 10 µM of the Aurora kinase small-molecule compound for 5, 10, or 60 minutes, demonstrated that Ser10 on histone H3 was no longer phosphorylated at 60 minutes posttreatment (Fig. 4D, lane i).
Immunofluorescence imaging of WM1158 MGP melanoma cells that had been treated with the Aurora kinase inhibitor for 2 hours and then were probed with an antibody to Aurora kinase A pT288 as well as an antibody to α-tubulin (Fig. 4E, panel b), or that had been incubated in the presence of nocodazole and thereafter were treated for 2 hours with the inhibitor and then stained with an antibody to pHisH3 as well as an α-tubulin antibody (Fig. 4F, panel b), revealed substantial perturbation of the microfilament structure when compared to cells that were not treated with the inhibitor (Fig. 4E and 4F, panel a). Furthermore, immunofluorescence imaging of nocodazole-treated WM1158 MGP melanoma cells that were treated for 2 hours with the Aurora kinase inhibitor and then probed with antibody to CREST to mark kinetochores, Aurora kinase A, Aurora kinase B, as well as α- or y-tubulin demonstrated disruption of the spindle checkpoint (Fig. 4G, panels b, d, f, h) compared to WM1158 MGP melanoma cells that had not been treated with the small-molecule agent (Fig. 4G, panels a, c, e, g).
Blocking the function of Aurora kinase A and B inhibits melanoma cell proliferation and causes melanoma cell cycle dysregulation and apoptosis
To determine whether, as in the case of downregulating the expression of the Aurora kinases by way of RNA interference, interfering with their functions would lead to inhibition of melanoma cell proliferation, we treated MGP melanoma cells with the Aurora kinase inhibitor for up to 5 days. As shown in Figure 5A, starting as early as 24 hours posttreatment, the proliferation of the melanoma cells was markedly inhibited and to a significantly greater extent than in the prior experimental setting where we had suppressed via siRNAs and the expression of Aurora kinase A and likewise of Aurora kinase B (Fig. 3B and 3C).
Figure 5.
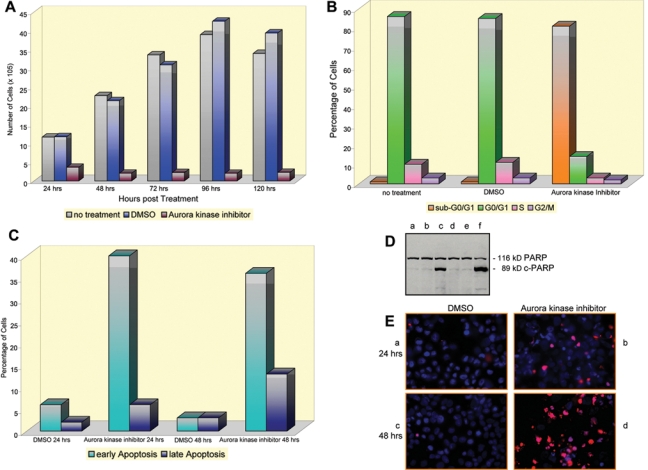
Inhibiting the function of Aurora kinases A and B leads to inhibition of melanoma cell proliferation, dysregulation of the melanoma cell cycle, and melanoma cell apoptosis. (A) Proliferation of WM1158 MGP melanoma cells at various time points following treatment with 10 µM of Aurora kinase inhibitor, PF-03814735. Controls were WM1158 MGP melanomas that were not treated or received only DMSO. (B) Following treatment with 10 µM of Aurora kinase inhibitor for 72 hours, WM1158 MGP melanoma cells were labeled with propidium iodide and subjected to flow cytometry. WM1158 MGP melanoma cells that were not treated or received only DMSO served as controls. (C) At 24 and 48 hours following treatment with 10 µM of the Aurora kinase inhibitor, WM1158 MGP melanoma cells were labeled with annexin V/propidium iodide and analyzed by flow cytometry. WM1158 MGP melanoma cells that had received only DMSO served as controls. (D) Immunoblot analysis of WM1158 MGP melanoma cells, treated with Aurora kinase inhibitor (10 µM) for 24 hours (lane c) or 48 hours (lane f) and probed with an antibody to c-PARP. (E) Immunofluorescence analysis of WM1158 MGP melanoma cells, treated with 10 µM of Aurora kinase inhibitor (panels b and d) or incubated in the presence of DMSO (panels a and c) for 24 hours (panels a and b) or 48 hours (panels c and d), that were analyzed by TUNEL staining. WM1158 MGP melanoma cells that had undergone apoptosis are pseudocolored red, and fluorescent DAPI-counterstained nuclei are pseudocolored blue.
To analyze whether, alongside with blocking the proliferation of melanoma cells, treatment with the Aurora kinase inhibitor also interfered with the cells’ progression through the cell cycle, we pursued experiments that involved propidium iodide as well as annexin V/propidium iodide–based flow cytometry. WM1158 MGP melanoma cells that were treated for 72 hours with 10 µM of the Aurora kinase inhibitor and then fixed and labeled with propidium iodide revealed a major accumulation of the cells in sub-G0/G1 (Fig. 5B), and flow cytometric analysis of annexin V/propidium iodide–labeled melanoma cells that had been treated for 24 or 48 hours with the small-molecule inhibitor documented that substantially more cells were arrested in the early rather than in the late stage of apoptosis (Fig. 5C). Additional experimental evidence, which documented that Aurora kinase inhibitor–treated melanoma cells underwent massive apoptosis, came from an immunoblot analysis, which demonstrated cleavage of PARP to cPARP within 24 hours following addition of the inhibitor to the cells (Fig. 5D), and from fluorescent imaging analysis of TUNEL-stained cells (Fig. 5E).
In vivo and ex vivo analysis of human melanoma xenografts of nude mice treated with Aurora kinase inhibitor
In light of the extreme resistance of advanced melanoma to standard regimens of treatment, and the fact that, to date, only limited information is available regarding genes that might constitute useful targets for molecular therapy of advanced melanoma, we next undertook a series of preclinical studies to determine whether molecular targeting of Aurora kinase A and/or Aurora kinase B would be efficacious for human MGP melanoma cells grown as subcutaneous tumors in nude mice.
The first set of these in vivo studies involved systemic treatment of nude mice, bearing WM983-B MGP human melanoma xenografts, with the Aurora kinase inhibitor PF-03814735 administered twice a week intraperitoneally (i.p.) at a dose of 30 mg/kg for a total period for 24 days (Fig. 6A). Until about the fifth i.p. injection of the inhibitor on day 14, the tumors did not substantially increase in volume. However, following day 14, it became apparent that the MGP melanoma xenografts in mice that continued to receive systemic treatment with the Aurora kinase inhibitor for another 10 days did grow at a slower rate compared to WM983-B MGP melanoma xenograft-bearing nude mice that were not given injections or that received only the Aurora kinase inhibitor delivery vehicle, dimethyl sulfoxide (DMSO) (Fig. 6A). Unlike some other currently available Aurora kinase small-molecule agents, PF-03814735 can be given orally. Thus, we also pursued WM983-B human melanoma xenograft studies that for a period of 24 days involved twice-weekly delivery of the Aurora kinase inhibitor (30 mg/kg) by oral gavage. As a third route of delivery, WM983-B human melanoma xenografts received twice-weekly intratumoral injections of the inhibitor at a dose of 2.5 mg/kg or at a 4-fold higher dose of 12 mg/kg. Both of these latter routes of treatment led to similar tumor-growth impairment (data not shown) as we observed in the case of the systemic i.p. route of delivery. Since extensive in vitro and in vivo pharmacokinetic (PK) and pharmacodynamic (PD) studies involving PF-03814735 were previously performed and recently published,9 we did not make PK and PD analyses a particular focus in the setting of this melanoma study. Furthermore, since it had been determined that when the small-molecule inhibitor was administered at a dose of 60 mg/kg, animals exhibited weight loss of >20%,9 we did not explore the impact of treating human melanoma xenograft-bearing mice with doses of PF-03814735 higher than the ones we administered, which were well tolerated by the animals.
Figure 6.
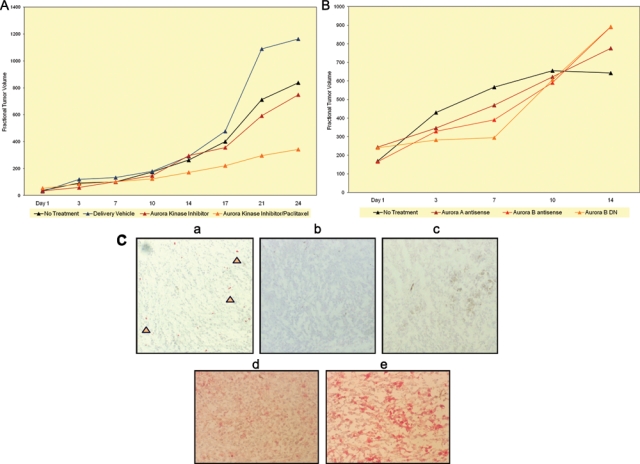
Aurora kinase small-molecule inhibitor treatment of human melanoma xenograft-bearing nude mice. (A) Nude mice, bearing subcutaneous WM983-B MGP melanoma xenografts, received the first i.p. injection of Aurora kinase inhibitor (30 mg/kg) or Aurora kinase inhibitor followed 24 hours later by i.p. injection of paclitaxel (10 mg/kg) on the day (day 1) a tumor(s) had reached 5 mm in any direction. Subsequent i.p. injections were given on day 3, 7, 10, 14, 17, 21, and 24. Controls depicted show the fractional tumor volume of mice that were not injected or received only the Aurora kinase inhibitor delivery vehicle, DMSO. Tumor volumes for each of the 2 experimental arms are the mean of 3 animals each and for each of the control arms of 2 animals each. (B) WM983-B MGP melanoma xenografts received the first intratumoral injection of Aurora kinase A or Aurora kinase B antisense vector, or Aurora kinase B dead-kinase (DN) vector, each mixed with DC-Chol liposomes, on the day (day 1) a tumor(s) had reached 5 mm in any direction. Subsequent intratumoral injections were given on day 3, 7, 10, and 14. Tumors that did not receive intratumoral injections served as controls. (C) pHisH3 immunohistochemical analysis of tissue sections, prepared from WM983-B MGP melanoma xenografts of nude mice that as described in A were injected i.p. with the delivery vehicle DMSO (panel a), the Aurora kinase inhibitor (30 mg/kg) (panel b), or the Aurora kinase inhibitor (30 mg/kg) combined with paclitaxel (10 mg/kg) (panel c). The arrows in the melanoma xenograft tissue section depicted in panel a point to some of the pHisH3-positive cells. Tissue sections, prepared from WM983-B MGP melanoma xenografts of nude mice that were injected i.p. with the Aurora kinase inhibitor (30 mg/kg) combined with paclitaxel (10 mg/kg) (panel d) or that did not receive injections (panel e), were probed with an antibody to human Ki67. The tumor sections, depicted in panels a to e, were counterstained with hematoxylin.
Since it is unlikely that a small-molecule inhibitor, regardless of its molecular target, when administered as a single agent, will ever be effective to the extent that it will be a cure for patients with advanced melanoma, we next determined whether a combination treatment would further enhance the impact of the Aurora kinase inhibitor on MGP melanoma xenografts. Thus, we administered, in the same setting of these in vivo studies, the Aurora kinase inhibitor (30 mg/kg) in combination with the cytotoxic drug paclitaxel (10 mg/kg), which via binding to tubulin, blocks the disassembly of microtubules. Using a similar schedule of twice-a-week systemic treatment, the inhibitor was injected i.p. followed 24 hours later by i.p. injection of paclitaxel. In comparison with the growth rate of the tumors in the nude mice that had only been treated with the inhibitor, the tumors in the animals that had received the combination treatment of Aurora kinase inhibitor and paclitaxel over a period of 24 days grew noticeably slower, suggesting that the combination treatment was more effective.
Our alternative experimental approach to determine to which extent targeting of Aurora kinase A and B would exhibit efficacy for human melanoma xenografts involved the use of an Aurora kinase A and likewise an Aurora kinase B antisense vector, and in addition, a pcDNA-HA dead kinase Aurora B plasmid.12 One hundred micrograms of each of these 2 Aurora kinase AS plasmids and, likewise, the pcDNA-HA dead-kinase Aurora B construct, which has the lysine at position 106 of Aurora kinase B substituted by an alanine,12 were mixed with the delivery vehicle DC-Chol liposomes and injected twice weekly into WM983-B MGP melanoma xenografts for a period of 2 weeks. The 3 respective controls (data not shown) were tumors that did not receive injections, were injected with a pcDNA plasmid not containing a cDNA, or were given intratumoral injections of a pcDNA-HA Aurora kinase B wild-type plasmid construct.12 Although these 3 different Aurora kinase–targeting vectors were not nearly as effective in slowing the growth of the MGP melanoma xenografts as the Aurora kinase small-molecule inhibitor administered in combination with paclitaxel (Fig. 6A), we did find that more prominently than the Aurora kinase A or the Aurora kinase B antisense vector, which block gene expression, the Aurora B dead-kinase (DN) vector, which inhibits the function of Aurora kinase B, did impact the growth of the tumors until about the third intratumoral injection but not thereafter (Fig. 6B).
Given the results of these in vivo molecular targeting studies, we next determined the extent to which the systemic i.p. treatment with the small-molecule inhibitor when administered alone or in combination with paclitaxel had blocked Aurora kinase function in the tumor cells. Probed with an antibody to pHisH3, tissue sections prepared from the periphery, as well as the center of human melanoma xenografts that had been resected from tumor-bearing nude mice that had been euthanized within 3 hours following the last i.p. injection of the inhibitor on day 24 (Fig. 6A), demonstrated numerous pHisH3-positive melanoma cells in the xenografts from the nude mice that had been injected with the small-molecule inhibitor delivery vehicle, DMSO (Fig. 6C, panel a). In contrast, melanoma xenografts from the mice that had been treated systemically with the Aurora kinase inhibitor (Fig. 6C, panel b) or with a combination of the inhibitor and paclitaxel (Fig. 6C, panel c) did not reveal any pHisH3-positive cells. Furthermore, an immunohistochemical analysis with an antibody to the cell proliferation marker, Ki67, revealed noticeable differences between WM983-B MGP melanoma xenografts from mice that were treated with a combination of the inhibitor and paclitaxel (Fig. 6C, panel d) and WM983-B MGP melanoma xenografts from mice that did not receive treatment (Fig. 6C, panel e).
Discussion
To date, little information is available regarding the regulation of G2/M phase progression of advanced melanoma. In the study summarized herein, we present evidence that the Aurora kinases A and B are upregulated to high levels with progression from early to advanced melanoma and that VGP and MGP melanoma cells are susceptible to molecular targeting that inhibits the expression or blocks the function of these 2 crucial regulators of mitosis.
Although our analyses of cryopreserved and FFPE tissues revealed strong expression of both Aurora kinases in VGP and MGP melanomas, it is interesting to note that a higher number of the TMA cores representing VGP and MGP melanoma demonstrated expression of Aurora kinase B rather than Aurora kinase A. Unlike Aurora kinase A, Aurora kinase B is guided through mitosis to cytokinesis by the 3 companion proteins INCENP, Survivin, and Borealin that constitute the chromosomal passenger complex (CPC).13 However, unlike as indicated in the case of the Aurora kinase B probe sets (Fig. 1), none of the probe sets for INCENP, Survivin, or Borealin that we analyzed in the context of our previously conducted whole-genome microarray analysis of nevus and melanoma tissues2 provided evidence that expression of these latter 3 genes increases with progression to VGP and MGP melanoma (data not shown). At present, we do not know the molecular cause(s) for the upregulation of the 2 Aurora kinases in advanced melanoma. However, we believe it is unlikely that amplification or rearrangement of their chromosomal loci is the reason because neither 20q13.2-q13.3, the locus of Aurora kinase A, nor 17p13.1, where Aurora kinase B resides, has been reported to be altered in advanced-stage melanomas. One aspect, however, that could be of relevance to melanoma and that in part may help unravel why VGP and MGP melanomas are refractory to radiotherapy is the recently published finding that Aurora kinase A overexpression inhibits the recruitment of RAD51 to DNA double-strand breaks (DSB) and decreases DSB repair by homologous recombination.14
Given the findings of this Aurora kinase–targeting study, it is not surprising that in vitro, melanomas, like other malignant cells, are inhibited in their proliferation, undergo cell cycle arrest, and thereupon, enter apoptosis in the presence of Aurora kinase A or Aurora kinase B siRNAs or when treated with an Aurora kinase inhibitor. However, in light of the fact that this disease in its advanced stages is refractory to virtually all standard therapies, it is very encouraging that, as we report here, systemic treatment with an Aurora kinase inhibitor demonstrates efficacy for human MGP melanoma xenografts when administered alone and even more effectively, as also shown in other cases,9,15 when combined with paclitaxel. Unlike in the case of malignancies such as breast or lung cancer, there is not a single gene that thus far has proven to be “the” driver of advanced melanoma, which in part is one of the reasons that phase I/II studies focusing upon molecular targeted therapy for patients with advanced melanoma are lacking behind that for other malignancies. Second, despite the fact that in recent years, high-throughput studies have identified several genes that are upregulated to high levels in advanced melanoma, not every one of them has turned out to be a useful target for molecular therapy. For example, as the result of whole-genome expression profiling studies of nevus and melanoma tissue specimens, osteopontin was found to be one of the most abundantly expressed genes in advanced melanoma2,16 and, as recent studies have suggested, a prognostic marker17 and predictor of reduced relapse-free survival of melanoma.18 However, none of our molecular targeting approaches have provided an indication that osteopontin would be a useful target for molecular therapy of advanced melanoma (Hershey and Becker, unpublished observation). Another example is the Ataxia Telangiectasia Mutated (ATM) gene, which like the Aurora kinases is expressed at high levels in advanced-stage melanomas; yet, our molecular targeting studies of this pivotal DNA damage sensor did not sensitize VGP or MGP melanomas to the effects of radiation treatment.19
Apart from the by now widely established fact that monotherapies do not lead to a long-lasting clinical response in patients with advanced melanoma, emerging evidence from BRAFV600E molecular targeting studies also suggests that melanoma cells become quite rapidly resistant to treatment with a BRAF small-molecule inhibitor.20 Although, for example, in the case of Aurora kinase B, its inhibition leads to mitotic slippage and, in turn, polyploidy and genetic instability, it is unlikely that Aurora kinase small-molecule inhibitor monotherapy will result in a major clinical response in patients with locally advanced or stage IV melanoma. However, as our preclinical in vivo studies document, when the Aurora kinase inhibitor is administered in sequence with a spindle toxin, the antimelanoma activity is noticeably enhanced. Since we believe that it is also essential to explore multimodality treatments for melanoma that, instead of relying on combinations with chemotherapeutic agents, use a combination of small-molecule inhibitors, we are currently determining whether small-molecule inhibitors targeting the Aurora kinases and genes that regulate G1/2 transition, or genes that are crucial for melanoma cell proliferation and angiogenesis, when administered sequentially or simultaneously, will be a powerful strategy for interfering with the aggressive growth and metastatic dissemination of this disease.
Materials and Methods
Melanoma cell lines, cryopreserved tissues, and TMAs
VGP (WM983-A, WM98-2) and MGP (WM373, WM852, WM983-B, WM1158) human melanoma cell lines were propagated in vitro as described.21 Standard immunohistochemistry of deidentified, postdiagnosis excess cryopreserved or FFPE tissue samples, representing normal human skin, benign and atypical nevi, and early and advanced melanomas, was performed as described,22 using a mouse antihuman Aurora kinase A antibody (Leica Microsystems Inc., Bannockburn, IL, USA) or an antihuman Aurora kinase B rabbit monoclonal antibody (Epitomics Inc., Burlingame, CA, USA). Following antigen retrieval, tissue cores of nevus > melanoma progression TMAs19,23,24 were probed by standard immunohistochemistry with the respective antibody to Aurora kinase A or Aurora kinase B.
RT-PCR and immunoblot analysis
RT-PCR analysis of MGP melanoma cells was performed with a set of primers (forward primer: 5′ GGGAGAGCTGAAGATTGCTGACTTCG 3′; reverse primer: 5′ GTTATGCCTGAGCAGTTTG GAGATGAGGTCC 3′) spanning nucleotides 694 to 994 of the human Aurora kinase B cDNA (NM_004217.2).
Protein lysates (30 µg/sample), separated on sodium dodecyl sulfate–polyacrylamide gels (SDS-PAGE) and transferred onto nylon membrane, were probed with antibody to human Aurora A (Epitomics), human Aurora B (Epitomics), pT288 Aurora A, pHisH3, or c-PARP (Cell Signaling Technology Inc., Danvers, MA, USA), followed by incubation with a horseradish peroxidase–conjugated secondary antibody and Luminol reagent (Millipore Corporation, Billerica, MA, USA). An antibody to human pFGFR-1 was obtained from Invitrogen Corporation (Carlsbad, CA, USA); an antibody to CREST was purchased from Promega (Madison, WI, USA), an actin antibody from Abcam Inc. (Cambridge, MA, USA), an antibody to α-tubulin from Cell Signaling Technology, and an antibody to y-tubulin from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
RNA interference assays, Aurora kinase inhibitor treatment, and immunofluorescence
siGENOME SMARTpool siRNAs to human Aurora kinase A (150 nM) or Aurora kinase B (150 nM) (Thermo Fisher Scientific Inc., Waltham, MA, USA) and likewise ON-TARGETplus nontargeting siRNAs (150 nM) (Thermo Fisher Scientific Inc.) were mixed with Lipofectamine 2000 (Invitrogen Corporation) as per the manufacturer’s instructions and transfected into subconfluent MGP melanoma cells.
MGP melanoma cells were incubated in the presence of Aurora kinase inhibitor, PF-3814735 (Pfizer Inc., New York, NY, USA) solubilized in DMSO, or only in the presence of DMSO. Where indicated, MGP melanoma cells were incubated, prior to addition of the Aurora kinase inhibitor, for 20 hours in the presence of either 20 ng/mL or 50 ng/mL of nocodazole (Sigma-Aldrich, St. Louis, MO, USA) solubilized in DMSO.
MGP melanoma cells, fixed with 4% paraformaldehyde, blocked with goat serum, probed with primary antibody and an Alexa Fluor or a Streptavidin Alexa Fluor–conjugated secondary antibody (Invitrogen Corporation), and counterstained with fluorescent 40-6-diamidino-2-phenylindole (DAPI) (Invitrogen Corporation) were imaged with an inverted, epifluorescent TE2000 Nikon microscope (Tokyo, Japan) and a charge-coupled device (CCD) camera (Roper Scientific Inc./Photometrics, Tucson, AZ, USA).
Flow cytometry analysis and TUNEL staining
Aurora kinase inhibitor–treated MGP melanoma cells, labeled with propidium iodide or Alexa Fluor 488 annexin V/propidium iodide (Vybrant Apoptosis Assay Kit #2, Invitrogen), were analyzed by flow cytometry as previously described.23 Cytospin preparations of MGP melanoma cells, treated with Aurora kinase inhibitor, were fixed, permeabilized, and labeled with the In Situ Cell Death Detection Kit, TMR red (Roche Applied Science, Indianapolis, IN, USA).
Melanoma xenograft studies
4-week-old, female nude mice (NCr-Hfh11) (Taconic Farms Inc., Hudson, NY, USA) were injected subcutaneously on their right dorsal site with WM983-B MGP human melanoma cells (8 × 106 cells per animal). When the tumors had reached a size of 5 mm in any direction, the animals were given twice-weekly i.p. injections of the PF-03814735 Aurora kinase inhibitor (30 mg/kg, dissolved in DMSO), either alone or in combination with paclitaxel (10 mg/kg, dissolved in a formulation of Cremophor-EL [Sigma-Aldrich], ethanol, saline at 12.5%/12.5%/ 75%), administered i.p. 24 hours following injection of the Aurora kinase inhibitor. For oral delivery, the Aurora kinase inhibitor, dissolved in DMSO, was administered twice a week by oral gavage at a dose of 30 mg/kg and for intratumoral delivery at 2.5 mg/kg or 12 mg/kg twice a week. Controls were WM983-B human melanoma xenograft-bearing nude mice that did not receive injections, were injected with only the delivery vehicle(s), or received only paclitaxel.
Using RT-PCR and a set of primers amplifying the sequence between nucleotides 558 and 945 of human Aurora A (NM_198433), and in the case of Aurora B (NM_004217), with primers amplifying the sequence between nucleotides 11 and 250, spanning in each case, the AUG codon, we amplified 2 nonhomologous cDNA fragments for subcloning in antisense (AS) orientation into a pcDNA mammalian expression vector (Invitrogen Corporation). One hundred micrograms of each of these 2 Aurora kinase AS plasmids and likewise a pcDNA-HA dead-kinase Aurora B construct12 and, serving as controls, a pcDNA plasmid (Invitrogen Corporation) not containing a cDNA, or a pcDNA-HA Aurora B wild-type plasmid construct,12 were mixed with 6 nmol of DC-Chol liposomes (Avanti Polar Lipids Inc., Alabaster, AL, USA) and injected twice weekly into the center of WM983-B MGP melanoma xenografts. Tumor length and width of all xenografts were measured twice a week with a caliper, and tumor volumes were calculated using the equation v = (lxw2)/2.
Tissue sections, prepared from the human melanoma xenografts, were fixed with paraformaldehyde, treated with Rodent Block M (Biocare Medical, Concord, CA, USA), probed with antibody to human S100 antigen (Dako North America Inc., Carpinteria, CA, USA), pHisH3 (Cell Signaling Technology), or Ki67 (Epitomics), incubated with Rabbit-on-Rodent Polymer (for AP) (Biocare Medical), and counterstained with hematoxylin.
Acknowledgments
The authors thank C. Fowst and J. Jakubczak for making the Aurora kinase inhibitor available via Compound Transfer Agreement, D. Skoufias for providing the pcDNA-HA Aurora B plasmids, and M. Acquafondata and D. Jukic for assistance with probing and scoring of the TMAs.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by a grant from the National Institutes of Health [R01CA091870] to D.B.
References
- 1. Trent JM. Cytogenetics of human malignant melanoma. Cancer Metastasis Rev. 1991;10:103-13 [DOI] [PubMed] [Google Scholar]
- 2. Smith AP, Hoek K, Becker D. Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer Biol Ther. 2005;4:1018-29 [DOI] [PubMed] [Google Scholar]
- 3. Jaeger J, Koczan D, Thiesen HJ, et al. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res. 2007;13:806-15 [DOI] [PubMed] [Google Scholar]
- 4. Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547-66 [DOI] [PubMed] [Google Scholar]
- 5. Keen N, Taylor S. Mitotic drivers: inhibitors of the Aurora B Kinase. Cancer Metastasis Rev. 2009;28:185-95 [DOI] [PubMed] [Google Scholar]
- 6. Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60-72 [DOI] [PubMed] [Google Scholar]
- 7. Perez de Castro I, de Carcer G, Montoya G, Malumbres M. Emerging cancer therapeutic opportunities by inhibiting mitotic kinases. Curr Opin Pharmacol. 2008;8:375-83 [DOI] [PubMed] [Google Scholar]
- 8. Emanuel S, Rugg CA, Gruninger RH, et al. The in vitro and in vivo effects of JNJ-7706621: a dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res. 2005;65:9038-46 [DOI] [PubMed] [Google Scholar]
- 9. Jani JP, Arcari J, Bernardo V, et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol Cancer Ther. 2010;9:883-94 [DOI] [PubMed] [Google Scholar]
- 10. Becker D, Lee PL, Rodeck U, Herlyn M. Inhibition of the fibroblast growth factor receptor 1 (FGFR-1) gene in human melanocytes and malignant melanomas leads to inhibition of proliferation and signs indicative of differentiation. Oncogene. 1992;7:2303-13 [PubMed] [Google Scholar]
- 11. Wang Y, Becker D. Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat Med. 1997;3:887-93 [DOI] [PubMed] [Google Scholar]
- 12. Scrittori L, Skoufias DA, Hans F, et al. A small C-terminal sequence of Aurora B is responsible for localization and function. Mol Biol Cell. 2005;16:292-305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798-812 [DOI] [PubMed] [Google Scholar]
- 14. Sourisseau T, Maniotis D, McCarthy A, et al. Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. EMBO Mol Med. 2010;2:130-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimomura T, Hasako S, Nakatsuru Y, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. 2010;9:157-66 [DOI] [PubMed] [Google Scholar]
- 16. Zhou Y, Dai DL, Martinka M, et al. Osteopontin expression correlates with melanoma invasion. J Invest Dermatol. 2005;124:1044-52 [DOI] [PubMed] [Google Scholar]
- 17. Rangel J, Nosrati M, Torabian S, et al. Osteopontin as a molecular prognostic marker for melanoma. Cancer. 2008;112:144-50 [DOI] [PubMed] [Google Scholar]
- 18. Conway C, Mitra A, Jewell R, et al. Gene expression profiling of paraffin-embedded primary melanoma using the DASL assay identifies increased osteopontin expression as predictive of reduced relapse-free survival. Clin Cancer Res. 2009;15:6939-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moschos SJ, Dodd NR, Jukic DM, Fayewicz SL, Wang X, Becker D. Suppressing the high-level expression and function of ATM in advanced-stage melanomas does not sensitize the cells to ionizing radiation. Cancer Biol Ther. 2009;8:1815-25 [DOI] [PubMed] [Google Scholar]
- 20. Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Becker D, Meier CB, Herlyn M. Proliferation of human malignant melanomas is inhibited by antisense oligodeoxynucleotides targeted against basic fibroblast growth factor. EMBO J. 1989;8:3685-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Rao U, Mascari R, et al. Molecular analysis of melanoma precursor lesions. Cell Growth Differ. 1996;7:1733-40 [PubMed] [Google Scholar]
- 23. Moschos SJ, Smith AP, Mandic M, et al. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene. 2007;26:4216-25 [DOI] [PubMed] [Google Scholar]
- 24. Watson-Hurst K, Becker D. The role of N-cadherin, MCAM and beta3 integrin in melanoma progression, proliferation, migration and invasion. Cancer Biol Ther. 2006;5:1375-82 [DOI] [PubMed] [Google Scholar]