

Abstract
Centrosome aberrations are frequently detected in c-MYC–associated human malignancies. Here, we show that c-MYC–induced centrosome and centriole overduplication critically depend on the protease tripeptidyl peptidase II (TPPII). We found that TPPII localizes to centrosomes and that overexpression of TPPII, similar to c-MYC, can disrupt centriole duplication control and cause centriole multiplication, a process during which maternal centrioles nucleate the formation of more than a single daughter centriole. We report that inactivation of TPPII using chemical inhibitors or siRNA-mediated protein knockdown effectively reduced c-MYC–induced centriole overduplication. Remarkably, the potent and selective TPPII inhibitor butabindide not only potently suppressed centriole aberrations but also caused significant cell death and growth suppression in aggressive human Burkitt lymphoma cells with c-MYC overexpression. Taken together, these results highlight the role of TPPII in c-MYC–induced centriole overduplication and encourage further studies to explore TPPII as a novel antineoplastic drug target.
Keywords: c-MYC, TPPII, centrosome, butabindide, cancer
Introduction
The c-MYC proto-oncogene encodes a transcription factor that is overexpressed in a wide range of human cancers. c-MYC contributes to tumorigenesis by altering a spectrum of cellular functions such as proliferation, differentiation, and sensitivity to apoptosis. c-MYC is rapidly induced in response to mitogenic stimuli and contributes to the regulation of normal cell cycle progression.1 Several lines of evidence suggest that c-MYC plays a role in both structural and numerical chromosomal instability by inducing DNA damage2-5 as well as centrosome aberrations,6 which can result in cell division errors and aneuploidy.7 Besides c-MYC, the related N-MYC protein as well as other basic helix-loop-helix transcription factors have previously been implicated in centrosome abnormalities.8,9
Centrosomes are the major microtubule organizing centers in most mammalian cells and orchestrate cell division through various functions.10,11 The single centrosome of a cell normally duplicates precisely once prior to mitosis, thus assuring bipolarity of the cell division process and proper chromosome segregation. During this process, the 2 pre-existing (maternal) centrioles disengage, and each nucleates the formation of a single new centriole (daughter centriole).12 It has recently been discovered that certain oncogenic stimuli can override this “one-and-only-one” rule of centriole duplication and stimulate the formation of more than one daughter at a single maternal centriole (centriole multiplication).11,13 Several key players have been identified that are involved in centriole multiplication including cyclin E/CDK2 complexes,6,13 hSAS-6,14 and PLK4.15-18
There is compelling evidence that centriole duplication control is regulated by proteolysis, and inhibitors of certain protein degradation systems have been found to rapidly cause centriole multiplication.6,13 Components of the 26S proteasome as well as several proteases have been found to localize to the centrosome and contribute to centriole duplication control.19-21 Besides microtubule organization, the centrosome has been suggested to play an important role in MHC class I–mediated antigen presentation.22 This function requires a specific peptide length for MHC loading, which has been suggested to depend on the concerted proteolytic action of the proteasome and, under certain conditions, aminopeptidases such as tripeptidyl peptidase II (TPPII).23-25 TPPII is a serine peptidase of the subtilisin family that sequentially removes tripeptides from the amino-terminal end of oligopeptides released by the proteasome. TPPII has also been reported to possess endoproteolytic activity, but this function remains poorly characterized. TPPII subunits can assemble into higher order, spindle-shaped giant peptidase complexes of 6 MDa, thus forming the largest known protease complex in eukaryotic cells.26 TPPII has been implicated in cancer and has been shown to be upregulated in c-MYC–associated Burkitt lymphoma cells.27
We have recently shown that c-MYC–overexpressing cells contain aberrant centrosome numbers.6,28 Here, we extend these findings by showing that c-MYC can stimulate centriole multiplication in a process that critically depends on TPPII. We show that TPPII localizes to the centrosome and that its overexpression, like c-MYC, stimulates centriole multiplication. Moreover, we demonstrate that inactivation of TPPII with either chemical inhibitors or by siRNA-mediated protein knockdown effectively suppresses c-MYC–induced centriole overduplication. Remarkably, the potent and selective TPPII inhibitor butabindide, which has originally been designed to control appetite and overeating,29 showed pronounced proapoptotic and growth-suppressive activities in highly aggressive c-MYC–driven Burkitt lymphoma cells. Our findings therefore highlight the role of TPPII in c-MYC–induced centriole overduplication and suggest targeting TPPII with the potent and selective inhibitor butabindide as a novel antineoplastic strategy.
Results
Overexpression of c-MYC induces centriole multiplication
The c-MYC oncogene has previously been implicated in centrosome duplication control.6,28 We found that transient overexpression of c-MYC led to an increase of cells with abnormal centrosome numbers (more than 2 per cell) in U-2 OS cells (Fig. 1A) from 4.9% in control-transfected cells to 11.5% in c-MYC–transfected cells (P ≤ 0.005) (Fig. 1B) as well as to an increase of cells with abnormal centriole numbers (more than 4 per cell with no specific arrangement of supernumerary centrioles) from 4.6% in controls to 12% in c-MYC–transfected U-2 OS/centrin-GFP cells (P ≤ 0.0005) (Fig. 1B). c-MYC was also found to stimulate abnormal centrosome numbers in nonimmortalized normal human keratinocytes from 2.6% in controls to 7.2% in c-MYC–transfected cells (P ≤ 0.05) (Fig. 1B). To corroborate our results, we engineered U-2 OS/centrin-GFP cells to stably overexpress empty control vector or c-MYC. Stable overexpression led to a statistically significant 2.8-fold increase of cells with abnormal centriole numbers from 6.3% in controls to 17.4% in c-MYC–expressing cells (P ≤ 0.05) (Fig. 1C).
Figure 1.
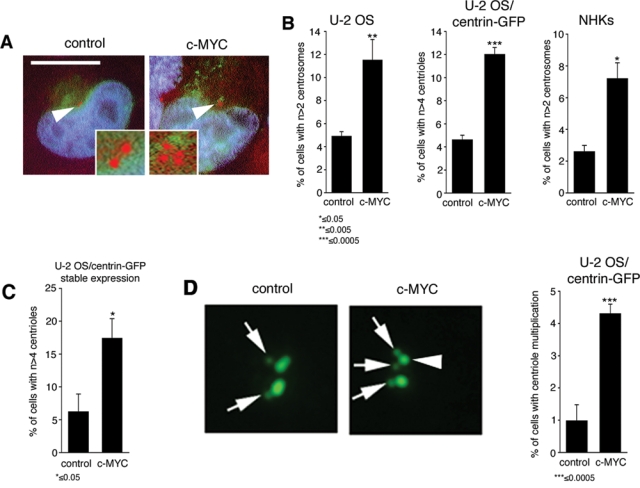
Overexpression of c-MYC induces abnormal duplication of centrosomes and centrioles. (A) Immunofluorescence microscopic analysis of U-2 OS cells for centrosome aberrations using an anti-γ-tubulin antibody. Note the abnormal centrosome numbers in a U-2 OS cell transiently transfected with c-MYC (right panel: arrowhead, inset) in comparison to an empty vector–transfected cell (left panel). Farnesylatable GFP is used as a transfection marker. Nuclei stained with DAPI. Scale bar indicates 10 µm. (B) Quantification of abnormal centrosome numbers in U-2 OS cells (left panel), abnormal centriole numbers in U-2 OS/centrin-GFP cells (middle panel), and abnormal centrosome numbers in normal human keratinocytes (NHKs) (right panel) following transient transfection (48 hours) with either empty vector (control) or c-MYC. Each bar indicates average ± standard error of at least 3 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences and P values. (C) Quantification of abnormal centriole numbers in U-2 OS/centrin-GFP cells manipulated to stably express either empty vector (control) or c-MYC. Each bar indicates average ± standard error of at least 3 independent counts of at least 100 cells. Asterisk indicates statistically significant differences and P value. (D) Fluorescence microscopic analysis of U-2 OS/centrin-GFP cells transiently transfected (24 hours) with empty vector (control) or c-MYC. Note the presence of 2 daughter centrioles (arrows) at a single maternal centriole (arrowhead) in the c-MYC–expressing cell (centriole multiplication). Quantification of centriole multiplication in U-2 OS/centrin-GFP cells after transient transfection (24 hours) of empty vector (control) or c-MYC (right panel). Each bar indicates average ± standard error of 5 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences and P values.
Remarkably, we found that a proportion of transiently c-MYC–transfected cells with abnormal centriole numbers showed a phenotype in which single maternal centrioles organize the concurrent formation of more than one daughter (referred to as centriole multiplication in contrast to centriole overduplication with no specific centriole arrangement) (Fig. 1D). At 24 hours posttransfection, the proportion of cells with centriole multiplication was significantly increased from 1% in controls to 4.3% in c-MYC–transfected cells (P ≤ 0.0005). These results confirm and extend previous findings by showing that c-MYC overexpression rapidly disrupts centriole duplication control, which in part involves centriole multiplication.
c-MYC–induced abnormal centriole duplication requires TPPII activity
There is compelling evidence that proteolysis plays a crucial role in centriole duplication control.6,13 Cells overexpressing c-MYC, in particular, Burkitt lymphoma cells, have previously been reported to have impaired ubiquitin-proteasome activity and to rely more on alternative proteolytic pathways, in particular, tripeptidyl peptidase II (TPPII)–mediated proteolysis.27,30,31 We therefore tested whether 2 inhibitors of TPPII activity, Ala-Ala-Phe-chloromethylketone (AAF-CMK) and butabindide, can interfere with c-MYC–induced abnormal centriole duplication (Fig. 2). Whereas AAF-CMK covalently binds to TPPII, butabindide is a selective and competitive inhibitor of TPPII.29 Treatment of c-MYC–transfected U-2 OS/centrin-GFP cells with 1 µM AAF-CMK for 24 hours resulted in a suppression of c-MYC–induced centriole overduplication from 12.4% in c-MYC–transfected, DMSO-treated controls to 8.9% in c-MYC–transfected, AAF-CMK–treated cells (P ≤ 0.05) (Fig. 2A). Treatment with increasing amounts of the potent and selective TPPII inhibitor butabindide led to a complete abrogation of c-MYC–induced centriole overduplication from 10.2% in untreated, c-MYC–transfected U-2 OS/ centrin-GFP cells to 4% in c-MYC–transfected treated cells with 10 µM butabindide for 24 hours (P ≤ 0.001), which is similar to empty vector–transfected controls (4.6%) (Fig. 2B). These results show that c-MYC–induced centriole overduplication is effectively abrogated by TPPII inhibitors.
Figure 2.

Inhibitors of TPPII suppress c-MYC–induced centriole overduplication. (A and B) Quantification of numerical centriole abnormalities in U-2 OS/centrin-GFP cells transiently transfected with empty vector (control) or c-MYC for 24 hours and either treated with 0.1% DMSO or 1 µM of the TPPII inhibitor Ala-Ala-Phe-chloromethylketone (AAF-CMK) for an additional 24 hours (A). (B) Cells were treated with the TPPII inhibitor butabindide for 24 hours at the indicated concentrations. Each bar indicates average ± standard error of at least 3 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences between DMSO and AAF-CMK–treated cells (A), control- and c-MYC–transfected cells (B), and c-MYC–transfected cells in comparison to c-MYC–transfected cells treated with butabindide (B) and P values.
Knockdown of TPPII suppresses c-MYC–induced centriole overduplication
To further corroborate the role of TPPII in c-MYC–induced centriole overduplication, we performed a series of siRNA and shRNA experiments (Fig. 3). c-MYC–induced centriole overduplication (10.6%) was significantly suppressed when TPPII was depleted (3.9%; P ≤ 0.0001) (Fig. 3A and 3B), whereas control siRNA duplexes had no effect (Fig. 3B). A similar significant inhibition of c-MYC–induced centriole overduplication was detected when TPPII was depleted by shRNA from 12.8% in c-MYC–transfected cells to 6.4% in c-MYC–transfected and TPPII-depleted cells (P ≤ 0.0001) (Fig. 3C and 3D).
Figure 3.

Knockdown of TPPII inhibits c-MYC–induced centriole overduplication. (A) Immunoblot analysis of TPPII expression following siRNA-mediated knockdown in U-2 OS/centrin-GFP cells in comparison to control siRNA-transfected cells (siCtrl). Immunoblot for actin shows protein loading. (B) Quantification of U-2 OS/centrin-GFP cells with centriole overduplication following transient overexpression (24 hours) of empty vector (control) or c-MYC alone or c-MYC in combination with control siRNA duplexes (siControl) or siRNA duplexes targeting TPPII (siTPPII). Centriole overduplication following transient overexpression (24 hours) of TPPII alone or in combination with control siRNA (siControl) or siRNA targeting TPPII (siTPPII) is also shown. Each bar indicates average ± standard error of 3 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences and P values. (C) Immunoblot analysis of TPPII expression following shRNA-mediated knockdown in U-2 OS/centrin-GFP cells in comparison to control shRNA-transfected cells (shCtrl). Immunoblot for actin shows protein loading. (D) Quantification of U-2 OS/centrin-GFP cells with centriole overduplication following transient overexpression (48 hours) of empty vector (control) or c-MYC alone or c-MYC in combination with an shRNA construct targeting TPPII (shTPPII). Centriole overduplication following transient overexpression (48 hours) of TPPII alone or in combination with shRNA targeting TPPII (shTPPII) is also shown. Each bar indicates average ± standard error of 3 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences and P values.
In both siRNA and shRNA experiments, we also ectopically expressed TPPII, which led to a significant increase of cells with centriole overduplication from 5.4% in empty vector controls to 11.5% (P ≤ 0.0001) (Fig. 3B) and 4.9% to 11% (P ≤ 0.0001) (Fig. 3D), respectively. siRNA- or shRNA-mediated knockdown of TPPII was found to efficiently abrogate the increase of cells with centriole overduplication (Fig. 3B and 3D).
However, we were not able to substantiate an increase of TPPII mRNA in cells transfected with c-MYC by quantitative real-time PCR, whereas a significant increase of ornithine decarboxylase (ODC), a known transcriptional target of c-MYC, was readily detectable (data not shown). As expected from these results, we did not find an increase of TPPII expression on the protein level (data not shown). Hence, although TPPII overexpression clearly stimulates centriole overduplication, c-MYC does not seem to induce its upregulation on the transcriptional or protein level. Nonetheless, c-MYC strictly requires TPPII for centriole overduplication, and our results therefore support the idea that additional c-MYC–associated oncogenic mechanisms may be involved in c-MYC–induced centriole overduplication.
TPPII is a centrosomal protein and rapidly subverts centriole duplication control when overexpressed
Having shown that TPPII is critically required for c-MYC–induced centriole overduplication and can stimulate centriole overduplication itself, we next investigated whether TPPII localizes to the centrosome. Immunofluorescence microscopic analysis of U-2 OS/centrin-GFP cells showed that TPPII is present at the centrosome throughout the cell division cycle (Fig. 4A). Based on the fact that daughter centrioles are commonly smaller in size compared to maternal centrioles, we observed a staining pattern suggestive of selective association with daughter centrioles in late mitosis (anaphase) and in the unduplicated state, whereas TPPII was detected mostly between mother and daughter centrioles during the G2 phase (Fig. 4A).
Figure 4.

TPPII localizes to the centrosome and stimulates centriole multiplication. (A) Immunofluorescence microscopic analysis of TPPII in U-2 OS/centrin-GFP cells. Note the presence of TPPII at centrioles throughout the cell division cycle. Nuclei stained with DAPI. Scale bar indicates 10 µm. (B) Fluorescence microscopic analysis of centrioles in a U-2 OS/centrin-GFP cells transiently transfected with empty vector (control) or TPPII. Note the presence of centriole multiplication in the TPPII-transfected cell as evidenced by the concurrent presence of 2 daughter centrioles (arrows) at a single maternal centriole (arrowhead). (C) Quantification of centriole overduplication (gray bars) and centriole multiplication (black bars) in U-2 OS/centrin-GFP cells transiently transfected with empty vector (control) or TPPII. Each bar indicates average ± standard error of 3 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences and P values.
We next found that ectopic TPPII expression was sufficient to cause centriole multiplication (Fig. 4B) similar to c-MYC overexpression. The proportion of cells with centriole overduplication (i.e., more than 4 centrioles without the characteristic arrangement of 2 or more daughters at single maternal centrioles) was 1.9% following TPPII overexpression, whereas 5.5% of cells showed centriole multiplication (Fig. 4B) compared to 0.1% of cells with centriole overduplication (P ≤ 0.001) and 1% of cells with centriole multiplication (P ≤ 0.0001) in empty vector–transfected control cells (Fig. 4C). These results extend a previous report that has implicated TPPII in centrosome overduplication32 by showing that TPPII upregulation causes a genuine centriole duplication defect and not merely centrosome accumulation, a possibility that has not rigorously been excluded in that previous study.32 In combination with our data that TPPII is required for c-MYC–induced centriole multiplication, these findings further support the notion that c-MYC and TPPII may function in the same pathway to induce centriole multiplication but cannot rule out that additional c-MYC–associated oncogenic mechanisms may be involved.
Butabindide suppresses centriole overduplication and malignant growth in Burkitt lymphoma cells
We next sought to explore whether the TPPII inhibitor butabindide may suppress centrosome aberrations in cells that constitutively overexpress c-MYC. We first compared the baseline levels of numerical centrosome aberrations between 2 low-grade malignant B cell lines U266 (multiple myeloma with undetectable c-MYC mRNA expression) and HBL2 (mantle cell lymphoma) and 3 EBV-negative Burkitt lymphoma cell lines with c-MYC overexpression (BJAB, DG75, BL2), respectively. Overall, cell lines with c-MYC overexpression showed a trend towards increased proportions of cells with numerical centriole aberrations (Fig. 5A). We next treated BJAB, DG75, and BL2 cells with 1 µM or 10 µM butabindide for up to 4 days. All drug treatments led to a significant reduction of the proportion of cells with centriole overduplication (Fig. 5B). In addition, treatment of each of the 3 Burkitt lymphoma cell lines with 10 µM butabindide caused a significant increase of cell death at 96 hours as evidenced by an increase in cells with sub-G1 DNA content by flow cytometry (Fig. 5C).
Figure 5.

The TPPII inhibitor butabindide suppresses centriole aberrations and has proapoptotic and growth-suppressive activities in Burkitt lymphoma cells. (A) Quantification of centriole abnormalities in 2 low-grade malignant B cell lines (U266 and HBL2) and 3 Burkitt lymphoma cell lines (BJAB, DG75, and BL2). Each bar indicates average ± standard error of 3 independent experiments with at least 100 cells counted per experiment. (B) Quantification of centriole abnormalities in BJAB, DG75, and BL2 Burkitt lymphoma cells either left untreated (c) or treated with butabindide diluted in sterile water at the concentrations indicated. Each bar indicates average ± standard error of 3 independent experiments with at least 100 cells counted per experiment. Asterisks indicate statistically significant differences compared to the corresponding untreated controls (c) and P values. (C) Flow cytometric analysis of BJAB, DG75, and BL2 cells treated with 10 µM butabindide for the time intervals indicated. Note the significant increase of apoptotic cells (sub-G1 population) after 24 hours and 96 hours. (D) Analysis of the proportion of viable GA-10 Burkitt lymphoma cells grown in the presence of 0.1% DMSO or 1 µM clastolactacystin β-lactone (CLBL). Number of viable cells was determined by counting trypan blue–negative cells in a hemocytometer on days in culture indicated. (E) Immunoblot analysis of TPPII and c-MYC expression in GA-10 cells adapted to grow in the presence of 0.1% DMSO or 1 µM clastolactacystin β-lactone (CLBL). Note the induction of TPPII in adapted cells. (F) Analysis of cell growth of GA-10 cells mock adapted to 0.1% DMSO and GA-10 cells adapted to 1 µM CLBL (GA-10/adapted) either untreated (black squares) or treated with butabindide as indicated (open symbols). Number of viable cells was determined by counting trypan blue–negative cells in a hemocytometer on days in culture indicated.
To further investigate whether TPPII inhibition restrains Burkitt lymphoma growth, we cultured the human Burkitt lymphoma cell line GA-10 in the presence of the proteasome inhibitor clastolactacystin β-lactone (CLBL; 1 µM) or 0.1% DMSO, respectively (Fig. 5D). Continuous culture in the presence of the proteasome inhibitor CLBL promotes the use of components of alternative proteolytic pathways, in particular, TPPII, to maintain a certain level of intracellular proteolysis.33 The GA-10 cell line was derived from a highly aggressive t(8;14)-positive, Epstein-Barr virus– negative Burkitt lymphoma with mutated p53 and was resistant to conventional chemotherapy.34 After exposure to CLBL, we first observed a marked decrease of cell viability, but proteasome inhibitor–resistant cells emerged within approximately 10 days (Fig. 5D). As expected from previous studies,33,35 GA-10 cells adapted to grow in the presence of CLBL exhibited increased TPPII protein levels (Fig. 5E). The levels of centrosome abnormalities did not differ significantly between parental GA-10 cells (18.7%), GA-10 cells mock adapted in DMSO (19.9%), and CLBL-adapted cells (19.4%), respectively. Importantly, however, adapted GA-10 cells were more sensitive to TPPII inhibition by butabindide compared to their nonadapted counterparts (Fig. 5F). Moreover, 10 µM butabindide reduced soft agar colony formation of the GA-10/adapted cell line by 6.5-fold (data not shown). To explore whether the growth-inhibitory effect of butabindide in adapted GA-10 cells may be related to interference with centrosome duplication, we specifically analyzed mitotic cells for the number of γ-tubulin–positive (i.e., centrosome associated) mitotic spindle poles. We did not observe a significant increase of monopolar spindles in butabindide-treated adapted GA-10 cells compared to vehicle-treated cells (4.6% v. 3.1%, respectively). However, we observed an increase of cells with multinucleation and enlarged nuclei, suggestive of abortive mitoses (data not shown) in line with previous results.32 Taken together, these results suggest potent antineoplastic activities of the TPPII inhibitor butabindide in aggressive, c-MYC–associated Burkitt lymphomas.
Discussion
The c-MYC proto-oncogene has been implicated in the induction of genomic instability through a number of mechanisms including aberrant centrosome duplication.6 Here, we show that c-MYC can rapidly stimulate centriole multiplication, a recently identified pathway of centriole overduplication, in which a single maternal centriole nucleates the concurrent formation of 2 or more daughters.13-15 We found that c-MYC–induced centriole overduplication critically depends on the giant protease TPPII. We identified TPPII as a centriolar protein that by itself can stimulate centriole multiplication when overexpressed. Furthermore, we show that siRNA-mediated knockdown of TPPII as well as treatment with the potent and selective TPPII inhibitor butabindide abolish c-MYC–induced centriole overduplication. To test butabindide as a potential anticancer agent, we treated highly aggressive, c-MYC–associated Burkitt lymphoma cells with butabindide and found not only suppression of centriole overduplication but also potent growth-suppressive and proapoptotic activities.
Our results go beyond the findings by Stavropoulou et al.32 in several ways. We not only identify centriole multiplication as the cellular mechanism of centrosome aberrations in the context of overexpressed TPPII but also demonstrate that a fraction of TPPII has a centrosomal localization. In addition, we provide evidence that TPPII overexpression phenocopies the centriole duplication defect detected in c-MYC–overexpressing cells and demonstrate the potential use of the potent and selective TPPII inhibitor butabindide as an antineoplastic agent.
The c-MYC oncogene has been suggested to transcriptionally regulate a large fraction of human genes. We were not able to substantiate an increase of TPPII expression in cells transfected with c-MYC by quantitative real-time PCR and immunoblot analysis (data not shown). Hence, c-MYC requires TPPII for centriole overduplication, but it is conceivable that other c-MYC–induced oncogenic mechanisms may also contribute to this phenotype.
The selective and potent TPII inhibitor butabindide had initially been designed to target a membrane-bound isoform of TPPII involved in cholecystokinin inactivation with the intention to control appetite and food intake.29 No major toxicities have been reported in rodents.29 Here, we show that this compound has promising antineoplastic activities in clinically aggressive Burkitt lymphoma. Although we show that butabindide suppresses centriole overduplication in Burkitt lymphoma cells, we do not have evidence that this activity is the cause of the pronounced apoptotic cell death in butabindide-treated Burkitt lymphoma cells. Nonetheless, we did find morphological signs of failed mitoses (data not shown) in accordance with previous experiments in which TPPII was knocked down in Burkitt lymphoma cells.32
The precise role of TPPII in aberrant centriole biogenesis remains to be determined, but there is convincing evidence that key regulators of centriole multiplication such as PLK4 are substrates for proteases.36 Our results furthermore warrant preclinical experiments to explore the use of butabindide as a novel anticancer or chemoprevention agent in tumors with c-MYC overexpression.
Materials and Methods
Cell culture, transfections, and inhibitor treatments
U-2 OS and U-2 OS/centrin-GFP cells (plasmid kindly provided by M. Bornens, Institut Curie, Paris, France)37 were maintained as previously described.13 Stable cell populations were generated as previously described.38 Normal human keratinocytes (NHKs) were isolated from neonatal foreskins and cultured as previously described.39 BJAB, DG75, BL2 (kindly provided by Elliott Kieff, Channing Laboratory, Brigham and Women’s Hospital, Boston, MA), U266, and HBL2 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 µg/mL streptomycin. GA-10 cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained according to the supplier’s recommendations. Proteasome inhibitor–adapted GA-10 cells were obtained by the culture of cells in the presence of 1 µM clastolactacystin β-lactone (CLBL; Sigma-Aldrich, St. Louis, MO), giving rise to the GA-10/ad cell line, whereas control GA-10 cells were grown in 0.1% DMSO. Expression plasmids used were encoding c-MYC (kindly provided by P. Leder, Harvard Medical School, Boston, MA) or TPP2 (obtained from OriGene, Rockville, MD). Empty vector controls were included in all experiments. In all transient overexpression experiments, cells were cotransfected with either farnesylatable green fluorescent protein (GFP) or DsRed fluorescent protein (Clontech, Mountain View, CA) as the transfection marker. The TPPII inhibitor H-Ala-Ala-Phe-chloromethylketone (AAF-CMK; Sigma-Aldrich) was dissolved in DMSO, whereas butabindide (Tocris Cookson, Bristol, UK) was diluted in sterile water and used at the concentrations indicated. Appropriate solvent controls (DMSO or water) were included in all experiments.
Immunological methods
Immunoblot analysis was performed as described previously.13 Antibodies used were directed against c-MYC (9E10; Covance, Princeton, NJ), TPPII (Proteintech Group, Chicago, IL), or β-actin (Millipore/Chemicon, Billerica, MA). For immunofluorescence analysis, cells were either grown on coverslips, or cytospin preparations were made from cells growing in suspension. For visualization of centrosomes, cells were fixed in 4% paraformaldehyde in PBS and permeabilized with 1% Triton-X 100 in PBS for 10 minutes each at room temperature. Cells were then blocked with 10% normal donkey serum (Jackson Immunoresearch, West Grove, PA) and incubated with a mouse monoclonal anti-γ-tubulin antibody (GTU-88; Sigma-Aldrich) at a 1:1,000 dilution overnight at 4°C followed by a donkey anti-mouse rhodamine red–conjugated secondary antibody (Jackson Immunoresearch) at a 1:100 dilution in PBS. For visualization of centrioles, cells were stained with an anti-centrin mouse monoclonal antibody (Abcam, Cambridge, MA) at a 1:200 dilution for 45 minutes at 37°C, followed by a donkey anti-mouse FITC-labeled secondary antibody (Invitrogen, Carlsbad, CA) at a 1:500 dilution. For visualization of TPPII, cells were pre-extracted with 1% Triton-X 100 in PBS for 5 minutes, fixed in 4% paraformaldehyde/PBS for 10 minutes at room temperature, and rinsed 3 times in PBS. Cells were then blocked as described above and incubated with an anti-TPP2 antibody at a 1:500 dilution (Proteintech Group) overnight at 37ºC. After 3 rinses in PBS, cells were incubated with secondary anti-rabbit antibody at a 1:1,000 dilution (Jackson Immunoresearch) for 1 hour at 37ºC and subsequently rinsed 3 times with PBS and stained with 4′,6′-diamidino-2-phenylindole (DAPI; Vector, Burlingame, CA). Cells were analyzed using a Leica (Wetzlar, Germany) or Olympus (Tokyo, Japan) epifluorescence microscope equipped with a digital camera system. Pictures were transferred to Adobe Photoshop (San Jose, CA) for printout.
siRNA and shRNA
U-2 OS cells were grown on coverslips to 60% confluence and transfected with the indicated plasmids (c-MYC or TPP2) together with siRNA duplexes targeting TPPII or a shRNA plasmid targeting TPPII (Thermo Scientific, Auburn, AL) and a DsRed-encoding plasmid as transfection marker using FuGENE 6 Transfection Reagent (Roche, Mannheim, Germany). Herring sperm DNA was used to normalize the total quantity of DNA transfected. After 24 hours, cells were stained for centrioles, and the percentage of cells displaying supernumerary centrioles was assessed.
Flow cytometry
DNA content of cells was analyzed by propidium iodide staining followed by flow cytometry (FACSCalibur; Becton Dickinson, Franklin Lakes, NJ). Data were acquired and analyzed using the CellQuest software (Becton Dickinson).
Statistical analysis
Statistical significance was assessed using the 2-tailed Student t test for independent samples.
Acknowledgments
The authors are grateful to Michel Bornens, Elliott Kieff, and Philip Leder for sharing important reagents. They also thank Alyce Chen for critical reading of the paper.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by PHS grant R01 CA112598 and a Research Scholar Grant from the American Cancer Society (to S. Duensing), PHS grant R01 CA066980 and a grant from AstraZeneca (to K. Münger), PHS grant R01 CA118880 and grants from Gabrielle’s Angel Foundation for Cancer Research and The Pittsburgh Foundation (to A.G. Brickner), a grant from the Deutsche Forschungsgemeinschaft (to N. Melquiot), and grants from the Studienstiftung des Deutschen Volkes and the Biomedical Exchange Program of the International Academy of Life Sciences (to S. Darr).
References
- 1. Soucek L, Evan GI. The ups and downs of Myc biology. Curr Opin Genet Dev. 2010;20:91-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mai S, Fluri M, Siwarski D, Huppi K. Genomic instability in MycER-activated Rat1A-MycER cells. Chromosome Res. 1996;4:365-71 [DOI] [PubMed] [Google Scholar]
- 3. Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031-44 [DOI] [PubMed] [Google Scholar]
- 5. Yin XY, Grove L, Datta NS, Long MW, Prochownik EV. C-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene. 1999;18:1177-84 [DOI] [PubMed] [Google Scholar]
- 6. Korzeniewski N, Zheng L, Cuevas R, et al. Cullin 1 functions as a centrosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res. 2009;69:6668-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duensing A, Spardy N, Chatterjee P, et al. Centrosome overduplication, chromosomal instability, and human papillomavirus oncoproteins. Environ Mol Mutagen. 2009;50:741-7 [DOI] [PubMed] [Google Scholar]
- 8. Slack AD, Chen Z, Ludwig AD, Hicks J, Shohet JM. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007;67:2448-55 [DOI] [PubMed] [Google Scholar]
- 9. Hasskarl J, Duensing S, Manuel E, Munger K. The helix-loop-helix protein ID1 localizes to centrosomes and rapidly induces abnormal centrosome numbers. Oncogene. 2004;23:1930-8 [DOI] [PubMed] [Google Scholar]
- 10. Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci. 2007;120:2139-42 [DOI] [PubMed] [Google Scholar]
- 11. Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17:215-21 [DOI] [PubMed] [Google Scholar]
- 12. Strnad P, Gonczy P. Mechanisms of procentriole formation. Trends Cell Biol. 2008;18:389-96 [DOI] [PubMed] [Google Scholar]
- 13. Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26:6280-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gonczy P. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell. 2007;13:203-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140-6 [DOI] [PubMed] [Google Scholar]
- 16. Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. Plk4-induced centriole biogenesis in human cells. Dev Cell. 2007;13:190-202 [DOI] [PubMed] [Google Scholar]
- 17. Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol. 2005;15:2199-207 [DOI] [PubMed] [Google Scholar]
- 18. Cunha-Ferreira I, Rodrigues-Martins A, Bento I, et al. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr Biol. 2009;19:43-9 [DOI] [PubMed] [Google Scholar]
- 19. Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Activity and regulation of the centrosome-associated proteasome. J Biol Chem. 2000;275:409-13 [DOI] [PubMed] [Google Scholar]
- 20. Wigley WC, Fabunmi RP, Lee MG, et al. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsou MF, Stearns T. Mechanism limiting centrosome duplication to once per cell cycle. Nature. 2006;442:947-51 [DOI] [PubMed] [Google Scholar]
- 22. Hung CF, Cheng WF, He L, et al. Enhancing major histocompatibility complex class I antigen presentation by targeting antigen to centrosomes. Cancer Res. 2003;63:2393-8 [PubMed] [Google Scholar]
- 23. Kawahara M, York IA, Hearn A, Farfan D, Rock KL. Analysis of the role of tripeptidyl peptidase II in MHC class I antigen presentation in vivo. J Immunol. 2009;183:6069-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guil S, Rodriguez-Castro M, Aguilar F, Villasevil EM, Anton LC, Del Val M. Need for tripeptidyl-peptidase II in major histocompatibility complex class I viral antigen processing when proteasomes are detrimental. J Biol Chem. 2006;281:39925-34 [DOI] [PubMed] [Google Scholar]
- 25. York IA, Bhutani N, Zendzian S, Goldberg AL, Rock KL. Tripeptidyl peptidase II is the major peptidase needed to trim long antigenic precursors, but is not required for most MHC class I antigen presentation. J Immunol. 2006;177:1434-43 [DOI] [PubMed] [Google Scholar]
- 26. Chuang CK, Rockel B, Seyit G, et al. Hybrid molecular structure of the giant protease tripeptidyl peptidase II. Nat Struct Mol Biol. 2010;17:990-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gavioli R, Frisan T, Vertuani S, Bornkamm GW, Masucci MG. c-myc overexpression activates alternative pathways for intracellular proteolysis in lymphoma cells. Nat Cell Biol. 2001;3:283-8 [DOI] [PubMed] [Google Scholar]
- 28. Duensing S, Lee BH, Dal Cin P, Munger K. Excessive centrosome abnormalities without ongoing numerical chromosome instability in a Burkitt’s lymphoma. Mol Cancer. 2003;2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rose C, Vargas F, Facchinetti P, et al. Characterization and inhibition of a cholecystokinin-inactivating serine peptidase. Nature. 1996;380:403-9 [DOI] [PubMed] [Google Scholar]
- 30. Geier E, Pfeifer G, Wilm M, et al. A giant protease with potential to substitute for some functions of the proteasome. Science. 1999;283:978-81 [DOI] [PubMed] [Google Scholar]
- 31. Glas R, Bogyo M, McMaster JS, Gaczynska M, Ploegh HL. A proteolytic system that compensates for loss of proteasome function. Nature. 1998;392:618-22 [DOI] [PubMed] [Google Scholar]
- 32. Stavropoulou V, Xie J, Henriksson M, Tomkinson B, Imreh S, Masucci MG. Mitotic infidelity and centrosome duplication errors in cells overexpressing tripeptidyl-peptidase II. Cancer Res. 2005;65:1361-8 [DOI] [PubMed] [Google Scholar]
- 33. Wang EW, Kessler BM, Borodovsky A, et al. Integration of the ubiquitin-proteasome pathway with a cytosolic oligopeptidase activity. Proc Natl Acad Sci U S A. 2000;97:9990-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rapoport AP, Simons-Evelyn M, Chen T, et al. Flavopiridol induces apoptosis and caspase-3 activation of a newly characterized Burkitt’s lymphoma cell line containing mutant p53 genes. Blood Cells Mol Dis. 2001;27:610-24 [DOI] [PubMed] [Google Scholar]
- 35. Princiotta MF, Schubert U, Chen W, et al. Cells adapted to the proteasome inhibitor 4-hydroxy-5-iodo-3-nitrophenylacetyl-Leu-Leu-leucinal-vinyl sulfone require enzymatically active proteasomes for continued survival. Proc Natl Acad Sci U S A. 2001;98:513-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. Totowa, NJ: Humana Press; 2005 [Google Scholar]
- 37. Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M. The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol. 2000;149:317-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075-82 [PubMed] [Google Scholar]
- 39. Duensing S, Lee LY, Duensing A, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002-7 [DOI] [PMC free article] [PubMed] [Google Scholar]