

Abstract
MCC is a potential tumor suppressor gene, which is silenced by promoter hypermethylation in a subset of colorectal cancers. However, its functions have remained poorly understood. In the present study, we describe a novel function of MCC in the DNA damage response. Several novel phosphorylation sites were identified by mass spectrometry, including 2 highly conserved ATM/ATR consensus sites at serine 118 and serine 120. In addition, exposure to ultraviolet radiation (UV), but not phleomycin, caused PI3K-dependent phosphorylation of MCC and its nuclear localization. Re-expression of MCC in HCT15 colorectal cancer cells led to a G2/M arrest, and MCC knockdown impaired the induction of a G2/M arrest following UV radiation. Finally, mutation of S118/120 to alanine did not affect MCC nuclear shuttling following UV but did impair MCC G2/M checkpoint activity. Thus, these results suggest that MCC is a novel target of the DNA damage checkpoint and that MCC is required for the complete cell cycle arrest in the G2/M phase in response to UV.
Keywords: colorectal cancer, DNA damage response, phosphorylation, G2/M checkpoint
Introduction
We have previously shown that MCC is silenced by promoter hypermethylation in a subset of colorectal cancers (CRC).1 MCC methylation occurs early in premalignant polyps and is particularly frequent in serrated polyps, which are thought to be the precursors of cancer in the putative serrated neoplasia pathway.1,2 Mutated MCC was recently identified as one of the “driver” genes of carcinogenesis in a mouse model of CRC using random transposon integration.3 Therefore, MCC is a putative tumor suppressor gene in CRC. Little is known about the function of MCC and more importantly how its loss of function leads to CRC tumorigenesis.
To maintain genomic integrity, eukaryotic cells activate an evolutionarily conserved set of checkpoint proteins that rapidly induce cell cycle arrest to prevent replication of damaged DNA. Consequently, defects in the DNA damage response are characteristic of many cancers, including CRC. The DNA damage checkpoints arrest the cell cycle either in G1 before DNA replication, S phase, or in G2 before mitosis. This allows repair of the damaged DNA prior to resumption of cell cycle progression.
The ATM/ATR/DNA-PK kinases are key components of DNA damage checkpoints. These proteins are members of the phosphatidylinositol-3 kinase–related kinase (PI3K) family and phosphorylate serine or threonine residues that are followed by a glutamine SQ/TQ. As such, ATM and ATR are often referred to as Ser/Thr-Gln–directed kinases. Unlike ATM, deletion of ATR in mice results in an embryonic lethal phenotype, indicating that ATR is an essential gene. Another feature that distinguishes these 2 kinases is their sensitivity to different types of mutagens. Cells lacking ATM are hypersensitive to ionizing radiation (IR) but not to UV radiation or hydroxyurea (HU), whereas cells overexpressing a kinase-inactive form of ATR are sensitive to UV and HU, as well as to IR.4,5
In initial mass spectrometry studies, we discovered that MCC is phosphorylated on serine 118 and serine 120, part of 2 ATM/ATR consensus sites. This finding led us to study the role of MCC in the DNA damage response. In this study, we used UV radiation of CRC cell lines as a model of DNA damage. UV mainly induces single-strand DNA breaks (SSB), while IR induces both SSB and double-strand breaks (DSB). Both types of DNA damage are relevant in colorectal carcinogenesis and its treatment. SSBs are more common in all cells, including in the colon, and are caused by spontaneous DNA decay or oxidative attack by reactive oxygen species (ROS) from intracellular metabolites.6 SSBs are found in the healthy colon but are increased with inflammation or certain dietary agents.7,8 Therefore, efficient repair of both types of DNA damage is important in the protection against proliferation of damaged cells. On the other hand, impaired DNA damage response is a major determinant of radiotherapy and cytotoxic drug responsiveness of cancer cells.9 Therefore, cells with defective repair of either SSBs or DSBs are potentially more efficiently eliminated through apoptosis by these treatments.
Results
MCC is phosphorylated on serines 115, 118, and 120 in colon cancer cells
To provide some insights into MCC regulation and function, we first set out to map the specific phosphosites in the human MCC protein. FLAG-tagged MCC was ectopically expressed in HCT15 colon cancer cells, where MCC is silenced due to promoter hypermethylation,1 before being immunoprecipitated and processed using LC-MS/MS. Several tryptic phosphopeptides were identified including pSELSQSQHEVNEDSR, SELpSQSQHEVNEDSR, and SELSQpSQHEVNEDSR (Fig. 1A and Suppl. Fig. S1). The probability of localization (PL) as calculated by MaxQuant was as follows: 80.7%, S115 (S(0.807)ELS(0.191)QS(0.002)QHEVNEDSR); 85.5%, S118 (S(0.007)ELS(0.855)QS(0.139)QHEVNEDSR); and 99.5%, S120 (SELS(0.004)QS(0.995)QHEVNEDSR).
Figure 1.
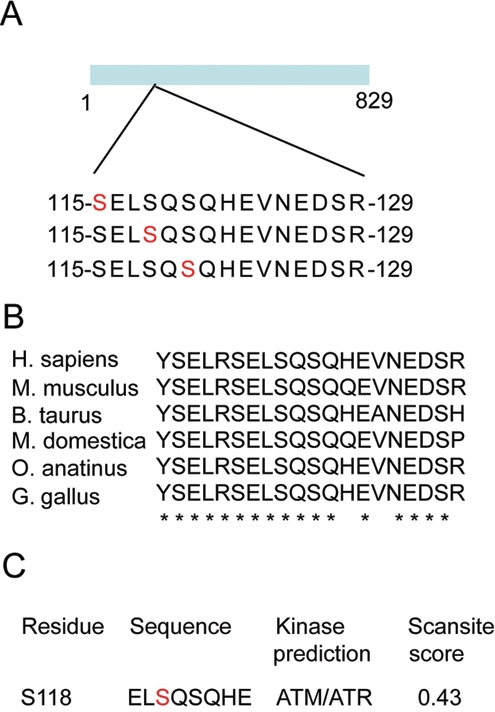
MCC is phosphorylated at the ATM/ATR consensus sites Ser118 and Ser120. (A) FLAG-tagged MCC was overexpressed in HCT15 and immunoprecipitated using M2-FLAG antibody. Endogenous MCC was immunoprecipitated from HCT116 cells using N-terminal mouse monoclonal antibody. MCC was excised from SDS gel and digested with trypsin, and peptides were analyzed by LC/MS/MS. The search was performed using Mascot software. (B) Sequence alignment of the tryptic phoshopeptide from a human, mouse, cow, opossum, platypus, and chicken. Identical residues are marked with a star. (C) Scansite search at high stringency predicts ATM/ATR kinases as the most likely kinases to phosphorylate serine at position 118.10
These serines were also phosphorylated on endogenous MCC from asynchronous HCT116 colon cancer cells. Serine residues 115, 118, and 120 and the adjacent sequences are strongly conserved in MCC among species, suggesting that these regions are functionally important (Fig. 1B). Using Scansite (Scansite 2.0, MIT, Cambridge, MA, USA) at high stringency, the serine at position 118 is predicted to be phosphorylated with a score of 0.43 by ATM/ATR kinase (Fig. 1C).10 The preferred phosphorylation motif for ATM/ATR has been determined.11 The consensus is Ψ-S/T-Q, where Ψ is a hydrophobic residue, which is in agreement with the L-pS-Q phosphosite found at position 118 in MCC. In addition, our mass spectrometry data revealed that this tryptic phosphopeptide can also be doubly phosphorylated at serine 120 (PL:1) and at serine 115 (PL:0.5) or 118 (PL:0.5). The equal probability of localization for serine 115 and serine 118 did not allow unambiguous assignment. ATM/ATR substrates commonly contain multiple phosphorylation sites known as SQ/TQ cluster domains (SCD).12 SCDs are thought to be advantageous for efficient phosphorylation by ATM/ATR kinases and to allow various degrees of activation.
MCC is phosphorylated following UV radiation of colon cancer cells
Western blots of endogenous MCC show 2 closely migrating bands. Disappearance of the upper band after pretreatment with lambda phosphatase indicated that the slow migrating band (upper band) is a phosphorylated form of MCC (Fig. 2A). We next investigated whether the phosphorylation-dependent shift in MCC electrophoretic mobility could be induced by DNA-damaging agents that activate ATM or ATR.
Figure 2.
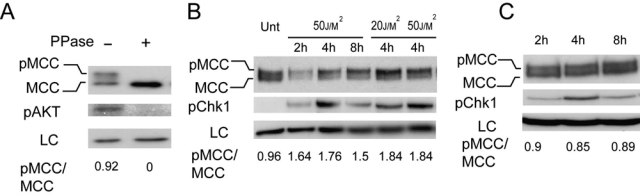
Exposure to UV but not phleomycin caused phosphorylation of MCC as shown by electrophoretic mobility shift. (A) Whole-cell lysate from HCT116 was untreated or lambda phosphatase treated for 1 hour before being run on 8% SDS gel and blotted for MCC, phospho-AKT, and GAPDH (LC) antibodies. (B) HCT116 cells were mock treated or UV irradiated at different doses and harvested after recovery for the indicated times. Lysates were run on 8% SDS gel and blotted for MCC, phospho-Chk1, and GAPDH (LC) antibodies. (C) HCT116 cells were treated with 100 ng/uL phleomycin and let to recover for the indicated amount of time before lysates were collected. Lysates were run on 8% SDS gel and blotted for MCC, phospho-Chk1, and GAPDH (LC) antibodies.
We observed that endogenous MCC was hyperphosphorylated following UV treatment but not by phleomycin-induced double-strand breaks in HCT116 cells (Fig. 2B and 2C) and SW620 cells (Suppl. Fig. S2A), with the ratio between phosphorylated and unphosphorylated MCC increasing 2-fold. Increased phosphorylation was observed as early as 30 minutes following UV treatment and remained strong for up to 8 hours (Fig. 2B and data not shown). This early activation is similar in timing to that observed for Chk1 phosphorylation, but MCC remained hyperphosphorylated for up to 8 hours after UV compared to 4 hours for Chk1 (Fig. 2B).
To initially test whether phosphorylation by ATM/ATR/DNA-PK kinases might account for the shift in MCC electrophoretic mobility, we pretreated the cells with wortmannin, a PI3K inhibitor. Wortmannin prevented MCC hyperphosphorylation following UV radiation (Fig. 3A). Since the major PI3K that mediates the cellular response to UV is ATR,13 whereas phleomycin principally activates ATM, this suggests that MCC may be preferentially targeted by ATR. Consistent with this conclusion, the peptide “E-L-S-Q-S-Q” from the 53BP1 protein (aa174-179), identical in sequence to MCC (aa116-121), is phosphorylated by the ATR kinase following UV treatment (Fig. 3B).14
Figure 3.
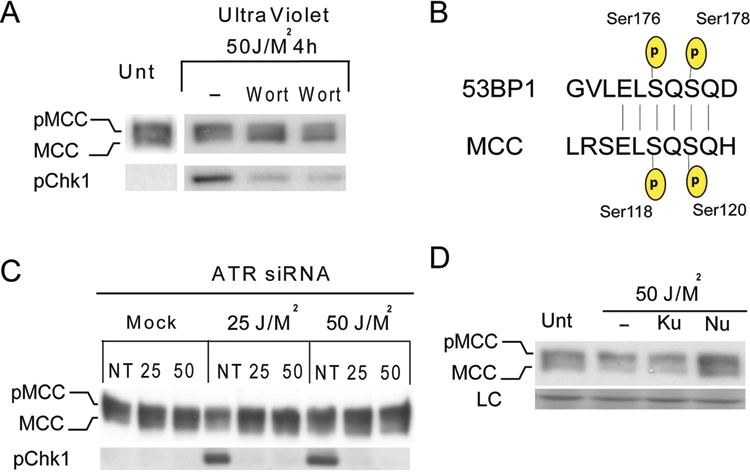
MCC is phosphorylated by ATR and DNA-PK in HCT116 cells following UV treatment. (A) HCT116 cells were untreated or preincubated with PI3K inhibitor wortmannin 1 hour prior to UV treatment. Lysates were collected 4 hours after UV radiation and run on 8% SDS gel before being blotted for MCC and phospho-Chk1. (B) Sequence similarity search found 53BP1 ATR and ATM-mediated phosphorylation sites (aa174-179) to be identical to MCC.14 (C) HCT116 cells were transfected with nontargeting siRNA or ATR siRNA for 48 hours before being mock or UV treated. Cells were allowed to recover for 4 hours, and lysates were collected and run on 8% SDS gel and blotted for MCC and phospho-Chk1 antibody. (D) HCT116 cells were incubated with DMSO (0.1% v/v) or with KU55933 (10 uM) or NU7041 (10 uM) for 1 hour before exposure to UV light (50 J/M2). Cells were allowed to recover for 4 hours, and lysates were collected and run on 8% SDS gel.
There are no specific inhibitors of ATR currently available. Therefore, to test whether ATR is necessary for UV-induced MCC phosphorylation, we treated HCT116 cells with ATR siRNA 48 hours prior to UV treatment. Chk1 phosphorylation was used as readout of ATR kinase activity.15 Functional ATR downregulation was confirmed by diminished Chk1 phosphorylation after UV treatment in the presence of ATR siRNA (Fig. 3C). CHK1 phosphorylation following UV was reduced by more than 90% in cells treated with ATR siRNA compared to nontargeting siRNA (NT) (Fig. 3C). However, the shift in MCC mobility was only partially reduced. This suggests that ATR does phosphorylate MCC following UV, but other PI3K-kinases might also be involved.
To test whether ATM or DNA-PK might also phosphorylate MCC following UV, we used a series of specific inhibitors. Partial reduction in UV-induced phosphorylation was observed when the cells were pretreated with NU7441, a specific inhibitor of DNA-PK, but not in the presence of KU55933, a specific inhibitor of ATM (Fig. 3D).16 Taken together, these results suggest that both ATR and DNA-PK contribute to MCC phosphorylation following UV treatment, but the involvement of other kinases could not be excluded.
MCC localizes to the nucleus following UV radiation
As the sensing of DNA damage occurs in the nucleus, we investigated MCC cellular localization before and after UV treatment. Previous work reported that MCC is present in the cytoplasm, and the nucleus and several potential nuclear localization sequences (NLS) have been described (Fig. 4A).17 Confocal microscopy showed that in untreated HCT116 cells, MCC is mainly cytoplasmic, with only 5% to 8% of the cells showing nuclear staining (Fig. 4B and 4C). This finding is consistent with a previous report in which MCC is predominantly detected in the cytoplasm (Fig. 6A).17 We next assessed whether MCC relocalizes to the nucleus following UV irradiation. Four hours after UV treatment, 60% to 80% of the cells showed strong nuclear staining compared to 5% to 8% in untreated cells, indicating that MCC relocalizes to the nucleus following DNA damage (Fig. 4B and 4C). UV-induced MCC nuclear shuttling was also observed by Western blot (Fig. 4D). Site-directed mutagenesis of serine 115, 118, and 120 to alanine did not prevent UV-induced nuclear localization, suggesting that phosphorylation at these sites is not required for MCC nuclear shuttling (data not shown).
Figure 4.
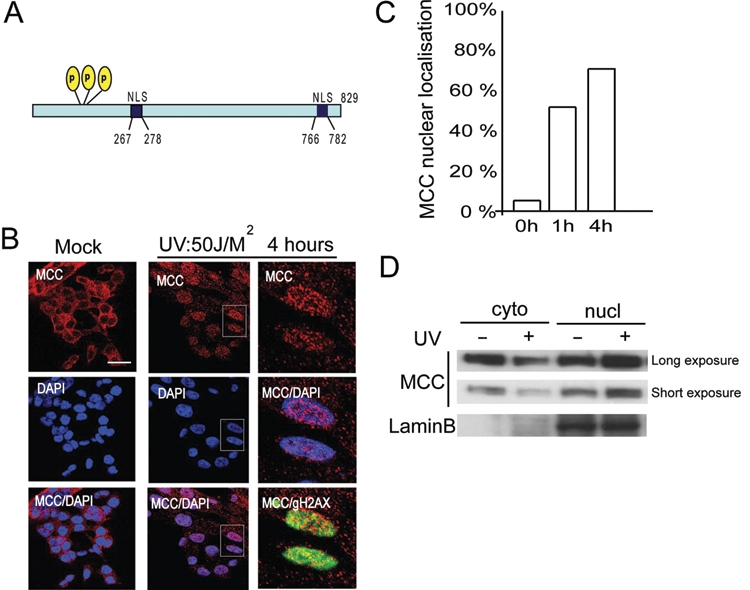
Following UV-induced DNA damage, MCC relocalizes to the nucleus. (A) Schematic representation of MCC, indicating the 3 identified phosphorylation sites ser115, ser118, and ser120 and 2 predicted NLS domains. (B) HCT116 cells were untreated or UV radiated at 50 J/M2 and let to recover for 4 hours before being fixed. Cells were permeabilized and stained for MCC, γH2AX, and DAPI. Right panels show a zoom from the UV-treated panel (middle). MCC signal is merged with DAPI, showing MCC nuclear localization or γH2AX(S139) site of DNA damage (bar: 15 µm). (C) HCT116 cells were untreated or UV radiated at 50 J/M2 and let to recover for 1 or 4 hours before being fixed. Cells were permeabilized and stained for MCC and DAPI. Cells were counted for MCC nuclear or cytoplasmic localization in mock (0 hours), 1, and 4 hours postirradiation, respectively. (D) HCT116 cells were UV treated at 50 J/M2 and let to recover for 4 hours before cytoplasmic and nuclear fractions were harvested and run on an SDS gel. Long and short exposure of MCC is shown, and lamin B is used as a marker of the nuclear fraction.
Figure 6.
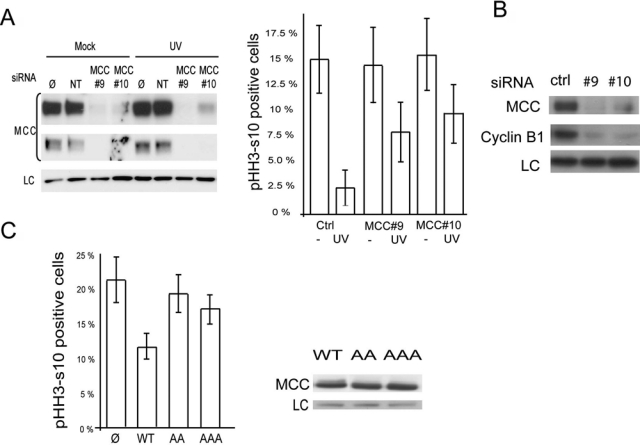
MCC knockdown impairs UV-induced G2/M arrest. (A) G2/M checkpoint arrest was monitored using the mitotic index.19 HCT116 cells were mock-transfected (Ø), nontargeting (NT), or MCC siRNA (#9 or #10) for 24 hours. Cells were untreated or irradiated with 2.5 J/M2 and incubated for 24 hours in the presence of 1.5 µM nocodazole, and the number of mitotic cells was assessed by counting pHH3-Ser10–positive cells. Two independent experiments with a minimum of 500 cells were counted per condition. The average of the 2 experiments is represented. Knockdown of MCC in mock or UV-treated cells was validated by Western blot. (B) Lysates of UV-treated HCT116 transfected with NT and MCC siRNA #9 or #10 were run on a gel and blotted for MCC, cyclin B1, and GAPDH (LC). (C) Cells expressing MCC Ser118A/120A shows impaired G2/M arrest following UV treatment compared to wild type. HCT15 cells were transfected with empty vector, MCC wild type, or MCC-bearing serine to alanine substitution at position 118-120 (AA) and 115-118-120 (AAA). The 48-hour posttransfection cells were irradiated with 2.5 J/M2 and incubated for 24 hours in the presence of 1.5 µM nocodazole, and the number of mitotic cells was assessed by counting MCC-positive and pHH3-positive cells. The average of 2 experiments is represented. The relative expression of MCC wild type, AA, and AAA is shown by Western blot.
To gain mechanistic insight into the role of MCC in the DNA damage response, we next assessed whether MCC is directly involved in DNA damage signaling by monitoring γH2AX formation following UV. We depleted MCC using 2 targeted siRNAs (referred to as #9 and #10) and assessed the intensity of DNA damage using γH2AX following UV treatment. γH2AX staining was strongly increased following UV treatment but was not noticeably changed in MCC knockdown cells compared to control siRNA, suggesting that MCC is not involved in early DNA damage recognition (Suppl. Fig. S3A). In addition, we did not observe any change in Chk1 phosphorylation following UV when MCC was knocked down or overexpressed (Suppl. Fig. S3B and S3C).
Ectopic expression of MCC induces G1/S and G2/M arrest
There is strong evidence suggesting that ATR controls cell cycle progression through phosphorylation of many proteins involved in DNA replication such as histone H2AX, RPA2, and Mcm2.18,19 ATR-mediated phosphorylation of CHK1 upon DNA damage, as well as DNA-PK activation, has also been shown to be critical for the G2/M checkpoint.15,20 It has also been suggested that MCC has a role in cell cycle progression.17,21 Therefore, we examined the effect of MCC re-expression on cell cycle progression in HCT15 cells. In agreement with previous reports describing the cell cycle inhibitory function of MCC, we found that re-expression of MCC, to a level similar to that of endogenous MCC in LIM1215 cells, strongly reduced the rate of proliferation of 2 separate HCT15 clonal cell lines compared with empty vector transfected or parental cells (Fig. 5A and 5C).
Figure 5.
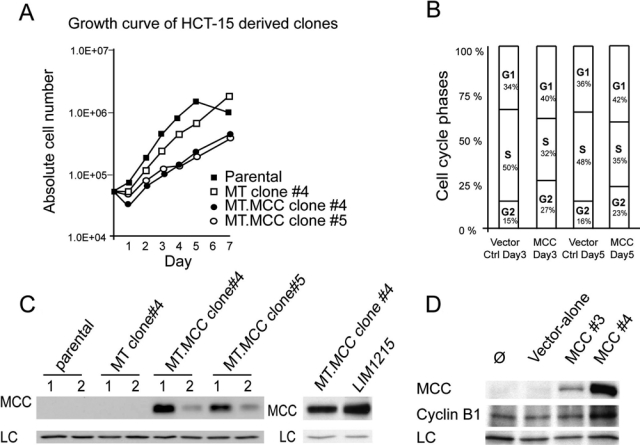
Re-expression of MCC in HCT15 cells slows proliferation rate and induces G1/S and G2/M arrest. (A) Stable MCC-expressing and control HCT15-derived clones were generated using the pDEST-MT vector system. HCT15 stably expressing MCC cells and control cells were seeded at 5.104 cells at day 0. Cells were harvested and counted in triplicate over a 7-day time course. Graphed values are the average number of cells counted ± SEM. (B) FACS analysis was performed at day 3 and day 5 of the growth curve. (C) Western blot shows MCC expression in the stable cell lines after 1 passage (1) and 2 passages (2). (D) Total lysates of cells harvested at day 5 were blotted for MCC, cyclin B1, and GAPDH (LC).
DNA analyses by flow cytometry on MCC-expressing HCT15 clones indicated an increase in the percentage of cells in G1-phase and G2/M-phase and decrease in the percentage of cells in S-phase (Fig. 5B), indicating effects in both G1 and G2/M. These results are consistent with Matsumine et al.,21 who showed that MCC overexpression in NIH3T3 cells caused cell cycle arrest at the G1/S transition. However, our findings also suggest an additional role at the G2/M transition. Accumulation of cells at the G2/M transition following MCC re-expression is also supported by an increase in cyclin B, as observed by Western blot (Fig. 5D). We also observed that after several passages, the level of MCC expression was strongly reduced in MCC-expressing HCT15 clones, and the proliferation rate was restored to a similar level to the parental nonexpressing cell line (Fig. 5C and data not shown), suggesting selection against MCC expression in these cultures.
Depletion of MCC impairs cell cycle arrest following UV irradiation
Based on the observed role of MCC at the G1/S and G2/M transition, we next assessed whether MCC is required for UV-induced cell cycle arrest in HCT116 cells. We used phosphohistone H3-s10 (pHH3-S10) as a marker of mitosis to address the role of MCC in the irradiation-induced G2 checkpoint delay.22 We depleted MCC using 2 targeted siRNAs (referred as #9 and #10) and scored the percentage of mitotic cells (pHH3-S10 positive) compared to control siRNA before and after UV irradiation, as previously described.19,23 We obtained 90% and 85% MCC knockdown efficiency with siRNA #9 and #10, respectively (Fig. 6A). Consistent with the decrease in MCC expression observed by Western blot, indirect immunofluorescence using anti-MCC antibody showed significant loss of MCC signal intensity in cells treated with MCC siRNA compared to control siRNA (Suppl. Fig. S3A). UV treatment in MCC-expressing cells reduced the percentage of pHH3-S10–positive cells from 15% to 2.4%, indicative of a proficient cell cycle arrest. Knockdown of MCC had no detectable effect on the number of pHH3-S10–positive cells in nonirradiated cells (Fig. 6A). However, following UV irradiation, the percentage of pHH3-positive cells only decreased to 7.7% and 9.2% in cells transfected with MCC-siRNA #9 and MCC-siRNA #10, respectively, compared to 2.4% in control cells (Fig. 6A). In addition, the level of cyclin B1, a G2/M marker, was reduced in cells treated with MCC siRNA following UV treatment compared to nontargeting control siRNA (Fig. 6B).
Re-expression of MCC phosphomutants does not arrest the cell cycle as efficiently as MCC wild type following UV radiation
We next assessed whether increased MCC expression affected G2 arrest after UV treatment and also whether phosphorylation at ser115, ser118, and ser120 was required. To test this, we ectopically expressed MCC-wild-type (WT) or MCC-bearing alanine substitution at ser118 and ser120, referred to as AA, or triple substitution of ser115, 118, and 120, referred to as AAA in HCT15 cells, which lack endogenous MCC expression. Cells were then UV treated and allowed to recover for 24 hours in the presence of nocodazole before being fixed and stained for the pHH3-S10 mitotic marker (Fig. 6C). More than 20% of the mock-untransfected cells remained in M phase following UV treatment as expected from their lack of MCC expression. However, expression of WT MCC efficiently prevented cells entering mitosis compared to untransfected HCT15 cells, with MCC reducing the number of pHH3-positive cells by almost half (21% to 12%). In comparison, the number of cells entering mitosis was only partially decreased from 21% to 19% and 17% for MCC mutants AA and AAA, respectively. MCC phosphomutants were approximately 60% to 70% less effective at blocking the cell cycle prior to mitotic entry following UV compared with cells expressing WT MCC, despite similar levels of expression between WT MCC and the phosphomutants (Fig. 6C).
Discussion
The ability to sense and respond to DNA damage is critical to the maintenance of genomic stability and the prevention of cancer. Our data show that MCC is a novel target of the DNA damage checkpoint and that depletion of MCC impairs UV-induced cell cycle arrest. In this study, we initially used a mass spectrometry phosphomapping strategy to unveil potential new functions of MCC.
Among the residues being phosphorylated, we identified 2 candidate ATM/ATR/DNA-PK phosphorylation sites (S/T-Q), which led us to investigate the potential role of MCC in the DNA damage response. In agreement with in silico prediction, we found that MCC is phosphorylated following UV radiation in a PI3K-dependent manner. Knockdown and specific inhibitor assays suggest that ATR and DNA-PK are the main kinases to phosphorylate MCC following UV. Intriguingly, 53BP1, a p53 binding partner and a major regulator of genome stability and DNA damage repair,24 has previously been shown to be phosphorylated by ATR following UV radiation on an identical phosphosite (E-L-S-Q-S-Q).14 This raises the possibility that MCC might also be involved in DNA damage repair. Chk1 and H2AX phosphorylation are immediate early events after DNA damage that trigger DNA repair processes. Our data suggest that Chk1 and H2AX phosphorylation at ser139 are not dependent on MCC, as we did not observe a reduction of γH2AX staining intensity or changes in the amount of Chk1 phosphorylation following UV in MCC-depleted cells. However, our data do not exclude the possibility that MCC functions directly in the repair process downstream of H2AX and Chk1 phosphorylation.
Earlier studies have described MCC as a cell cycle inhibitory protein, and the main function of ATR following DNA damage is to induce cell cycle arrest to allow DNA repair or induce apoptosis.21 Therefore, we next investigated whether MCC is involved in UV-induced cell cycle arrest. Using HCT15 clones stably re-expressing MCC, we first confirmed the cell cycle inhibitory role of MCC. We found that the reduced proliferation was characterized by a G1/S delay and a previously unreported G2/M transition delay. Conversely, we saw a reduced G2/M arrest following UV radiation in MCC-depleted cells. Involvement of MCC in 2 different cell cycle checkpoints may indicate that MCC silencing has multiple effects in colon cancer cells. Defects in both the G1/S and G2/M checkpoints may lead to increased proliferation, while the impairment of G2/M transition may also allow cells with damaged DNA to enter mitosis.
Our data strongly suggest that MCC phosphorylation at the candidate ATM/ATR consensus site is necessary for MCC to induce G2/M arrest. We mutated phosphoserines S115, S118, and S120 to nonphosphorylable alanine and tested the ability of these phosphomutants to block the cells following UV radiation. We found that double (S118A-S120A) and triple (S115A-S118A-S120A) phosphomutants fail to arrest the cell cycle as efficiently as MCC WT in UV-treated cells. Our work shows that MCC phosphorylation at serines 118 and 120 contributes to the DNA damage response following UV treatment. There are 5 other ATM/ATR/DNA-PK consensus sites (S/T-P) present along the MCC protein that could also be targeted by these kinases. Although these sites were not identified as phosphosites by mass spectrometry in the absence of UV treatment, it is possible that they may be phosphorylated after DNA damage.
The mechanism by which MCC mediates its UV-induced G2/M arrest is as yet unknown. However, a recent study showed that MCC is a regulator of the β-catenin pathway.17 In addition to its well-known role as transcriptional coactivator of T-cell factor/lymphocyte enhancer factor-1 (TCF/Lef-1), and cell-cell adhesion,25 β-catenin is also associated with the regulation of the cell cycle. Hence, β-catenin localizes at the centrosome during mitosis and participates in establishing the bipolar spindle.26 In addition, the level of β-catenin, its cellular localization, and its phosphorylation status have been shown to play a role in G2/M arrest.27
MCC is emerging as a multifunctional protein that affects several cellular processes and pathways. In addition to regulating cell proliferation in many cancer cell lines,17,21 it has been suggested that MCC is involved in differentiation,28 epithelial cell migration,29 and inhibition of NFκB activation or Wnt signaling.17,30 Importantly, mutated MCC is now recognized as a driver of carcinogenesis in a mouse model of colon cancer,3 but it is poorly understood how the loss of MCC function promotes carcinogenesis. Our study suggests that MCC has a novel function in the DNA damage response at the G2/M checkpoint, which could be important in the malignant progression of polyps with silenced MCC. Impaired DNA damage response is also a major determinant of radiotherapy and cytotoxic drug responsiveness. A subset of rectal cancers respond remarkably well to preoperative radiotherapy, but the molecular basis of this is unknown. Therefore, it should be investigated whether MCC silencing in primary cancers makes them more responsive to radiotherapy or chemotherapy treatment.
In summary, we have shown that MCC relocalizes to the nucleus and is phosphorylated in response to DNA damage in a PI3K-dependent manner. We have also identified 2 phosphorylation sites required for the full induction of UV-induced cell cycle arrest. We propose a model in which following UV radiation, MCC relocalizes to the nucleus, where it is phosphorylated by ATR and DNA-PK, most likely at serines 118 and 120. We have also identified a new cell cycle function of MCC at the G2/M transition and show that Ser118-120 phosphorylation is required to mediate UV-induced cell cycle arrest. Future work will assess whether MCC silencing is associated with impaired DNA damage response in primary tumors. This could be important in the treatment of CRC as these cancers are likely to be more sensitive to certain chemotherapy drugs and radiotherapy.
Materials and Methods
Cell culture
HCT116, SW620, and HCT15 human colorectal cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and propagated in RPMI 1640 media supplemented with fetal calf serum (10%), HEPES 10 mM, insulin 0.3 U/mL, 100 U/mL penicillin, and 100 ug/mL streptomycin.
Antibodies
The following antibodies were used: anti-MCC (610740, BD Transduction Laboratories, Franklin Lakes, NJ, USA), anti-FLAG (M2, Sigma-Aldrich, St. Louis, MO, USA), anti-phosphorylated-AKT (#9271, Cell Signaling Technology, Danvers, MA, USA), anti-phosphorylated-Chk1 (S345 #2341, Cell Signaling Technology), anti-phosphorylated-γH2AX (S139, JBW103, Upstate Biotechnology, Waltham, MA, USA), anti-cyclin B1 (#4135, Cell Signaling Technology), anti-phosphorylated-histone (H3-S10, H0412, Sigma-Aldrich), GAPDH (4300, Ambion, Austin, TX, USA), horseradish peroxidase-conjugated anti-mouse, or anti-rabbit IgG (Amersham, GE Healthcare, Rydalmere, Australia).
siRNA and transfection
MCC siRNA ON-target plus (#9, J-010523-09; and #10, J-010523-10) (Dharmacon, Lafayette, CO, USA) were added at a final concentration of 5 nM. ATR siRNA (J-003203-19, J-003203-20, J-003203-21, J-003203-22, Dharmacon) were added at the final concentration of 25 or 50 nM for 48 hours prior to UV treatment. Nontargeting #2 control siRNA (D-001810-02-20) was used as a control. Transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The empty pDEST vector or pDEST containing full-length MCC cDNA (amplified from clone MGC:12731, Invitrogen) was transfected into HCT15 cells, and stable clones were selected with G418. Site-directed mutagenesis was carried out as previously described.31
In-gel protein tryptic digestion, LC-MS/MS
FLAG MCC was overexpressed in HCT15 and immunoprecipitated using FLAG antibody and released from G-beads using competitive FLAG peptide. Alternatively, endogenous MCC was immunoprecipitated from HCT116 using anti-MCC monoclonal antibody (BD Transduction Laboratories). Immunoprecipitated endogenous MCC or overexpressed FLAG MCC was run on 10% polyacylamide gel, and SYPRO-stained (Invitrogen) MCC band was excised and destained in 1 mL of 50% acetonitrile and 250-mm ammonium bicarbonate at room temperature for 45 minutes with shaking. The gel slice was dehydrated by incubation in 1 mL of 100% acetonitrile for 10 minutes at room temperature. All solution was carefully removed prior to the addition of modified trypsin (12.5 ng/µL) in 100-mm NH4HCO3 and incubation overnight at 37°C. Peptides were extracted by the addition of 0.1 mL of 5% formic acid and incubation at 37°C for 1 hour. Peptides were further extracted by the addition of 0.1 mL of 100% acetonitrile and incubation at 37°C for 1 hour. The gel slice was completely dehydrated by the addition of 0.5 mL of 100% acetonitrile and incubation at 37°C for 10 minutes. The entire supernatant was then vacuum dried. The peptides were redissolved in 20 µL of 5% formic acid for LC-MS/MS analysis. Data were processed, searched, and quantified using the MaxQuant software version 1.0.13.13 package (Max Planck Institute of Biochemistry, Martinsried, Germany), and searches were performed using Mascot server version 2.2 (Matrix Science Ltd, London, UK) and against the entire International Protein Index (IPI) database. The settings used for the Mascot search were as follows: 2 missed cleavages were allowed; enzyme was trypsin cleaving after arginine and lysine; variable modifications were methionine oxidation, propionamide cysteine, and phosphorylation of serine, threonine, or tyrosine; no fixed modifications were used; a mass tolerance of 6 ppm was used for precursor ions; and a MS/MS tolerance of 0.5 Da was used for fragment ions. False recovery rate was less than 1%, and localization score cutoff was greater than 75%.
Immunofluorescence microscopy
HCT116 cells were cultured as described above on glass coverslips. Cells were then fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature (RT). The cells were then processed for immunolabeling by permeabilization and labeling in PBS containing 0.5% triton for 5 minutes at RT and 2% BSA using standard procedures. Primary antibodies were detected with Alexa Fluor 488- or Cy3-conjugated secondary antibodies; DNA was stained with 4’,6-diamindino-2-phenylindole (DAPI). Optical sections were analyzed by confocal microscopy on a Leica inverted microscope (Wetzlar, Germany). The contrast was adjusted for all images with the same settings.
Induction of DNA damage and drug treatment
For UV treatment of cells, the media were removed and stored, and cells were irradiated in a UV Stratalinker (254 nm, Stratalinker, Stratagene, La Jolla, CA, USA). Stored media were added back, and cells were left at 37°C to recover for the indicated amount of time before being harvested. Phleomycin (#D1515, Sigma-Aldrich) was added at 100 µg/mL for the indicated amount of time before the cell being harvested. The ATM inhibitor (KU55933) and the DNA-PK inhibitor (NU7441) (Tocris Bioscience, Bristol, UK) were used at 10-µM final concentration, and wortmannin (#W1628, Sigma-Aldrich) was added to the cell at a final concentration of 10 nM 4 hours prior to treatment and left on for the duration of the experiment. Quantitation of Western blot intensity was performed using ImageJ software (ImageJ 1.43, NIH, Bethesda, MD, USA).
Mitotic index
For mitotic index measurements, the cells were treated with or without UV radiation 2.5 J/M2 and incubated for 24 hours in the presence of 1.5 µM nocodazole to trap any cells that had overcome UV radiation–induced G2/M arrest and entered mitosis.19 HCT116 cells were transfected with nontargeting siRNA, MCC siRNA #9, or MCC siRNA #10 and incubated for 48 hours. HCT15 cells were transfected with MCC wild-type or MCC-bearing missense mutation and incubated for 24 hours. Mitotic cells were identified as histone H3-p-Ser10 and MCC-positive cells by immunofluorescence. The graph represents the mean of 2 separate experiments.
Supplementary Material
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The authors thank the Cancer Institute NSW, Cancer Council NSW, the Australian Cancer Research Foundation, and the Ramaciotti Foundation for financial support. M.K.C. and E.A.M. are Cancer Institute NSW Career Development Fellows, and M.K.C. is a recipient of the Cancer Institute NSW Program Grant SCRIPT for Colorectal Cancer.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Kohonen-Corish MR, Sigglekow ND, Susanto J, et al. Promoter methylation of the mutated in colorectal cancer gene is a frequent early event in colorectal cancer. Oncogene. 2007;26:4435-41 [DOI] [PubMed] [Google Scholar]
- 2. Jass JR, Whitehall VL, Young J, Leggett BA. Emerging concepts in colorectal neoplasia. Gastroenterology. 2002;123:862-76 [DOI] [PubMed] [Google Scholar]
- 3. Starr TK, Allaei R, Silverstein KA, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cliby WA, Roberts CJ, Cimprich KA, et al. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wright JA, Keegan KS, Herendeen DR, et al. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci U S A. 1998;95:7445-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619-31 [DOI] [PubMed] [Google Scholar]
- 7. Boland CR, Luciani MG, Gasche C, Goel A. Infection, inflammation, and gastrointestinal cancer. Gut. 2005;54:1321-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Toden S, Bird AR, Topping DL, Conlon MA. High red meat diets induce greater numbers of colonic DNA double-strand breaks than white meat in rats: attenuation by high-amylose maize starch. Carcinogenesis. 2007;28:2355-62 [DOI] [PubMed] [Google Scholar]
- 9. Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008;68:7466-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538-43 [DOI] [PubMed] [Google Scholar]
- 12. Traven A, Heierhorst J. SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays. 2005;27:397-407 [DOI] [PubMed] [Google Scholar]
- 13. Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13:225-31 [DOI] [PubMed] [Google Scholar]
- 14. Jowsey P, Morrice NA, Hastie CJ, McLauchlan H, Toth R, Rouse J. Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair. 2007;6:1536-44 [DOI] [PubMed] [Google Scholar]
- 15. Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen BP, Uematsu N, Kobayashi J, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582-7 [DOI] [PubMed] [Google Scholar]
- 17. Fukuyama R, Niculaita R, Ng KP, et al. Mutated in colorectal cancer, a putative tumor suppressor for serrated colorectal cancer, selectively represses beta-catenin-dependent transcription. Oncogene. 2008;27:6044-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006;281:39517-33 [DOI] [PubMed] [Google Scholar]
- 19. Stiff T, Cerosaletti K, Concannon P, O’Driscoll M, Jeggo PA. Replication independent ATR signalling leads to G2/M arrest requiring Nbs1, 53BP1 and MDC1. Hum Mol Genet. 2008;17:3247-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arlander SJ, Greene BT, Innes CL, Paules RS. DNA protein kinase-dependent G2 checkpoint revealed following knockdown of ataxia-telangiectasia mutated in human mammary epithelial cells. Cancer Res. 2008;68:89-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsumine A, Senda T, Baeg GH, et al. MCC, a cytoplasmic protein that blocks cell cycle progression from the G0/G1 to S phase. J Biol Chem. 1996;271:10341-6 [DOI] [PubMed] [Google Scholar]
- 22. Juan G, Traganos F, James WM, et al. Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry. 1998;32:71-7 [DOI] [PubMed] [Google Scholar]
- 23. Alderton GK, Joenje H, Varon R, Borglum AD, Jeggo PA, O’Driscoll M. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet. 2004;13:3127-38 [DOI] [PubMed] [Google Scholar]
- 24. Fernandez-Capetillo O, Chen HT, Celeste A, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993-7 [DOI] [PubMed] [Google Scholar]
- 25. Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaplan DD, Meigs TE, Kelly P, Casey PJ. Identification of a role for beta-catenin in the establishment of a bipolar mitotic spindle. J Biol Chem. 2004;279:10829-32 [DOI] [PubMed] [Google Scholar]
- 27. Olmeda D, Castel S, Vilaro S, Cano A. Beta-catenin regulation during the cell cycle: implications in G2/M and apoptosis. Mol Biol Cell. 2003;14:2844-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Senda T, Matsumine A, Yanai H, Akiyama T. Localization of MCC (mutated in colorectal cancer) in various tissues of mice and its involvement in cell differentiation. J Histochem Cytochem. 1999;47:1149-58 [DOI] [PubMed] [Google Scholar]
- 29. Arnaud C, Sebbagh M, Nola S, et al. MCC, a new interacting protein for Scrib, is required for cell migration in epithelial cells. FEBS Lett. 2009;583:2326-32 [DOI] [PubMed] [Google Scholar]
- 30. Bouwmeester T, Bauch A, Ruffner H, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97-105 [DOI] [PubMed] [Google Scholar]
- 31. Morris JR, Pangon L, Boutell C, Katagiri T, Keep NH, Solomon E. Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum Mol Genet. 2006;15:599-606 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.