

Abstract
Rhabdomyosarcoma (RMS) is the most frequent soft-tissue sarcoma in children. Embryonal rhabdomyosarcoma (E-RMS) represents the most common RMS subtype, but the molecular events driving this tumor are still largely unknown. The hedgehog (HH) pathway, a major signal transduction cascade, is linked with many cancers, including RMS. As we previously have detected loss of heterozygosity of PTCH1 in E-RMS, we now examined 8 E-RMS tumor samples and 5 E-RMS cell lines for the presence of PTCH1 mutations, but none was detected. However, in the E-RMS cell lines, a variable pattern of up-regulated expression of certain HH signaling target genes, including HHIP, PTCH1, SFRP1, and GLI1, was observed. Moreover, treatment with the small molecule HH signaling inhibitors cyclopamine and GANT61 inhibited cell proliferation in all E-RMS cell lines analyzed. Interestingly, GANT61 was more effective, and this was accompanied by increased apoptosis, while cyclopamine promoted necrotic events. Specific knockdown of SMO had no effect on the proliferation of E-RMS cells, indicating the presence of an SMO-independent HH signaling pathway in the E-RMS cell lines. Furthermore, in an in vivo xenograft model, tumor growth was significantly reduced by GANT61 treatment of E-RMS cells. Additionally, siRNA experiments provided evidence that inhibition of GLI1 or GLI3 but not GLI2 was sufficient to reduce proliferation of these cell lines. As GANT61 is known to block GLI1/GLI2 transcriptional activity, the inhibition of E-RMS growth by GANT61 is likely to be mediated through GLI1. In conclusion, our findings implicate that GLI1 could constitute an effective therapeutic target in pediatric E-RMS.
Keywords: hedgehog signaling, embryonal rhabdomyosarcoma, GLI, GANT
Introduction
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma in children under the age of 15 years, with approximately 350 new cases per year in the United States.1 RMS is thought to derive from cells along the skeletal muscle lineage2 and is divided into 2 major subtypes, embryonal (E-RMS) and alveolar (A-RMS), which are characterized by distinct histological features.3,4 The E-RMS subtype constitutes approximately two thirds of all RMS and predominantly affects children aged 0 to 4 years. The A-RMS subtype is more aggressive, occurs throughout childhood, and is associated with a chromosomal translocation event fusing PAX3 or PAX7 to FOXO1A.4 Both subtypes have deregulations of the p53, RAS, and MYC pathways, but although E-RMS has frequent loss of heterozygosity (LOH) and loss of imprinting at the 11p15.5 locus, which may result in overexpression of the IGF2 gene,5 the specific molecular events driving E-RMS pathogenesis are largely unknown.
The hedgehog (HH) signaling pathway is fundamental in embryonic development, controlling cell fate and proliferation.6 HH is a secreted ligand that is received and transduced at the membrane by its receptor, the patched homolog 1 (PTCH1). HH binding relieves the PTCH1 inhibition on the signaling molecule smoothened (SMO), triggering a cascade of downstream events, which culminate in the activation of the glioma-associated oncogene (GLI) transcription factors, GLI2 and GLI3. The HH target genes include GLI1, which further amplifies the initial signal at the transcriptional level. Other target genes include PTCH1 and human hedgehog interacting protein (HHIP), a HH sequestering receptor, thereby creating negative feedback loops, as well as genes affecting proliferation, cell survival, and angiogenesis.
HH signaling has been shown to be deregulated in many sporadic tumor types, including basal cell carcinoma, medulloblastoma, small-cell lung cancer, and digestive tract tumors.7 Tumors related to the HH pathway can either be derived from ligand-dependent (by HH stimulation/overexpression) or ligand-independent mechanisms, such as mutations in components of the pathway, resulting in constitutively activated HH signaling. Hence, the HH cascade might be a good target for cancer therapy. The first known inhibitor of the HH pathway to be identified was the teratogenic alkaloid cyclopamine, which binds and inhibits SMO.8 Recently, derivatives of cyclopamine and other small molecule antagonists targeting SMO are entering clinical phase I/phase II trials.9,10 However, these drugs will not be effective in tumors with activation downstream of SMO, such as GLI amplification/mutation. A significant step in this direction is the discovery of a novel HH signaling inhibitor, GANT61 (GLI-ANTagonist), which successfully blocked cell growth in a xenograft model using human prostate cancer cells.11 In contrast with other known HH pathway inhibitors, GANT61 reduces GLI1/GLI2 transcriptional activity and was found to interfere with GLI1 DNA binding in the nucleus.
We have previously shown that sporadic RMS has activated HH signaling and that LOH of 2 tumor suppressor genes in this pathway, PTCH1 and suppressor of fused (SUFU), is a frequent event in E-RMS.12 In accordance, an earlier study also revealed that E-RMS has a 33% LOH at 9q22, including the PTCH1 locus.13 Additionally, knockout mouse models targeting members of the HH signaling pathway, Ptch1+/− and Sufu+/−, as well as a transgenic mouse model with the activated SmoM2 allele, develop RMS at variable frequencies.14-17 Altogether, these data indicate that HH signaling is deregulated during E-RMS development. In this study, we further evaluated the importance of HH signaling for E-RMS tumor growth by examining the effects of inhibition of this pathway by small molecule antagonists, specifically GANT61, in cell lines and in a xenograft tumor model.
Results
HH signaling activity in human E-RMS
The mechanisms of deregulated HH signaling in E-RMS tumorigenesis are not fully understood. Humans and mice carrying germ-line mutations in the PTCH1 gene are predisposed to develop fetal rhabdomyoma (FRM) and RMS.14,18 We have previously found LOH of PTCH1 in sporadic E-RMS and FRM,12 and thus, we examined whether these tumors also harbored mutations in the PTCH1 gene. Direct sequencing of exons 2 to 23 of the PTCH1 gene in 8 E-RMS and 4 FRM tumor samples, 5 E-RMS cell lines, 1 A-RMS cell line, and 1 Ewing sarcoma (EWS) cell line revealed polymorphisms but no mutations (Suppl. Table S2).
Despite these observations, we wanted to further examine whether the HH signaling pathway may play a functional role in E-RMS tumors. We therefore analyzed the 5 E-RMS cell lines (JR-1, RD, Rh36, CCA, and CT-TC) and the A673 EWS cell line for expression of the HH pathway target genes PTCH1, HHIP, and secreted frizzled-related protein 1 (SFRP1). At least one of these target genes was up-regulated in the cell lines compared to normal human fetal skeletal muscle cells (HFSMC), with Rh36 and CCA cells exhibiting higher mRNA levels for all 3 genes (Fig. 1A). Moreover, the signaling molecule SMO and the transcriptional effectors GLI1, GLI2, and GLI3 were expressed in all the cell lines, and the relative amounts were higher compared to HFSMC for at least 3 of these components (Fig. 1B). Interestingly, expression of GLI1, which also acts as a marker of HH signaling activation, was variable in the cell lines. Furthermore, we could not detect expression of Sonic HH or Desert HH (data not shown), while Indian HH expression could only be detected in the JR-1 and CT-TC cell lines at very low levels (20-fold less than human control cDNA) (Suppl. Fig. S1). This might be indicating the lack of an autocrine stimulation of the pathway in the cell lines. Taken together, the results suggest the presence of an apparently ligand-independent HH signaling activity in these E-RMS cell lines.
Figure 1.
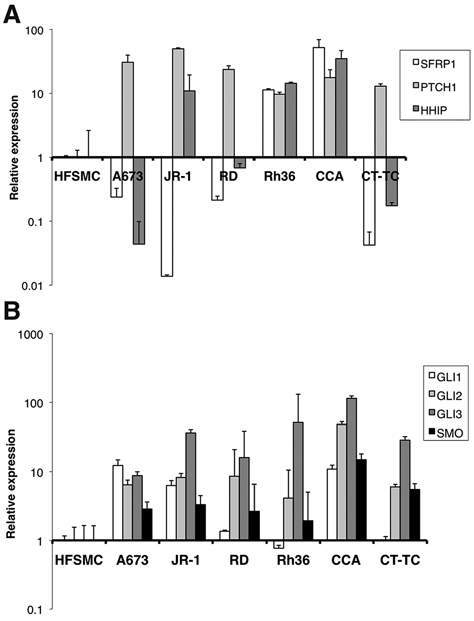
E-RMS cells express HH signaling target genes and components of the pathway. (A) Real-time RT-PCR analysis of the HH pathway target genes PTCH1, HHIP, and SFRP1 expression in the E-RMS cell lines JR-1, RD, Rh36, CCA, and CT-TC and the EWS cell line A673. (B) Real-time RT-PCR analysis of GLI1, GLI2, GLI3, and SMO expression in the E-RMS cell lines and the EWS cell line. For both (A and B), the expression levels relative to human fetal skeletal muscle cells (HFSMC), after normalization for the housekeeping gene GAPDH, are shown.
Treatment with HH signaling inhibitors leads to reduced growth/proliferation in E-RMS cells
To examine the significance of HH signaling in the E-RMS cell lines by means other than target gene activation, the cells were treated with the HH inhibitors cyclopamine and GANT61. Cyclopamine blocks HH signal transduction by direct binding to SMO,8 while GANT61 is an inhibitor of the GLI1 and GLI2 proteins.11 Treatment of all the E-RMS cells with 10 µM concentrations of either compound for 7 days led to reduced growth and widespread cell death (Fig. 2A and Suppl. Fig. S2). On the other hand, the EWS cell line A673 was not responsive to HH inhibition at all. To get further insight on the effects of the 2 inhibitors, the CCA and Rh36 cell lines were treated with 1, 3, 10, and 30 µM concentrations of either compound and observed for up to 5 days (Suppl. Fig. S3). At day 3, the cellular growth was suppressed by the 10 µM doses, whereas the 30 µM doses proved cytotoxic to the cells, while no effect was seen with the lower doses.
Figure 2.
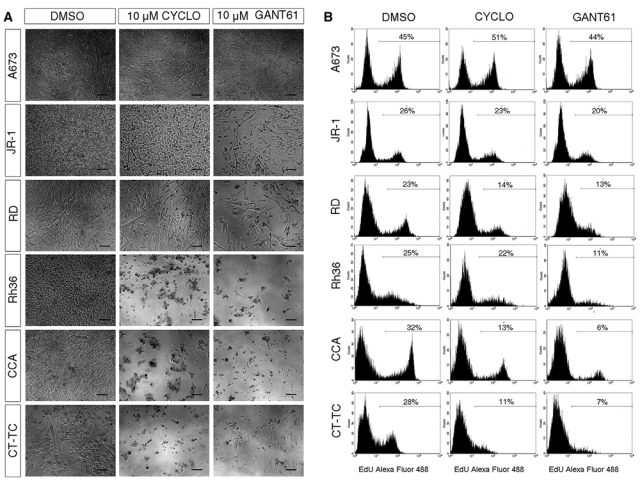
E-RMS cells are sensitive to treatment with HH signaling inhibitors. (A) Phase contrast micrographs depicting the morphology of the JR-1, RD, Rh36, CCA, and CT-TC E-RMS and the A673 EWS cell lines following HH signaling inhibition. The cells were treated with 10 µM concentrations of the inhibitors for 7 days and compared with the DMSO control. Scale bars, 100 µm. Note that cyclopamine and GANT61 inhibit at variable degrees the growth of E-RMS but not EWS cells. (B) Cyclopamine or GANT61 treatment for 72 hours inhibits the proliferation of the E-RMS but not the EWS cells. Proliferation was assayed by EdU incorporation for 2 hours. The percentage of cells labeled with Alexa fluor 488 azide and detected by flow cytometry is shown.
Furthermore, the cells were subjected to measurements of DNA replication by an EdU (BrdU analog) incorporation assay. All E-RMS cell lines showed reduced cell proliferation after 72-hour treatment with either GANT61 or cyclopamine at 10 mM concentrations, while no such effects were seen in the A673 EWS cells (Fig. 2B). GANT61 reduced proliferation to a higher extent than cyclopamine, most notably in CCA, Rh36, and CT-TC cells. These results are therefore suggestive that HH signaling has a role in E-RMS cells, as their proliferation/growth is sensitive to typical inhibitors of the pathway.
Treatment with GANT61 shows specificity towards the HH pathway and induces apoptosis in CCA and Rh36 E-RMS cells
The E-RMS cell lines Rh36, CCA, and CT-TC were found to be most sensitive to HH inhibition (Fig. 2A and 2B and Suppl. Fig. S2). Additionally, Rh36 and CCA cells up-regulated all 3 HH target genes analyzed (Fig. 1A); however, the levels of GLI1 were not increased in Rh36 (or CT-TC) cells, in contrast to the CCA cell line (Fig. 1B). Thus, we decided to further analyze the role of HH signaling in Rh36 and CCA cells. First, the expression of the HH pathway target genes HHIP and GLI1, following treatment with 10 µM of either cyclopamine or GANT61 for 24 hours, was examined. Real-time RT-PCR analysis revealed a down-regulation of HHIP and GLI1, which was most notable in the GANT61-treated cells, highlighting the HH signaling specificity of the GANT61 effects (Fig. 3A). In contrast, in the A673 cells, the expression levels of HHIP or GLI1 were not decreased upon GANT61 or cyclopamine treatment.
Figure 3.
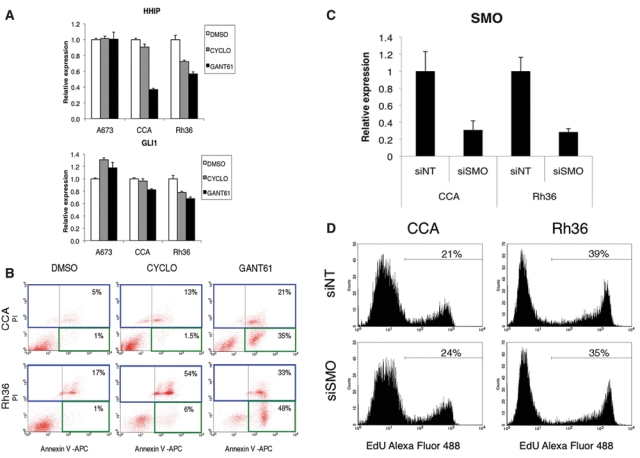
The HH signaling inhibitor GANT61 has an impact on HH target gene expression and reduces proliferation of E-RMS cells through apoptotic mechanisms, while SMO is dispensable for cellular growth. (A) Real-time RT-PCR analysis of the expression of the HH pathway target genes HHIP and GLI1 following HH signaling inhibition. A673, CCA, and Rh36 cells were treated for 24 hours with 10 µM concentrations of cyclopamine or GANT61. Shown are the mRNA levels relative to the DMSO control normalized to the mean expression of the housekeeping genes TBP and GAPDH. (B) GANT61 treatment significantly increases apoptosis in CCA and Rh36 cells. Shown are the apoptotic (annexin V–positive and PI-negative) cells within the green square and the necrotic (PI-positive) cells within the blue square, analyzed by flow cytometry. The percentage of total cells in the green and blue squares is indicated. (C) Real-time RT-PCR analysis of the SMO expression levels in CCA and Rh36 cells 48 hours after transfection with siRNAs directed against SMO (siSMO) or a nontargeting control (siNT). The mRNA levels are normalized to the mean expression of the housekeeping genes TBP and GAPDH. (D) Proliferation of CCA and Rh36 cells is unaffected by siSMO treatment. Cells were cultured for 72 hours after siRNA transfection followed by EdU incorporation for 3 hours. The percentage of cells labeled with Alexa fluor 488 azide and detected by flow cytometry is shown.
Additionally, as the morphological changes of E-RMS cells observed by treatment with the HH signaling inhibitors revealed that cell death had occurred (Fig. 2A), we investigated the mechanism whereby growth was prevented. CCA and Rh36 cells were treated with 10 µM doses of cyclopamine or GANT61 for 72 hours followed by determination of early apoptosis by annexin V–positive and propidium iodide (PI)–negative stained cells (Fig. 3B). GANT61 induced apoptosis at a significantly higher rate than cyclopamine. On the other hand, necrosis, indicated by PI-positive stained cells, was mainly increased after cyclopamine treatment. Thus, our findings clearly demonstrate that the GANT61 growth inhibition of CCA and Rh36 cells is mediated through apoptosis.
Moreover, specific down-regulation of SMO by a siRNA pool did not significantly alter the proliferation of the CCA and Rh36 cells (Fig. 3C and 3D), in line with the reduced effects of cyclopamine relative to GANT61. These results indicate a significant role for an SMO-independent HH signaling pathway in E-RMS cells.
GANT61 treatment reduces E-RMS tumor cell growth in vivo
To investigate whether treatment with GANT61 could affect tumor cell growth in vivo, the chick chorioallantoic membrane (CAM) assay was used. The chicken CAM is a readily accessible tissue, which is rich in blood vessels, allowing rapid vascularization, survival, and development of tumor cells.19 Initially, we examined if E-RMS cells were able to form tumors in this system, and indeed, solid, well-demarcated, and proliferative tumors were produced, as determined by immunohistochemical analysis of Ki67 (Suppl. Fig. S4). Also, the EWS cell line A673 grew large tumors. We then pretreated CCA, Rh36, and A673 cells with 10 µM or 30 µM concentrations of GANT61 at the time of introduction to the chick CAM. After 7 days, tumors were resected, weighed, and photographed. GANT61 clearly reduced the growth of the E-RMS tumors (Fig. 4A and 4B). Hematoxylin and eosin staining verified the complete removal of the tumors (data not shown). For Rh36 cells, tumor weights were reduced up to 50% with the 30 µM dose of GANT61 (P = 0.0173) (Fig. 4B). For CCA cells, the reduction was about 30% and did not reach statistical significance, but this is probably due to the difficulties in resecting clean tumors without retaining some CAM tissue, leading to big variations in tumor weights. The A673 cells grew large tumors irrespective of treatment. Immunohistochemical staining of tumors with Ki67 revealed no differences in the degree of proliferation (Fig. 4C), and no change in the apoptotic rate was detected by cleaved PARP (data not shown). It is therefore likely that the surviving cells after the initial treatment with the inhibitor are able to proliferate and retain their tumorigenic potential. In addition, apoptosis is an early event, and after 7 days, these effects are no longer detectable. These results therefore indicate an efficient reduction of E-RMS tumor growth in vivo by a single dose of GANT61.
Figure 4.
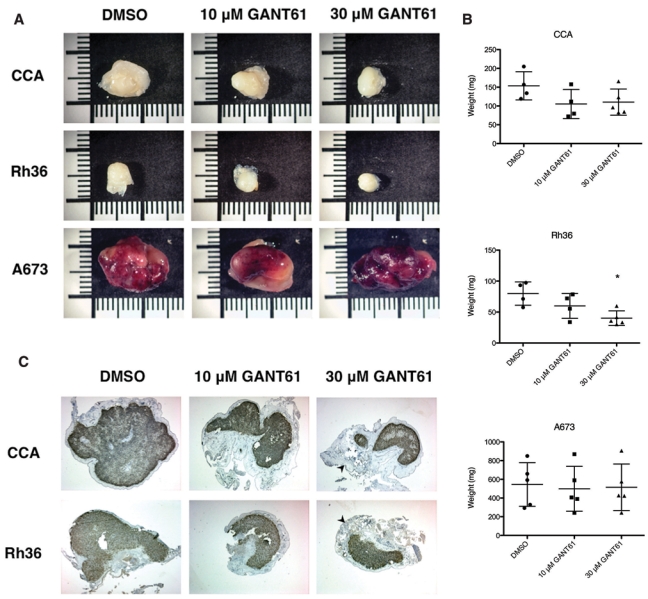
GANT61 reduces growth of E-RMS in vivo. (A) Five × 106 cells from the CCA, Rh36, or A673 cell lines were mixed with DMSO or 10 µM or 30 µM GANT61, introduced into chick CAMs, and allowed to grow for 7 days. After resection and trimming, tumors were photographed, with representative ones from each group shown. Scale bar, 1 cm. The CAM experiments were repeated 3 times with similar outcomes. (B) Tumor weights are represented as scatter plots, with the lines denoting mean ± standard deviation. The decrease in tumor weight of 30 µM GANT61-treated relative to DMSO-treated Rh36 cells reached statistical significance (P = 0.0173), indicated in the plot by an asterisk. (C) Immunohistochemical staining of the tumors for Ki67 (brown precipitate) revealed a comparable degree of proliferation despite the different sizes of the tumor mass. The unstained region surrounding the tumors is the CAM tissue (arrowhead).
Role of GLI1, GLI2, and GLI3 for the proliferation of CCA and Rh36 E-RMS cells
The relative expression levels of GLI3 were the highest among the GLI factors in the E-RMS cells analyzed (Fig. 1B). GLI3 is known to be proteolytically cleaved from a 190-kD precursor, which functions as an activator, into an 83-kD transcriptional repressor, and this processing is regulated by HH signaling.20 Western blot analysis indicated that the full-length 190-kD activator is the predominant GLI3 form in A673, CCA, and Rh36 cells (Fig. 5A). However, a full-length GLI3 expression construct, cotransfected with the 12xGLIBS-luc reporter into Hek293 cells, did not reveal a pronounced activation capacity, in line with an earlier report.21 Additionally, no significant effects on this weak activation by 10 µM or 30 µM GANT61 treatment were observed (Suppl. Fig. S5A). Furthermore, no changes in the GLI3 protein levels or the ratios of the activator/repressor forms could be seen after GANT61 treatment in A673, CCA, and Rh36 cells (Suppl. Fig. S5B). Thus, these findings suggest that GLI3 has a weak activating capacity, with GANT61 not capable of eliciting a major repression on this activity.
Figure 5.
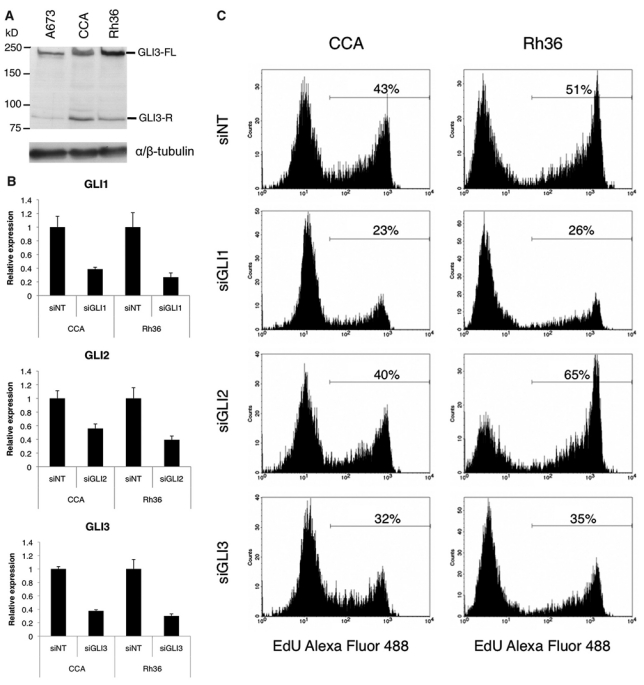
Role of GLI1, GLI2, and GLI3 for the proliferation of E-RMS cells. (A) Western blot analysis of extracts from A673, CCA, and Rh36 cells. The full-length (FL) 190-kD activator and the repressor (R) 83-kD GLI3 forms are indicated. αβ-tubulin was used as a loading control. (B) Real-time RT-PCR analysis of the GLI1, GLI2, and GLI3 expression levels in CCA and Rh36 cells 48 hours after transfection with siRNAs directed against GLI1 (siGLI1), GLI2 (siGLI2), GLI3 (siGLI3), or a nontargeting control (siNT). The mRNA levels are normalized to the mean expression of the housekeeping genes TBP and GAPDH. (C) Proliferation of CCA and Rh36 cells is reduced by siGLI1 or siGLI3 but not siGLI2 treatment. Cells were cultured for 72 hours after siRNA transfection followed by EdU incorporation for 2 hours. The percentage of cells labeled with Alexa fluor 488 azide and detected by flow cytometry is shown.
To examine the role of the 3 individual GLI transcription factors in E-RMS cellular proliferation, the RNA interference technology was used. CCA and Rh36 cells were transfected with siRNAs directed against GLI1, GLI2, or GLI3. After 48 hours, the expression levels of GLI1, GLI2, and GLI3 mRNAs were decreased, respectively (Fig. 5B). However, the EdU incorporation assay revealed that knockdown of either GLI1 or GLI3 leads to reduced proliferation in both cell lines, while knockdown of GLI2 had no such effects (Fig. 5C). Taken together, these experiments indicate that in these E-RMS cells, inhibition of GLI1 or GLI3 but not GLI2 is sufficient to reduce proliferation. Moreover, since GLI2 is apparently dispensable and GANT61 is not an effective inhibitor of the weak activating capacity of GLI3, the GANT61 effects in these cell lines are probably mediated through GLI1 inhibition.
Discussion
Although treatment options for RMS tumors have improved in the last decade, metastasizing and locally invasive E-RMS continue to have a poor prognosis. The specific molecular events driving E-RMS tumor development still remain elusive; however, comparative genomic hybridization studies have detected both loss at 9q22, where PTCH1 is located, as well as gain of 12q13, which is the GLI1 locus.22 In a previous report, we found consistent overexpression of HH pathway components in primary RMS tumor samples and LOH of PTCH1 specifically in E-RMS.12 In this study, we focused on E-RMS cell lines in addressing the functional consequences of dysregulated HH signaling. Even though no HH ligand expression or PTCH1 mutations were identified, the HH signaling pathway components and certain target genes were generally expressed in the E-RMS tumor cell lines at levels higher than normal HFSMC cells.
To functionally address the role of the HH signaling cascade in E-RMS tumor cells, we evaluated 2 different inhibitors of the HH pathway: cyclopamine, which acts on SMO, and GANT61, which targets the GLI1 and GLI2 transcription factors. Both inhibitors decreased tumor cell growth, but GANT61 proved to be more effective than cyclopamine, suggesting a rather downstream activation of HH signaling in E-RMS. This correlates well with a study in which RMS from Ptch+/− mice treated with cyclopamine revealed minor effects on tumor growth reduction, implying the possibility of additional, SMO-independent, events.23 Moreover, no growth inhibition of the E-RMS cells could be seen after siSMO transfection, which further supports the involvement of an SMO-independent HH pathway.
The antiproliferative properties of GANT61 are thought to be mediated through GLI1/GLI2 inhibition.11 Additionally, we have now shown that the weak transcriptional activation capacity of GLI3 is not particularly sensitive to GANT61. Furthermore, we have provided evidence that GANT61 induced apoptosis in CCA and Rh36 E-RMS cell lines, whereas cyclopamine treatment mainly resulted in necrotic events. The fact that SMO proved to be nonessential for the growth E-RMS cells, as indicated by the siRNA experiments, is in line with the minimal effects of the low doses of cyclopamine on E-RMS cell growth. On the other hand, the high doses could induce cell death through mechanisms that apparently do not involve SMO. Therefore, SMO inhibition, which blocks the canonical HH pathway, would not be efficient in E-RMS treatment, whereas inhibiting GLI activity may represent a much better therapeutic approach.
We also evaluated the effects of GANT61 in an in vivo tumor model system. Although the chicken CAM assay is mainly used to study angiogenesis, it also gives a rapid and efficient assessment of tumor growth. Treatment of CCA or Rh36 E-RMS cells with a single dose of GANT61 reduced tumor growth up to 50% in this in vivo model. One reason for the GANT61 effects not reaching statistical significance at all doses could be the pharmacokinetics of the CAM system, with rapid metabolism and degradation of the inhibitor. At the start of the CAM experiment, GANT61 has the capacity to reduce proliferation and increase apoptosis, but at day 7, such effects are no longer detectable. The A673 EWS cells, in contrast, were not responsive to GANT61 treatment in any setting. EWS is caused by a chromosomal translocation event that creates the EWS-FLI1 transcription factor, which is essential for the survival and growth of tumor cells. Among the EWS-FL1 multiple target genes is GLI1.24,25 Inhibition of HH/GLI1 activity by cyclopamine or another GLI-antagonist, GANT58, has been reported to lead to decreased proliferation in certain EWS cell lines. The GANT58 antiproliferative effects were observed in A673 cells,26 which in our hands are not responsive to GANT61 treatment. Although a different GANT molecule and a different method to evaluate proliferation have been used, the results are not in strict accordance to our observations. Also, in our hands, siGLI1 treatment of A673 cells leads to a small decrease in proliferation and reduced HHIP target gene expression (data not shown). The effect is not as significant as in the E-RMS cells, indicating that the A673 cells are not solely dependent upon GLI1 transcriptional activity. One plausible explanation for the lack of GANT61 antiproliferative effects in the A673 cell line is that other growth signals are taking over upon HH inhibition or that GANT61 in A673 cells is not efficiently uptaken/rapidly degraded.
Our results, demonstrating the high effectiveness of GANT61 in reducing E-RMS growth/proliferation, and the lack of growth inhibition by knockdown of SMO, suggest that in E-RMS cells, a noncanonical, SMO-independent HH pathway prevails. The canonical HH signaling pathway leads to activation of GLI2, which in turn activates HH target genes including GLI1,27 but in CCA and Rh36 E-RMS cells, GLI2 is dispensable for proliferation, as is seen by siRNA knockdown experiments.
Interestingly, even though the Rh36 cell line expresses GLI1 at apparently lower levels than normal HFSMC cells, the cellular proliferation is sensitive to siRNA-mediated GLI1 knockdown. Therefore, it is conceivable that other pathways converge on the GLI1 protein, increasing its transcriptional capacity. A recent example for this scenario has been observed in pancreatic ductal carcinoma, where TGFβ/RAS signaling was found to up-regulate GLI1 transcriptional activity.28 Worth noting is that in the same setting, TGFβ increased GLI3 mRNA levels >25-fold.
Furthermore, high levels of GLI3 were detected in the E-RMS cell lines analyzed, with a predominance of the activator relative to the repressor form. Since the GLI3 activator is quickly degraded following Sonic HH treatment of responsive cells,29 it is likely that noncanonical, ligand-independent mechanisms elicit the increased stabilization of the activator form. Moreover, GLI3 was found to have a role in the proliferation of CCA and Rh36 cells, as indicated by siRNA knockdown experiments. However, GANT61 has a minimal influence on the capacity of GLI3 to act as a transcriptional activator. The GANT61 growth-inhibitory effects on the E-RMS cells may therefore be mediated through GLI1.
Thus, GLI1 appears to be an effective target for inhibition of the growth of E-RMS, as indicated by the reduced proliferation following siRNA knockdowns. Additionally, the general concern for pediatric cancers that HH signaling inhibition might lead to bone growth retardation30 could be circumvented, as Gli1–/– mice do not show a phenotype and in particular bone growth defects.31 Thus, selective targeting of GLI1 may prove to be a rather optimal therapeutic strategy.
In conclusion, we have shown that HH signaling regulatory events are critical for the maintenance/survival of human E-RMS tumor cell lines. Additionally, inhibition of GLI1 by small molecule antagonists, such as GANT61, could be an effective therapeutic option in pediatric embryonal rhabdomyosarcoma. Further evaluation of the precise mechanism for action of GANT61 will in that case be needed.
Materials and Methods
Cell lines and reagents
The Ewing sarcoma (EWS) cell line A673 and the E-RMS cell line RD were purchased from ATCC (Manassas, VA). The E-RMS cell lines JR-1 and Rh36 were kind gifts from P. Houghton (St. Jude Children’s Research Hospital, Memphis, TN),32,33 CCA was a kind gift from P.-L. Lollini (University of Bologna, Italy),34 and CT-TC was established by and was a kind gift from H. Hosoi (Kyoto Prefectural University of Medicine, Kyoto, Japan). A673, RD, and CCA cells were cultured in DMEM supplemented with L-glutamine and 10% fetal bovine serum (FBS) from Saveen Werner (Limhamn, Sweden), while JR-1, Rh36, and CT-TC cells were cultured in RPMI-1640 with 10% FBS. All cell lines were maintained in a 5% CO2 humidified incubator. All media and supplements were from Invitrogen (Paisley, United Kingdom). The myogenic origin of the E-RMS cells was verified by desmin staining.
RNA from human fetal skeletal muscle cells (HFSMC) was purchased from Cell Applications Inc. (San Diego, CA), and human control cDNA was purchased from BD (Franklin Lakes, NJ). Cyclopamine was purchased from Sigma-Aldrich (St. Louis, MO), and GANT61 was custom synthesized, with both dissolved in DMSO. The following antibodies were used: rabbit anti-Ki67 (Sp6) from Thermo Fisher Scientific (Fremont, CA), rabbit anti-cleaved PARP (Asp214) from Cell Signaling Technology (Danvers, MA), and mouse anti-desmin from DakoCytomation (Glostrup, Denmark).
Real-time RT-PCR
Total RNA from cells was prepared with the RNeasy kit (Qiagen, Hamburg, Germany) followed by cDNA synthesis with random (N6) primers (New England Biolabs, Ipswich, MA) and Superscript II (Invitrogen). Real-time RT-PCR was performed with Power SYBR Green (Applied Biosystems, Foster City, CA) on a 7500 Fast real-time PCR system (Applied Biosystems) with the primers used shown in Supplementary Table S1.
Cell proliferation assay
Cells were seeded in 24-well plates in duplicate and maintained in 2.5% FBS containing media with 10 µM cyclopamine or GANT61 for 7 days. As a negative control, the cells were also treated with vehicle (DMSO) alone. Cells were photographed with an Axiovert inverted microscope connected to a CCD camera, and pictures were managed in the AxioVision software (Carl Zeiss, Oberkochen, Germany).
EdU incorporation assay
Cells were seeded in 6-well plates, treated with 10 µM cyclopamine or GANT61 for 72 hours, followed by a 2-hour 10 µM EdU (5-ethynyl-2’deoxyuridine) incubation. As a negative control, the cells were treated with vehicle (DMSO) alone. EdU was detected by fluorescent-azide coupling reaction (Click-iT, Invitrogen, Eugene, OR), and subsequently, 10,000 cells were counted on a FACS calibur machine (BD Biosciences, Stockholm, Sweden) to determine the percentage of cells in the population that are in the S-phase. Gating was performed to eliminate aggregated cells, and nonstained cells were used to set/define on the FACS plot the area with the cells that have incorporated EdU.
Apoptosis assay
Cells were seeded in 6-well plates, treated with 10 µM cyclopamine or GANT61 for 72 hours, followed by annexin V-APC (BD Biosciences) and propidium iodide staining. As negative controls, cells were also treated with vehicle (DMSO) alone. All cells were analyzed on a FACS calibur machine (BD Biosciences). The experiments were performed in duplicate and repeated twice.
Chick chorioallantoic membrane (CAM) assay
Fertilized chicken eggs were purchased from Ova Production AB (Vittinge, Sweden). Ten-day-old chicken embryos were prepared as described previously.35 Single-cell suspensions of 5 × 106 tumor cells were applied to the CAMs, and the eggs were sealed. After 7 days, the tumors were resected and trimmed free of surrounding CAM tissue, measured, and fixed in 4% paraformaldehyde. Digital photographs were taken with a Nikon COOLPIX 5000 (Tokyo, Japan) fitted to a stereomicroscope. Following embedding in paraffin and sectioning, the tumors were stained with hematoxylin and eosin. Histological examination was made to ensure the presence of tumor tissue.
For the GANT61 treatments, the cells were premixed with the inhibitor at final concentrations of either 10 µM or 30 µM, or with only DMSO as a vehicle control, and applied onto the chick CAM. The ANOVA and Dunnett multiple comparison test were used to analyze statistical significance (GraphPad Prism software, La Jolla, CA).
Western blot
Proteins were extracted and separated on 7% SDS-PAGE essentially as described.36 GLI3 was detected with a rabbit polyclonal antibody (H-280) from Santa Cruz Biotechnology (Santa Cruz, CA), and α/β-tubulin antibody from Cell Signaling Technology was used as a loading control.
siRNA
Predesigned siRNAs targeting human SMO, GLI1, GLI2, and GLI3 were purchased from Dharmacon (SiGenome SMART pools, Thermo Scientific, Stockholm, Sweden). As controls, non-targeting siRNA pools were used. Cells were plated in 6-well dishes at 30% to 50% confluency, and transfections were performed with Lipofectamine 2000 (Invitrogen) and 100 pmol siRNA. Transfection efficiencies of the siRNAs were confirmed by siGLO (green transfection indicator, Dharmacon) incorporation. After 48 hours, RNA was prepared, followed by cDNA synthesis and real-time PCR. For the proliferation analysis, the EdU incorporation assay after 72 hours was used.
Immunohistochemistry
Cells were fixed on 4-well slides for desmin staining. Paraffin-embedded tumors were sectioned and dewaxed. After heat-induced antigen retrieval and blocking, slides were incubated overnight at +4°C with primary antibodies, followed by incubation with secondary horseradish peroxidase-conjugated antibodies. Slides were subsequently developed with 3,3-diaminobenzidine tetrahydrochloride (Zymed Laboratories, Carlsbad, CA), followed by counterstaining with Mayer’s hematoxylin.
Supplementary Material
Acknowledgments
The authors thank Matthias Lauth for helpful comments and Minna Thullberg, Staffan Strömblad, and Åsa Bergström for technical advice and help.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the Swedish Research Council, Swedish Cancer Fund, Swedish Childhood Cancer Foundation, Magnus Bergvall and Robert Lundberg memorial foundations.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Ries L, Smith M, Guerney J, et al. Cancer incidence and survival among children and adolescents: United States SEER program 1975-1995. NIH Pub. No. 99-4649 Bethesda, MD: National Cancer Institute; 1999 [Google Scholar]
- 2. Merlino G, Helman LJ. Rhabdomyosarcoma: working out the pathways. Oncogene. 1999;18:5340-8 [DOI] [PubMed] [Google Scholar]
- 3. Parham D, Barr F. Embryonal rhabdomyosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and genetics: tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 146-9 [Google Scholar]
- 4. Parham D, Barr F. Alveolar rhabdomyosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and genetics: tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 150-2 [Google Scholar]
- 5. Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest. 1994;94:445-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454-72 [DOI] [PubMed] [Google Scholar]
- 7. Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805:181-208 [DOI] [PubMed] [Google Scholar]
- 8. Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002;16:2743-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-72 [DOI] [PubMed] [Google Scholar]
- 10. Scales SJ, de Sauvage FJ. Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303-12 [DOI] [PubMed] [Google Scholar]
- 11. Lauth M, Bergström A, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104:8455-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208:17-25 [DOI] [PubMed] [Google Scholar]
- 13. Bridge JA, Liu J, Weibolt V, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer. 2000;27:337-44 [DOI] [PubMed] [Google Scholar]
- 14. Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619-22 [DOI] [PubMed] [Google Scholar]
- 15. Lee Y, Kawagoe R, Sasai K, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442-7 [DOI] [PubMed] [Google Scholar]
- 16. Svärd J, Rozell B, Toftgård R, Teglund S. Tumor suppressor gene co-operativity in compound Patched1 and suppressor of fused heterozygous mutant mice. Mol Carcinog. 2009;48:408-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao J, Ligon KL, Rakhlin EY, et al. A novel somatic mouse model to survey tumorigenic potential applied to the hedgehog pathway. Cancer Res. 2006;66:10171-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore). 1987;66:98-113 [DOI] [PubMed] [Google Scholar]
- 19. Hagedorn M, Javerzat S, Gilges D, et al. Accessing key steps of human tumor progression in vivo by using an avian embryo model. Proc Natl Acad Sci U S A. 2005;102:1643-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423-34 [DOI] [PubMed] [Google Scholar]
- 21. Tsanev R, Tiigimagi P, Michelson P, Metsis M, Osterlund T, Kogerman P. Identification of the gene transcription repressor domain of Gli3. FEBS Lett. 2009;583:224-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bridge JA, Liu J, Qualman SJ, et al. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer. 2002;33:310-21 [DOI] [PubMed] [Google Scholar]
- 23. Ecke I, Rosenberger A, Obenauer S, et al. Cyclopamine treatment of full-blown Hh/Ptch-associated RMS partially inhibits Hh/Ptch signaling, but not tumor growth. Mol Carcinog. 2008;47:361-72 [DOI] [PubMed] [Google Scholar]
- 24. Zwerner JP, Joo J, Warner KL, et al. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene. 2008;27:3282-91 [DOI] [PubMed] [Google Scholar]
- 25. Beauchamp E, Bulut G, Abaan O, et al. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem. 2009;284:9074-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joo J, Christensen L, Warner K, et al. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS One. 2009;4:e7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915-24 [DOI] [PubMed] [Google Scholar]
- 28. Nolan-Stevaux O, Lau J, Truitt ML, et al. GLI1 is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of hedgehog signaling is controlled by the dynamic association between suppressor of Fused and the Gli proteins. Genes Dev. 2010;24:670-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kimura H, Ng JM, Curran T. Transient inhibition of the hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13:249-60 [DOI] [PubMed] [Google Scholar]
- 31. Park HL, Bai C, Platt KA, et al. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593-605 [DOI] [PubMed] [Google Scholar]
- 32. Roberts I, Gordon A, Wang R, Pritchard-Jones K, Shipley J, Coleman N. Molecular cytogenetic analysis consistently identifies translocations involving chromosomes 1, 2 and 15 in five embryonal rhabdomyosarcoma cell lines and a PAX-FOXO1A fusion gene negative alveolar rhabdomyosarcoma cell line. Cytogenet Cell Genet. 2001;95:134-42 [DOI] [PubMed] [Google Scholar]
- 33. Clayton J, Pincott JR, van den Berghe JA, Kemshead JT. Comparative studies between a new human rhabdomyosarcoma cell line, JR-1 and its tumour of origin. Br J Cancer. 1986;54:83-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Giovanni C, Nanni P, Nicoletti G, et al. Metastatic ability and differentiative properties of a new cell line of human embryonal rhabdomyosarcoma (CCA). Anticancer Res. 1989;9:1943-9 [PubMed] [Google Scholar]
- 35. Brooks PC, Montgomery AM, Rosenfeld M, et al. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157-64 [DOI] [PubMed] [Google Scholar]
- 36. Shimokawa T, Svard J, Heby-Henricson K, Teglund S, Toftgard R, Zaphiropoulos PG. Distinct roles of first exon variants of the tumor-suppressor Patched1 in hedgehog signaling. Oncogene. 2007;26:4889-96 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.