

Abstract
Drug resistance remains a clinical challenge in cancer treatment due to poor understanding of underlying mechanisms. We have established several drug-resistant prostate cancer cell lines by long-term culture in medium containing chemotherapeutic drugs. These resistant lines displayed a significant increase in side population cells due to overexpression of drug efflux pumps including ABCG2/BCRP and MDR1/Pgp. To uncover potential mechanisms underlying drug resistance, we performed microarray analysis to identify differentially expressed genes in 2 drug-resistant lines. We observed that POU5F1/OCT4, a transcription factor key to regulating pluripotency in embryonic stem cells, was upregulated in drug-resistant lines and accompanied by transcriptional activation of a set of its known target genes. Upregulation of OCT4 in drug-resistant cells was validated by RT-PCR and sequencing of PCR products as well as confirmation by Western blot and specific shRNA knockdown. Analysis of the regulatory region of POU5F1/OCT4 revealed a reduction of methylation in drug-resistant cell lines. Furthermore, these drug-resistant cells exhibited a significant increase in tumorigenicity in vivo. Subcutaneous inoculation of as few as 10 drug-resistant cells could initiate tumor formation in SCID mice, whereas no detectable tumors were observed from the parental line under similar conditions, suggesting that these drug-resistant cells may be enriched for tumor-initiating cells. Knocking down OCT4 expression by specific shRNAs attenuated growth of drug-resistant cells. Our data suggest that OCT4 re-expression in cancer cells may play an important role in carcinogenesis and provide one possible mechanism by which cancer cells acquire/maintain a drug-resistant phenotype.
Keywords: drug resistance, prostate cancer, OCT4, POU5F1
Introduction
Although screening and early detection have dramatically increased survival rates, prostate cancer remains the second leading cause of cancer deaths among men in the Western world. Hormone ablation is often achieved through surgical or pharmacological means as a standard treatment approach for most advanced prostate cancer patients. This therapy aims to reduce androgen levels, which are believed to support prostate cancer cell growth and proliferation. A majority of patients initially respond well to this treatment but inevitably relapse and develop an incurable castration-resistant disease within a few years.1
Few treatment options are currently available for castration-resistant prostate cancer. Interchalating agents, alkylating compounds, and microtubule stabilizers are the main classes of drugs used in the clinic at this stage.2 While most offer only palliative effects, the latter group that includes taxanes has shown promising clinical response in combination with other drugs like prednisone.3,4 However, intrinsic or acquired resistance can often lead to failure of this secondary treatment. Taxane resistance has been attributed to a number of factors including increased efflux pump activity, selection of cells with increased survival advantages, or enhanced DNA-damage repair. Overexpression of ATP-binding cassette (ABC) transporter efflux pumps can offer a protective effect from chemotherapeutic drugs as a result in enhanced efflux of xenobiotics out of the cell. Taxanes such as docetaxel are direct substrates for P-glycoprotein (Pgp), so they can easily be exuded by cancer cells expressing a high level of this protein.5,6 Because docetaxel exerts its mode of action by preventing microtubule depolymerization, alterations in β-tubulin expression or signaling can also lead to decreased patient sensitivity to the drug.7 Understanding such pathways of chemoresistance is critical not only for resensitizing tumor cells to therapeutic drugs but also for prevention of this process.
OCT4/POU5F1 is a well-established transcription factor critical for maintaining pluripotency in embryonic stem cells. It remains unclear what roles if any OCT4 serves in somatic cells or during carcinogenesis. A novel function of OCT4 in tumorigenesis was proposed when its ectopic expression induced dysplastic growth of epithelial tissue.8 It has also been described as a marker for germ cell tumors.9 OCT4 was reported to be reactivated in cancer cell lines,10,11 yet other groups have not been able to detect such upregulation.12,13 One possible explanation for these discrepancies is that the POU5F1/OCT4 gene encodes 2 protein isoforms, named OCT4A and OCT4B, the latter of which has no known biological role currently.14 Some confusion has also arisen from OCT4 pseudogenes existing within human and mouse genome, which may produce false positives in PCR detection.15 One former pseudogene, POU5F1B, is now believed to encode a protein with 95% homology to OCT4A and resides on chromosome 8q24, which is frequently amplified in prostate cancers.16 These hurdles underscore the importance of proper primers and controls utilized for analysis to avoid false-positive detection. Although a number of target genes of OCT4 have been identified in embryonic stem cells,17,18 many remain to be discovered, potentially uncovering novel roles for this protein.19
We previously established drug-resistant derivatives from the CWR-R1 prostate cancer cell line.20 In an effort to characterize these lines, we examined the differentially expressed genes that may underlie their drug resistance and enhanced tumorigenicity. Side population and microarray analysis indicated that several stem-like genes were preferentially expressed in drug-resistant cells. Of interest, we noted that embryonic stem cell marker OCT4 was overexpressed compared to the drug-sensitive parental line. We confirmed OCT4 expression using multiple analyses. We further showed that OCT4 in drug-resistant lines is functional in regulating transcription of its known target genes. Knockdown of OCT4 in drug-resistant lines demonstrated its biological significance as it drastically attenuated tumor growth. This study provides an interesting insight into how prostate cancer cells may function during advanced disease progression and how they may be targeted to increase survival rates during secondary chemotherapy.
Results
Drug-resistant prostate cancer cells overexpress drug efflux pumps and exhibit an increased side population
In our previous publication, we developed drug-resistant prostate cancer cell lines to examine the role of Pim-1 kinases in chemoresistance.20 These cells developed resistance to docetaxel (DTX) or mitoxantrone (MX) after maintaining clinically relevant doses in culture media. Both of these chemotherapeutic drugs are commonly used to treat castration-resistant prostate cancer. Side population (SP) was first analyzed to characterize these lines and determine what percentage of cells possesses a drug-resistant phenotype (Fig. 1). The SP is defined as the cells that exhibit enhanced efflux of DNA-binding dye Hoechst 33342 dye and is often considered to have similar characteristics to stem cells.21 As expected, both docetaxel-resistant (R1/DTX) and mitoxantrone-resistant (R1/MX) lines exhibited a significant increase in SP percentage as a result of their constant exposure to drugs. The SPs of R1/DTX and R1/MX after adding drug efflux pump blockers verapamil or Ko143, respectively, were effectively eliminated. Conversely, there was little change in SP when treating R1/DTX with Ko143 or R1/MX with verapamil. Treating the parental CWR-R1 cells with either verapamil or Ko143 had no significant effect on the already small SP. This suggested that docetaxel-resistant cells preferentially utilize MDR1 as their dominant efflux pump, whereas mitoxantrone cells predominantly use BCRP. This observation was confirmed by flow cytometry and immunofluorescence (data not shown), which agreed with current literature regarding known substrates of these efflux pumps.22,23
Figure 1.
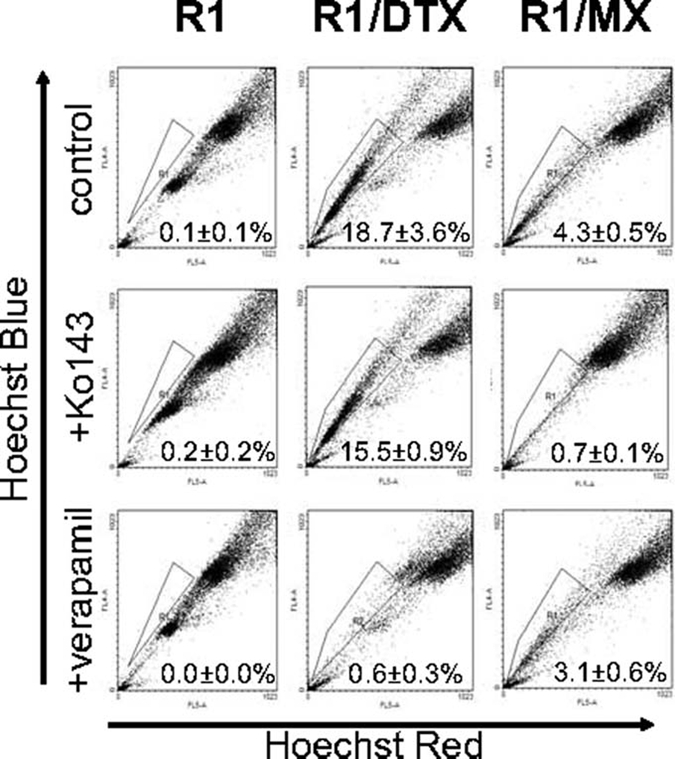
Drug-resistant prostate cancer cells have an expanded side population. Flow cytometric side population analysis was performed as described in Materials and Methods. Verapamil and Ko143 were used as blockers of MDR- and BCRP-expressing populations, respectively. A significant change of SP population in drug-resistant lines (R1/DTX and R1/MX) compared to the parental line CWR-R1 (R1) was detected. P < 0.05.
Drug-resistant prostate cancer cells overexpress embryonic stem cell marker POU5F1/OCT4
Microarray analysis was performed to identify unique genes commonly altered in both drug-resistant cell lines compared to their drug-sensitive parental line. Only genes that were altered in both lines at least 2-fold with P values of at least <0.05 were considered to be of interest. A summary of some relevant genes is listed in Table 1. Interestingly, we observed that a stem cell marker POU5F1, also known as OCT4, was among the genes significantly upregulated at the transcript level in both R1/DTX and R1/MX cell lines. In addition, several OCT4 target genes were also upregulated. These findings were particularly intriguing given that OCT4 expression is believed to be restricted to self-renewable pluripotent embryonic stem cells. Therefore, we further investigated whether OCT4 could serve a role in drug resistance of prostate cancer cells. To validate the microarray data, we performed RT-PCR using OCT4 specific primers, which do not amplify pseudogene sequences.12 Furthermore, these primers are located in exon 1 and 4 and thus do not amplify the OCT4B isoform. We obtained a 748-bp PCR product for both of the drug-resistant cell lines, while very little was detected in the parental line, suggesting that OCT4 is upregulated in these cell lines (Fig. 2A). We also performed Western blot to confirm that OCT4 protein is also expressed in drug-resistant cells (Fig. 2B). It should be noted that this monoclonal OCT4 antibody recognizes an N-terminal epitope between amino acids 1 to 134, which are not present in the OCT4B isoform. To further confirm that the protein band detected by the OCT4 antibody was indeed encoded by POU5F1, we treated drug-resistant cells with lentivirus encoding 2 independent short hairpin RNA (shRNA) constructs specific for OCT4. As shown in Figure 2C, the level of OCT4 was significantly decreased by treatment with either shOCT4 in both resistant lines. These data support the possibility that OCT4 is re-expressed in R1/DTX and R1/MX lines.
Table 1.
Differentially Expressed Genes in Drug-Resistant Cell Lines
| Upregulateda | Downregulateda | |
|---|---|---|
| Cell signaling | GK | JAKMIP1 |
| NUCKS1 | KLK8 | |
| HUNK | MLKL | |
| HMOX1 | MAPK13 | |
| MYB | RASSF4 | |
| Stress related | MDM2 | PTEN |
| MGMT6 | PARP8 | |
| ANXA1 | PTPN20B | |
| Metastasis and cytoskeleton | CDH2 | ITGB2 |
| KRT4, KRT5, KRT19 | VIM | |
| MMP1 | COL1A2 | |
| PLAT | MMP2 | |
| MID1 | CDH11, CDH12 | |
| MARK1 | ||
| GJA1 | ||
| TUB4A | ||
| MAPT | ||
| Stem cell related | POU5F1 | WIF1 |
| AID | SMO | |
| SNAI1 | WNT5A, WNT6, WNT11 | |
| GDF6 | WISP1 | |
| KLF5 | BMP2, BMP4, BMP7 | |
| GATA6 | ADFP | |
| IL1RN | GATA4 | |
| ZIC1 | ||
| SOX11 | ||
| Growth factors and receptors | FGFR | FGF7, FGF14 |
| OXTR | IGF1, IGF2 | |
| LDLR | VEGFA | |
| FOLR1, FOLR3 | PPARG | |
| TLR4 | PGF | |
| PPARGC1A | HLF | |
| LIFR | ||
| GDF15 | ||
| ADRB2 | ||
| Transcription factors | EBF3 | SNAI2 |
| SOX6 | FOXL2 | |
| E2F8 | HOXB13 | |
| DIDO1 | MAF | |
| ELF1 | TCF7 | |
| CUGBP2 | ||
| TEAD4 |
Partial list of genes that were altered in both resistant lines at least 2-fold with P < 0.05.
Figure 2.
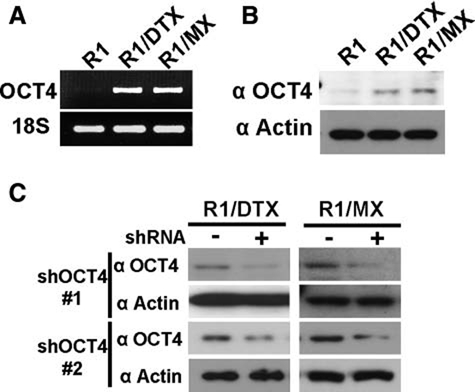
OCT4 expression is upregulated in drug-resistant cells. (A) Total RNA was isolated from CWR-R1 and its drug-resistant derivatives, and then RT-PCR analysis was performed using OCT4-specific primers as described in Materials and Methods. 18S was used as a control. (B) Total cell lysates from CWR-R1 and its drug-resistant derivatives were subjected to Western blot using monoclonal OCT4 antibody. Actin served as a loading control. (C) Drug-resistant R1/DTX and R1/MX cells were treated with lentivirus encoding a control shRNA (Control) or 2 independent OCT4 shRNAs (shOCT4) for 48 hours. Expression of OCT4 protein was determined by Western blot as above.
OCT4 is transcriptionally active in drug-resistant lines
Because OCT4 is known to be silenced through genomic methylation within its regulatory regions during differentiation of embryonic stem cells,24 we proceeded to test whether methylation status of regulatory regions of the POU5F1 gene was altered in these drug-resistant cell lines. This would provide a mechanism by which OCT4 could be re-expressed at an increased level in these cells. Genomic DNA from each cell line was collected and digested with either HpaII or MspI restriction enzymes to examine methylation status in a similar approach described by Yeo et al.25 These isoschizomers cleave DNA at the same sequences, but the former is methylation sensitive, whereas the latter is not. Therefore, a methylated region of DNA should remain intact after HpaII digestion and could be amplified by PCR. Conversely, MspI serves as a negative control to ensure complete DNA digestion. Primers in the POU5F1/OCT4 proximal promoter and distal enhancer region were used, each containing one HpaII/MspI cleavage site. Following enzyme digestion, real-time PCR was utilized to quantify the digestion efficiency in each cell line. Ntera cells were used as a negative control since they have been shown to exhibit low levels of methylation at the POU5F1/OCT4 locus.25 As shown in Figure 3A, there was a significant decrease in PCR product formation in both drug-resistant lines compared to the parental line. This suggested that OCT4 methylation is decreased at both the enhancer in promoter regions of POU5F1/OCT4 in the resistant lines. An analysis of the expression of AID, a demethylase associated with demethylation of the POU5F1/OCT4 locus in induced pluripotent stem (iPS) cells,26 supported this notion as it was increased in both resistant lines compared to the CWR-R1 control (Suppl. Fig. S1). To further validate the microarray data and assess the functionality of OCT4 in the drug-resistant cell lines, expression of several target genes regulated by OCT4 was analyzed by real-time RT-PCR. MID1, MYB, IL1RN, RPS27, and CUGBP2 have been previously shown to be OCT4 direct target genes in ChIP assays.18 As shown in Figure 3B, these OCT4 target genes were upregulated at the transcription level in drug-resistant cells compared to their drug-sensitive parental line. Taken together, these results suggest that not only is OCT4 overexpressed in these drug-resistant R1/DTX and R1/MX cells, but it may also play a functional role by regulating expression of its target genes.
Figure 3.
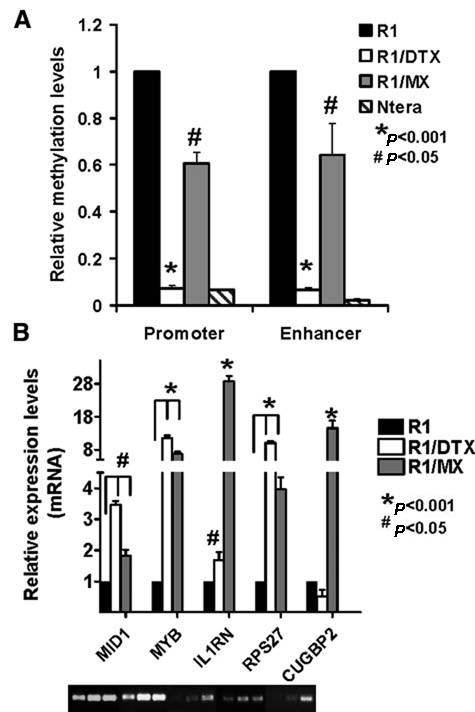
OCT4 is transcriptionally active in drug-resistant cells. (A) Reduced methylation of regulatory regions of OCT4 using HpaII-sensitive PCR. Genomic DNA isolated from CWR-R1 and its drug-resistant derivates were digested with control (ddH2O), HpaII, or MspI as described in Materials and Methods. The digested DNA was used as the template for PCR amplification using primers specific for the promoter or enhancer site of POU5F1/OCT4 gene. The level of methylation was determined by normalizing with undigested DNA from each cell line and set relative to the parental CWR-R1 line, whose value was set as 1. P < 0.001 and 0.05 as labeled. (B) Expression of OCT4 target genes in CWR-R1 or its drug-resistant derivatives was determined by real-time PCR analysis as described in Materials and Methods. The value in the parental line CWR-R1 was set as 1. The PCR products were shown at the bottom. P < 0.001 and 0.05 as labeled.
R1/DTX and R1/MX exhibit enhanced tumorigenicity in vitro and in vivo
To test whether the drug-resistant cells with re-expressed OCT4 protein have tumorigenic growth advantages compared to the parental line, we first examined their ability to self-expand in soft agar assays. As shown in Figure 4A, R1/DTX and R1/MX were more clonogenic than their drug-sensitive parental line. Furthermore, inoculation of as few as 10 R1/DTX or R1/MX cells into castrated male SCID mice was sufficient to form tumors after 8 weeks (Fig. 4B), while the parental R1 cells could not induce detectable tumor growth under similar conditions. These data suggested that the resistant lines are enriched for tumor-initiating cells. We hypothesized some of the genes differentially regulated in these drug-resistant lines, such as those identified in our microarray analysis, may be responsible for their enhanced tumorigenicity.
Figure 4.
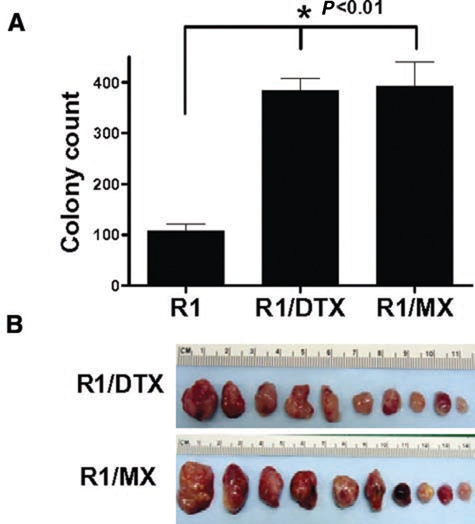
Drug-resistant cells are more tumorigenic. (A) Clonogenicity of CWR-R1 and its drug-resistant derivatives was examined using soft agar assay as described in Materials and Methods. Colonies between 50 and 200 µm were counted on an automated plate reader. Representative data of triplicate experiments are presented. P < 0.01. (B) Tumorigenicity of CWR-R1 and its drug-resistant derivatives was tested by subcutaneous inoculation of approximately 10 cells from each line into the flanks of castrated SCID mice (5 mice/line). After 8 weeks, mice were sacrificed and tumors harvested from R1/DTX and R1/MX xenograft. CWR-R1 cells did not form any detectable tumors under these conditions.
Expression of OCT4 plays a critical role in growth and survival of drug-resistant prostate cancer cells
To test whether OCT4 re-expression plays a role in drug-resistant activity in R1/DTX and R1/MX cells, we treated the resistant cells with either control shRNA or OCT4 shRNA and examined the effect of OCT4 knockdown on their growth in vitro. As shown in Figure 5A, knockdown of OCT4 in both drug-resistant lines reduced growth by more than 50% (Fig. 5A). Similar inhibitory effects on growth of drug-resistant lines were also detected in xenograft models (Fig. 5B). The parental CWR-R1 cells formed significantly smaller and fewer tumors than the drug-resistant cell lines under our experimental conditions, supporting our earlier clonogenicity experiments. In addition, immunohistochemical analysis was performed on the collected tumors using the monoclonal OCT4 antibody (which only recognizes OCT4A). Consistent with Western blot analysis, the xenograft tumors derived from drug-resistant cell lines showed an increase of OCT4 staining compared to their parental line (Fig. 5C).
Figure 5.
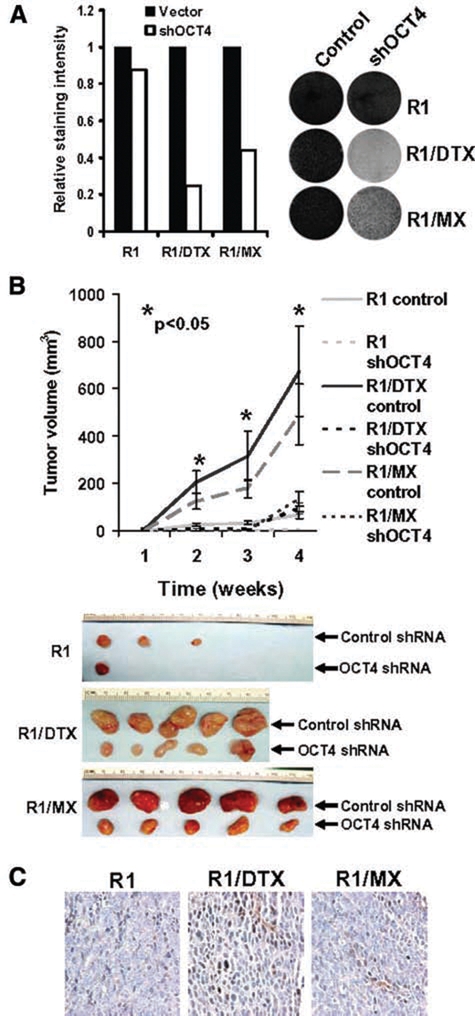
Expression of OCT4 plays a critical role in growth and survival of drug-resistant cells. (A) CWR-R1 and its drug-resistant derivates were treated with lentivirus encoding control or shOCT4 for 48 hours and allowed to grow for 6 days before being fixed and stained with Coomassie blue (left). The density of cells was quantified using a densitometer (right). Representative data from triplicate experiments were shown. (B) R1/DTX and R1/MX cells were treated with lentivirus encoding either control shRNA or shOCT4, and approximately 1 × 104 cells were mixed with Matrigel (BD Biosciences) and then inoculated subcutaneously into flanks of castrated SCID mice at 48 hours postinfection. Tumors were measured weekly (left) before the animals were sacrificed, and tumors were harvested at 4 weeks postinoculation (right). (C) Immunohistochemical analysis using a monoclonal OCT4 antibody in tumors collected from B.
Discussion
Treatment of prostate cancer remains a clinical challenge since there are limited options for patients with castration-resistant tumors. In addition, these patients will inevitably develop resistance to cytotoxic agents used during advanced stage. Our data suggest that this process may enrich more aggressive tumor-initiating cells in recurrent tumors that are increasingly difficult to target. Therefore, a better understanding of mechanisms of drug resistance is needed to improve treatment efficacy for secondary prostate cancer therapy. Identifying pathways underlying chemoresistance to use as targets offers an opportunity for patients to maintain or regain sensitivity to drugs used during combination therapy.
In this study, we have characterized the CWR-R1 cell line and its derivatives, which have developed resistance to docetaxel and mitoxantrone, two different agents commonly utilized to treat advanced recurrent prostate cancer. These drug-resistant cell lines were established by maintaining clinically relevant doses of the chemotherapeutic drugs in culture media over a period of months. As expected by virtue of their acquired drug resistance, these cell lines exhibited an enhanced SP compared to their parental line. Analysis of other tumor types by flow cytometry suggests potential cancer stem cells or tumor-initiating cells may reside within the SP.27 Soft agar assays and in vivo tumorigenicity experiments support this notion, as R1/DTX and R1/MX were found to be significantly more tumorigenic than their parent counterparts. As few as 10 unsorted cells were sufficient to form xenograft tumors in NOD/SCID mice, while the parental cells did not form any detectable tumors under similar conditions. This suggests that during the process by which cells acquire drug resistance, those cells with enhanced tumor-initiating ability were selected for.
A number of genes differentially regulated in the drug-resistant lines were identified by microarray analysis. Although DTX and MX have quite different mechanisms of action, both resistant lines shared numerous changes over their derivative CWR-R1. We focused on several stem cell–related genes following SP analysis and found that POU5F1/OCT4 was among those upregulated in R1/DTX and R1/DTX compared to their parental line. Although POU5F1/OCT4 expression in cancer remains controversial, we demonstrated that OCT4 expression was enhanced in both drug-resistant cell lines using multiple analyses including RT-PCR, sequencing, Western blot, and shRNA knockdown. We also showed that the “re-expression” of OCT4 was associated with reduced methylation in the regulatory region of POU5F1 locus, possibly due to the elevated expression of AID. AID is required for promoter demethylation and induction of OCT4 (also known as POU5F1) and NANOG gene expression in iPS cells. However, we did not detect a change in NANOG expression in our drug-resistant lines. It is possible that AID may demethylate different regulatory regions depending on cell context. It is still unclear whether the observed “re-expression” of POU5F1/OCT4 in drug-resistant cells was due to de novo demethylation of the POU5F1/OCT4 locus or enriched rare OCT4-positive cells pre-existing in the drug-sensitive parental line. It is also possible that chemotherapy may induce neuroendocrine differentiation analogous to that shown in radiation therapy.28 OCT4 is expressed in a subpopulation of prostate neuroendocrine cells,29 and therefore, the increased OCT4 expression may reflect an increase in this population. Nevertheless, we confirmed that OCT4 was transcriptionally active by analyzing the expression of several OCT4 target genes using real-time RT-PCR. Although these genes have not been previously confirmed as direct targets of OCT4 transcription, they were identified as associated with OCT4 through ChIP analyses. Some of the more commonly known OCT4 targets associated with stem cell function such as NANOG, KLF4, and GATA6 were unchanged in R1/DTX and R1/MX. Further, SOX2 was only modestly increased in these resistant lines (data not shown).
Consistent with our observations, a recent genome-wide study on prostate cancer showed that POU5F1 is overexpressed in over 25% of castrated metastatic prostate cancer cases but less than 11% of primary prostate cancer cases,30 implying a potential role of POU5F1 overexpression in disease progression. We propose that OCT4 may serve a novel role in mediating growth and survival of drug-resistant cancer cells as in our model. Targeting protein expression by shRNA led to a dramatic decrease in cell growth assays and tumorigenicity in vivo. The exact mechanisms by which OCT4 regulates growth in these drug-resistant prostate cancer cells are not yet known, so further studies will explore these precise pathways and mechanisms. Posttranslational modification of OCT4, such as phosphorylation, may determine its transcriptional specificity.31 Therefore, it is possible that OCT4 re-expression in cancer cells may regulate previously unidentified pathways supporting carcinogenesis. A ChIP-seq analysis may provide more insight into which OCT4 target genes are relevant within this context.
The implications of this study are of great significance to prostate cancer therapy. Unfortunately, the mortality rate of advanced prostate cancer remains high since patients often relapse and become resistant to androgen deprivation therapy. Chemotherapeutic drugs such as taxanes remain a viable option for treatment, but resistance to cytotoxic drugs can become another hurdle to overcome. Therefore, combination treatment targeting both the bulk tumor as well as chemoresistant tumor-initiating cells could provide a greater increase in survival rates. There is a critical need to better understand mechanisms of drug resistance in order to identify novel targets to improve treatment efficacy. Our data suggest that OCT4 may be one such target, and its use may be especially relevant to aggressive drug-resistant cancers.
Materials and Methods
Cell culture and constructs
CWR-R1 lines were cultured in RPMI 1640 medium (Mediatech Inc., Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin/streptomycin, and maintained in a 37°C incubator at 5% CO2. Drug-resistant cell lines were developed as previously described20 and maintained in medium containing 100 nM docetaxel (Fluka, Sigma-Aldrich, St. Louis, MO) for R1/DTX or 20 nM mitoxantrone (Sigma, Sigma-Aldrich) for R1/MX. Lentiviral constructs expressing OCT4 shRNAs were purchased from Sigma, which contains target sequence CCGGTCATTCACTAAGGAAGGAATTCTCGAGAATTCCTT CCTTAGTGAATGATTTTT or Addgene plasmid 12197 (Cambridge, MA) with target sequence GGATGTGGTCCGAGTGTGGTT.
Side population analysis
The method previously described by us for analyzing side population was utilized.32 One million cells were suspended in 1 mL DMEM containing 2% fetal calf serum (FCS) and 10 mM HEPES. The cell suspension was incubated with 1 mg/mL Hoechst 33342 (Sigma) at 37°C in the presence or absence of Pgp inhibitor verapamil (50 µM) or BCRP inhibitor Ko143 (1 µM). After 90 minutes of incubation, cells were centrifuged at 4°C and resuspended in cold HBSS with 2% FCS and 10 mM HEPES. Prior to analysis, propidium iodide (2 µg/mL) (Sigma) was added to discriminate dead cells. Samples were then analyzed by BD LSR II 4-laser flow cytometer (BD Biosciences, San Jose, CA). The Hoechst dye was excited at 355 nm, and the fluorescence profile was measured in dual wavelength analysis (405/30 nm and 670/40 nm). The side population was analyzed as mentioned in references.33,34 Two independent measurements were performed, and a significant change in SP population was determined by the Student t test at a P value less than 0.05.
Microarray analysis
Total RNA was collected from cells and analyzed using the RNA 6000 Nano kit on an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA). RNA was fluorescently labeled according to the standard protocol designed for the Agilent Human Whole Genome Expression System. Fluorescent dyes (Cye5 and Cye3) were incorporated into the amplified cRNA during a linear amplification step. Approximately 2 µg total RNA from drug-resistant lines was labeled with the Cye5 dye. RNA from the R1 parental cells was labeled with the Cye3 dye and served as a common reference, while the other labeled samples were cohybridized in a classic 2-color hybridization scheme. Hybridization was carried out using the conditions specified by the manufacturer. The Agilent microarrays contained 44k 60mers in sense orientation. Scanned images were processed for quality assessment using the Agilent Feature Extraction software. Preprocessed, normalized expression values were imported into the BRB-ArrayTools (National Cancer Institute, Bethesda, MD) software for visualization and analysis. A list of genes was compiled that composed of statistically significant changes that were least 2-fold increases or decreases in expression.
Real-time PCR
Total RNA was extracted from confluent 100-mm plates of CWR-R1 and drug-resistant cells using TRIzol reagent (Invitrogen, Carlsbad, CA). Approximately 5 µg total RNA was treated with DNase I (Ambion, Austin, TX) and then used for reverse transcription using the iScript kit (Bio-Rad Laboratories, Hercules, CA). PCR amplification of the resultant was carried out using FastStart Taq Polymerase (Roche, Basel, Switzerland), following the program: 10-minute denaturation at 94°C, 35 cycles at 94°C for 30 seconds, 58°C for 30 seconds, and 72°C for 45 seconds. PCR products were run on 1% agarose (Invitrogen) gel with a 100-bp DNA ladder. OCT4 primers used were the following: ACACCTGGCTTCGGATTTCG (forward) and GGCGATGTGGCTGATCTGCT (reverse). Bio-Rad GelDoc imagining system was used to capture images of PCR products separated by 1% agarose gels. HpaII-sensitive PCR was carried out as described by Yeo et al.25 Approximately 0.5 µg genomic DNA was incubated with HpaII or MspI for 48 hours, with Ntera DNA serving as a negative control. Digested DNA was amplified using real-time PCR using the undigested DNA for normalization. Real-time PCR was performed as described previously.35 18S primers used were TTGACGGAAGGGCACCACCAG (forward) and GCACCACCACCCACGGAATCG (reverse); OCT4 target gene primers: MID1 caccgtgtggaacaagtgtc (forward) and atttcgggacacttctggtg (reverse); IL1RN ggaagatgtgcctgtcctgt (forward) and ccttcgtcaggcatattggt (reverse); MYB ggcagaaatcgcaaagctac (forward) and gcagggagttgagctgtagg (reverse); RPS27 gacctacgcacacgagaaca (forward) and cgtttgtgcatggctaaaga (reverse); CUGBP2 ccatacgcatccacacactc (forward) and gcaaacatcgacggaatctt (reverse).
Western blot and antibodies
Immunoblotting was carried out as described previously.36 Mouse monoclonal anti-OCT4 antibody was from Santa Cruz Biotechnology (sc5279) (Santa Cruz, CA). Monoclonal anti-actin antibody (sc8432) were both from Santa Cruz Biotechnology. All experiments shown are representative of at least 3 experiments.
Soft agar assay
Soft agar assays were set up as described previously.37 Briefly, approximately 1 × 104 cells from CWR-R1 or its drug-resistant derivatives were plated in a 24-well plate and allowed to incubate for 8 days. Plates were stained with iodonitro tetrazolium blue (2 mg/mL) and quantified using an automated reader from Microbiology International (Frederick, MD) under an inverted microscope. Only colonies measuring between 50 to 200 µm were counted by the reader.
In vivo xenograft models
The tumor growth of CWR-R1 and its derivates in xenograft models was carried out as described previously,38 using the cell number as indicated in the figures. For tumor initiation experiments, 10 cells suspended in 25 µL medium were mixed with 25 µL of Matrigel (BD Biosciences, Franklin Lakes, NJ) and then subcutaneously (s.c.) injected in flanks of castrated NOD/SCID mice. For knockdown experiments, 104 cells were mixed with 100 µL of Matrigel (BD Biosciences) at 48 hours postinfection and then s.c. injected as above. Tumor volumes were measured weekly and calculated using the formula 0.5236 × r12 × r2, wherein r1 < r2. Differences in tumor sizes formed on both flanks were compared by the paired t test.
Immunohistochemical analysis
The Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) was used for immunohistochemical staining according to the protocol supplied by the manufacturer using a monoclonal OCT4 antibody as described previously.39
Supplementary Material
Acknowledgments
The authors thank Dr. C.W. Gregory for kindly providing CWR-R1 cells and Dr. Douglas Ross for supplying Ko143 for SP analysis.
Footnotes
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported in part by grants from DOD (W81XWH-08-1-0174) and Maryland Stem Cell Research Fund to Y.Q. and DOD Pre-doctoral Fellowship (W81XWH-08-1-0068) to D.E.L.
References
- 1. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34-45 [DOI] [PubMed] [Google Scholar]
- 2. Lee JT, Lehmann BD, Terrian DM, et al. Targeting prostate cancer based on signal transduction and cell cycle pathways. Cell Cycle. 2008;7:1745-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513-20 [DOI] [PubMed] [Google Scholar]
- 4. Oudard S, Banu E, Beuzeboc P, et al. Multicenter randomized phase II study of two schedules of docetaxel, estramustine, and prednisone versus mitoxantrone plus prednisone in patients with metastatic hormone-refractory prostate cancer. J Clin Oncol. 2005;23:3343-51 [DOI] [PubMed] [Google Scholar]
- 5. Ueda K, Cornwell MM, Gottesman MM, et al. The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem Biophys Res Commun. 1986;141:956-62 [DOI] [PubMed] [Google Scholar]
- 6. Hopper-Borge E, Chen ZS, Shchaveleva I, Belinsky MG, Kruh GD. Analysis of the drug resistance profile of multidrug resistance protein 7 (ABCC10): resistance to docetaxel. Cancer Res. 2004;64:4927-30 [DOI] [PubMed] [Google Scholar]
- 7. Moreno-Aspitia A, Perez EA. Treatment options for breast cancer resistant to anthracycline and taxane. Mayo Clin Proc. 2009;84:533-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465-77 [DOI] [PubMed] [Google Scholar]
- 9. de Jong J, Looijenga LH. Stem cell marker OCT3/4 in tumor biology and germ cell tumor diagnostics: history and future. Crit Rev Oncog. 2006;12:171-203 [DOI] [PubMed] [Google Scholar]
- 10. Monk M, Holding C. Human embryonic genes re-expressed in cancer cells. Oncogene. 2001;20:8085-91 [DOI] [PubMed] [Google Scholar]
- 11. Zangrossi S, Marabese M, Broggini M, et al. Oct-4 expression in adult human differentiated cells challenges its role as a pure stem cell marker. Stem Cells. 2007;25:1675-80 [DOI] [PubMed] [Google Scholar]
- 12. Cantz T, Key G, Bleidissel M, et al. Absence of OCT4 expression in somatic tumor cell lines. Stem Cells. 2008;26:692-7 [DOI] [PubMed] [Google Scholar]
- 13. Liedtke S, Enczmann J, Waclawczyk S, Wernet P, Kogler G. Oct4 and its pseudogenes confuse stem cell research. Cell Stem Cell. 2007;1:364-6 [DOI] [PubMed] [Google Scholar]
- 14. Lee J, Kim HK, Rho JY, Han YM, Kim J. The human OCT-4 isoforms differ in their ability to confer self-renewal. J Biol Chem. 2006;281:33554-65 [DOI] [PubMed] [Google Scholar]
- 15. Liedtke S, Stephan M, Kogler G. Oct4 expression revisited: potential pitfalls for data misinterpretation in stem cell research. Biol Chem. 2008;389:845-50 [DOI] [PubMed] [Google Scholar]
- 16. Panagopoulos I, Moller E, Collin A, Mertens F. The POU5F1P1 pseudogene encodes a putative protein similar to POU5F1 isoform 1. Oncol Rep. 2008;20:1029-33 [PubMed] [Google Scholar]
- 17. Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greco SJ, Liu K, Rameshwar P. Functional similarities among genes regulated by OCT4 in human mesenchymal and embryonic stem cells. Stem Cells. 2007;25:3143-54 [DOI] [PubMed] [Google Scholar]
- 19. Kang J, Shakya A, Tantin D. Stem cells, stress, metabolism and cancer: a drama in two Octs. Trends Biochem Sci. 2009;34:491-9 [DOI] [PubMed] [Google Scholar]
- 20. Xie Y, Xu K, Linn DE, et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. 2008;283:3349-56 [DOI] [PubMed] [Google Scholar]
- 21. Wu C, Alman BA. Side population cells in human cancers. Cancer Lett. 2008;268:1-9 [DOI] [PubMed] [Google Scholar]
- 22. Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. 2003;22:7340-58 [DOI] [PubMed] [Google Scholar]
- 23. Tran A, Jullien V, Alexandre J, et al. Pharmacokinetics and toxicity of docetaxel: role of CYP3A, MDR1, and GST polymorphisms. Clin Pharmacol Ther. 2006;79:570-80 [DOI] [PubMed] [Google Scholar]
- 24. Freberg CT, Dahl JA, Timoskainen S, Collas P. Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell. 2007;18:1543-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yeo S, Jeong S, Kim J, Han JS, Han YM, Kang YK. Characterization of DNA methylation change in stem cell marker genes during differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2007;359:536-42 [DOI] [PubMed] [Google Scholar]
- 26. Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207-19 [DOI] [PubMed] [Google Scholar]
- 28. Deng X, Liu H, Huang J, et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sotomayor P, Godoy A, Smith GJ, Huss WJ. Oct4A is expressed by a subpopulation of prostate neuroendocrine cells. Prostate. 2009;69:401-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saxe JP, Tomilin A, Scholer HR, Plath K, Huang J. Post-translational regulation of Oct4 transcriptional activity. PLoS One. 2009;4:e4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakanishi T, Chumsri S, Khakpour N, et al. Side-population cells in luminal-type breast cancer have tumour-initiating cell properties, and are regulated by HER2 expression and signalling. Br J Cancer. 2010;102:815-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu K, Shimelis H, Linn DE, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell. 2009;15:270-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim O, Jiang T, Xie Y, Guo Z, Chen H, Qiu Y. Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene. 2004;23:1838-44 [DOI] [PubMed] [Google Scholar]
- 37. Fiebig HH, Maier A, Burger AM. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur J Cancer. 2004;40:802-20 [DOI] [PubMed] [Google Scholar]
- 38. Guo Z, Dai B, Jiang T, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309-19 [DOI] [PubMed] [Google Scholar]
- 39. Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.