

Abstract
Constitutively activated mutant FLT3 has emerged as a promising target for therapy for the subpopulation of acute myeloid leukemia (AML) patients who harbor it. The small molecule inhibitor, PKC412, targets mutant FLT3 and is currently in late-stage clinical trials. However, the identification of PKC412-resistant leukemic blast cells in the bone marrow of AML patients has propelled the development of novel and structurally distinct FLT3 inhibitors that have the potential to override drug resistance and more efficiently prevent disease progression or recurrence. Here, we present the novel first-generation “type II” FLT3 inhibitors, AFG206, AFG210, and AHL196, and the second-generation “type II” derivatives and AST487 analogs, AUZ454 and ATH686. All agents potently and selectively target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. Cross-resistance between “type I” inhibitors, PKC412 and AAE871, was demonstrated. While cross-resistance was also observed between “type I” and first-generation “type II” FLT3 inhibitors, the high potency of the second-generation “type II” inhibitors was sufficient to potently kill “type I” inhibitor-resistant mutant FLT3-expressing cells. The increased potency observed for the second-generation “type II” inhibitors was observed to be due to an improved interaction with the ATP pocket of FLT3, specifically associated with introduction of a piperazine moiety and placement of an amino group in position 2 of the pyrimidine ring. Thus, we present 2 structurally novel classes of FLT3 inhibitors characterized by high selectivity and potency toward mutant FLT3 as a molecular target. In addition, presentation of the antileukemic effects of “type II” inhibitors, such as AUZ454 and ATH686, highlights a new class of highly potent FLT3 inhibitors able to override drug resistance that less potent “type I” inhibitors and “type II” first-generation FLT3 inhibitors cannot.
Keywords: neoplasia, FLT3 inhibitor, AML, leukemia, structure affinity, drug resistance, drug potency
Introduction
Acute myelocytic leukemia (AML) has an incidence of around 10,000 new cases annually in the United States and is characterized by abnormal proliferation of myeloid progenitor cells, as well as a total or incomplete block in cellular differentiation.1 Approximately 30% of AML patients, and a subset of ALL patients, harbor a mutant form of the class III receptor tyrosine kinase, FLT3 (Fms-Like Tyrosine kinase-3; STK-1 [human Stem Cell Tyrosine Kinase-1]; or FLK-2 [Fetal Liver Kinase-2]).2 Constitutively activated FLT3 occurs most frequently as internal tandem duplications (ITD) within the juxtamembrane domain3 and is observed in approximately 20% to 25% of AML patients but in below 5% of patients with myelodysplastic syndrome (MDS).3-8 The transplantation of murine bone marrow cells infected with a retrovirus expressing an FLT3-ITD mutant has been shown to lead to the development of a rapidly lethal myeloproliferative disease in mice.8
Also occurring are point mutations within the “activation loop” of FLT3.9 These are thought to alter the conformation of the domain, forcing it into an “activated” configuration. Approximately 7% of AML cases harbor this type of mutation, with the majority with a missense mutation in the aspartic acid residue at position 835. Other point mutations in the kinase domain occurring less frequently have been identified, including N841I10 and Y842C.11
The N-indolocarbazole PKC412 (midostaurin, N-benzoyl-staurosporine; Novartis Pharma AG, Basel, Switzerland) is a broad-spectrum small molecule inhibitor that is effective against mutant FLT3-expressing cells.12 A phase Ib clinical trial was conducted in which newly diagnosed AML patients were treated with PKC412 (at 50 mg po bid) in sequential and simultaneous combinations with daunorubicin and cytarabine induction and high-dose cytarabine consolidation. Patients in this trial experienced transient and/or reversible side effects, and clinical complete responses (CR) were observed in 100% of patients harboring mutant FLT3.13 PKC412 is currently in late-stage phase III clinical trials.
While small molecule inhibitors like PKC412 are showing promise in the clinic, up to now, none has achieved sustained cytogenic responses as a single agent in AML patients. In addition, a growing problem for the treatment of acute leukemia is the development of resistance due to acquired point mutations in the molecular targets.14,15 Indeed, the detection of drug-resistant leukemic blast cells in AML patients undergoing PKC412 therapy has sparked the screening and characterization of novel, structurally diverse inhibitors of FLT3 that, if used in combination with antileukemic agents, are predicted to prevent the development of drug resistance.
Pharmacological inhibitors of kinase activity are characterized as being in 1 of 3 main classes: “type I,” “type II,” or “non-ATP competitive” inhibitors.16 Type I, or “DFG-in” ATP competitive inhibitors, directly compete with ATP in the ATP binding site. Type II, or “DFG-out” ATP competitive inhibitors, in addition to binding the ATP binding site, also engage an adjacent hydrophobic binding site that is only accessible when the kinase is in an inactivated configuration. Non-ATP competitive inhibitors bind at sites outside the ATP binding site that affect the activity of the kinase.
We report here initial characterization of first-generation FLT3 inhibitors, AFG206, AFG210, and AHL196, assumed to be of the “type II” class, and second-generation derivative analog “type II” FLT3 inhibitors, AUZ454 and ATH686. All are potent and selective inhibitors of mutant FLT3 protein kinase activity. We show that these compounds, via inhibition of FLT3 kinase activity, selectively kill leukemic cells harboring mutant FLT3 with no apparent effect on cells harboring wild-type FLT3. However, the structural composition of the second-generation inhibitors allows these agents to exhibit higher potency than PKC412 and thus renders them able to override resistance to PKC412, as well as another FLT3 inhibitor, AAE871, assumed to be of the “type I” classification. These results highlight the emergence of a novel class of FLT3 inhibitors that exhibit high selectivity and potency toward mutant FLT3 as an oncogenic target and as such can override resistance to “type I” FLT3 inhibitors, such as PKC412.
Results
Inhibition of mutant FLT3 kinase by first-generation FLT3 inhibitors, AFG206, AFG210, and AHL196
An important class of “type II” kinase inhibitors is based on the diphenyl urea chemical motif.19 Because their core structure is based on this motif, the novel first-generation FLT3 inhibitors, AFG206, AFG210, and AHL196, fall into the “type II” ATP competitive inhibitor class of kinase inhibitors (see chemical structures in Fig. 1). Consequently, the binding mode of these inhibitors was modeled according to the “type II” phamacophore.20 This is illustrated in Figure 2 for AFG210, where the main interactions of the inhibitor with the ATP pocket of FLT3 in a “type II” binding mode are represented. With no crystal structure of FLT3 in the “DFG-out” conformation (the conformation to which “type II” inhibitors bind) being available, we resorted to homology modeling. A model of FLT3 in the “DFG-out” conformation was constructed using the WHAT IF program, a molecular modeling and drug design program (G. Vriend, 1990), based on the coordinates of the B-Raf kinase in complex with BAY-43-9006, a diphenyl urea “type II” inhibitor (PDB code: 1UWH). This model was subsequently used to dock AFG210 and its analogs. The binding mode of such inhibitors in the ATP pocket of FLT3 is illustrated with a model of the enzyme AFG210 complex in Figure 2. The pyridine moiety binds in the hinge region of the pocket, establishing a hydrogen bond with the backbone NH group of residue C694, while the trifluoromethyl-phenyl moiety occupies the adjacent hydrophobic site lined by residues M665, I674, L802, M664, and C807. In addition, the urea function is involved in 2 hydrogen bonds with the kinase that are characteristic of “type II” inhibitors: one with the aspartic acid residue of the DFG motif (D829), and the other with the glutamic acid residue of helix C (E661). The synthesis of these compounds has been previously described.21
Figure 1.
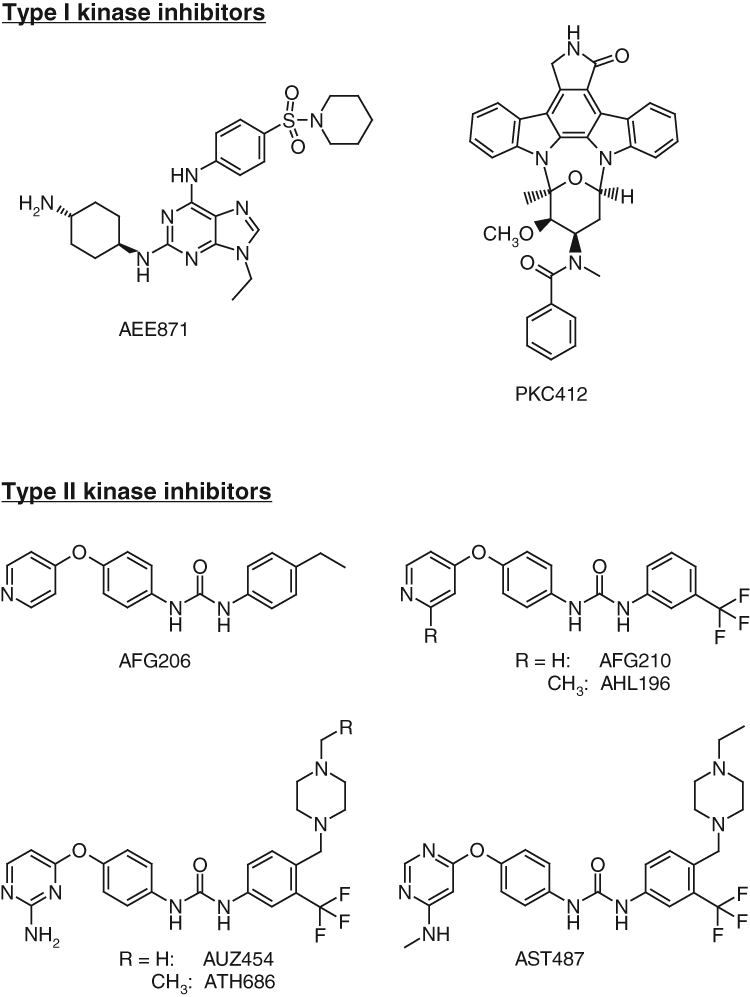
Chemical structures and classification. Chemical structures of type I kinase inhibitors (AAE871 and PKC412) and type II inhibitors (AFG206, AHL196, AUZ454, and ATH686).
Figure 2.
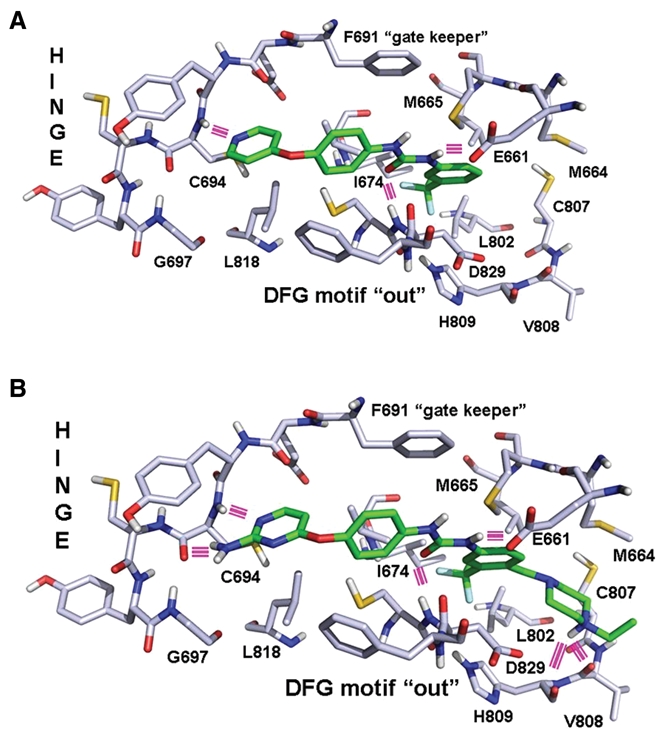
Structural models of enzyme-drug complexes. (A) Binding model of AFG210 in the ATP pocket of FLT3. Hydrogen bonds appear as pink lines. (B) Binding model of ATH686 in the ATP pocket of FLT3. Hydrogen bonds appear as pink lines.
Treatment of FLT3-ITD-Ba/F3 cells and D835Y-Ba/F3 cells with AFG206, AFG210, or AHL196 potently inhibited cell proliferation (IC50 around 0.1 µM) via induction of apoptosis (Fig. 3). Parental Ba/F3 cells were not affected by up to 1 µM of each agent (Fig. 3). Supplementation of culture media with WEHI, used as a source of IL-3, led to rescue of the cells from inhibition of proliferation and induction of apoptosis (Fig. 3 and Suppl. Figs. S1, S2, and S5). This suggests that these compounds selectively inhibit FLT3-ITD, with no effect on IL-3 signaling. The antiproliferative activity of AFG206, AFG210, and AHL196 was only modestly affected by the addition of human serum (Suppl. Fig. S10).
Figure 3.
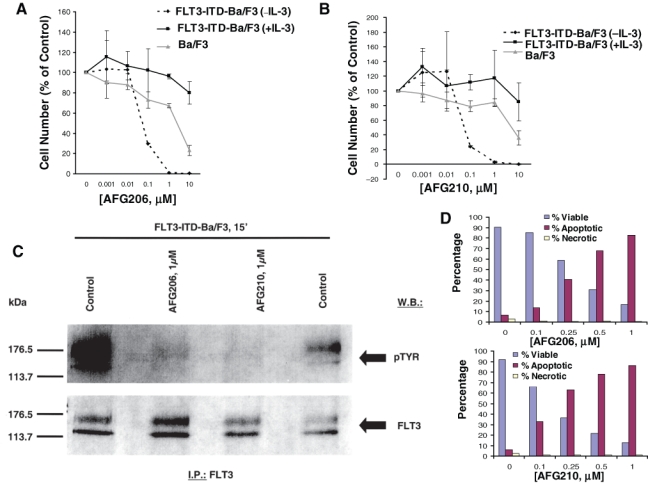
Inhibition of mutant FLT3 kinase by first-generation type II FLT3 inhibitors, AFG206 and AFG210. Approximately 3-day treatment of Ba/F3 cells (+ IL-3) and FLT3-ITD-Ba/F3 cells (+/− IL-3) with AFG206 (A) and AFG210 (B). (C) FLT3 I.P./Western. (D) Induction of apoptosis following an approximately 3-day treatment of FLT3-ITD-Ba/F3 cells with AFG206 (upper panel) and AFG210 (lower panel). Staining cells simultaneously with FITC-Annexin V and the nonvital dye propidium iodide allows (bivariate analysis) the discrimination of intact cells (FITC–PI–), early apoptotic cells (FITC+PI–), and late apoptotic or necrotic cells (FITC+PI+). Results shown in the bar graph are derived from cytograms in which early apoptotic cells are in the lower right quadrant that are Annexin V positive and PI negative; late apoptotic or necrotic cells are in the upper right quadrant that are PI positive and Annexin V positive; live cells in the lower left quadrant are negative for both fluorescent probes. Necrotic cells show up in the upper left-hand quadrant.
Treatment of FLT3-ITD- and D835Y-expressing cells with AFG206, AFG210, or AHL196 inhibited autophosphorylation of mutant FLT3 in these cells, with no apparent reduction in levels of the FLT3 protein (Fig. 3 and Suppl. Figs. S3A-C and S4). Whole cell lysate Western analysis showed strong inhibition of pSTAT5 and pMAPK expression (with no apparent effects on total STAT5 or MAPK expression levels), following 1.5-hour treatments with AFG206 (1 µM) or AFG210 (1 µM) (Suppl. Fig. S3D and S3E). These results suggest that FLT3 kinase is a target of AFG206, AFG210, and AHL196, and inhibition of mutant FLT3 kinase activity leads to loss of growth factor independence and consequent cell death.
The ability of the “type II” FLT3 inhibitors, AFG206 and AFG210, to positively combine with PKC412 was tested. Dose-response curves corresponding to combination treatments were shifted to the left as compared to dose-response curves corresponding to single-agent treatments (Suppl. Figs. S6 and S7). This suggests that “type II” FLT3 inhibitors like AFG206 and AFG210 could potentially be used in combination with “type I” inhibitors like PKC412 to possibly improve clinical results.
Inhibition of mutant FLT3 kinase by second-generation FLT3 inhibitors, AUZ454 and ATH686
A class of second-generation derivatives was developed based on the structural motif of “type II” inhibitors. AUZ454 and ATH686 are structural analogs of previously described AST487.22 They fall into the “type II” ATP-competitive inhibitor class of kinase inhibitors (see chemical structures in Fig. 1). These compounds fill the ATP extended pocket existing in a certain form of inactive conformation of the target kinase yet also bind to residues that interact with type I inhibitors in the so-called hinge-adenine region of the pocket. Compared to the first-generation “type II” inhibitors, AFG206, AFG210, and AHL196, additional structural features interacting favorably with the ATP pocket of FLT3 have been introduced in these molecules as shown in Figure 2B, where a model of the ATH686 kinase complex is represented. These consist of an amino group placed in position 2 of the pyrimidine ring to form an additional hydrogen bond with the backbone of hinge residue C694 and a piperazine moiety able to form 2 novel hydrogen bonds with the backbone carbonyl groups of residues H809 and V808 at the bottom of the extended pocket. The synthesis of these compounds has been previously described.21,23,24
Treatment of FLT3-ITD-Ba/F3 cells and D835Y-Ba/F3 cells with AUZ454 or ATH686 very potently inhibited cell proliferation (IC50 around 0.001 µM) via induction of apoptosis (Fig. 4 and Suppl. Fig. S9). Parental Ba/F3 cells were not affected by up to at least 0.1 µM of each agent (Suppl. Fig. S9). Supplementation of culture media with WEHI, used as a source of IL-3, led to rescue of the cells (Fig. 4). As with the first-generation inhibitors, the antiproliferative activity of AUZ454 and ATH686 was only modestly affected by the addition of human serum (Suppl. Fig. S10). As was observed previously with AST487,22 efficacy was similarly observed for AUZ454 and ATH686 against mutant FLT3-positive AML patient samples (Suppl. Fig. S11).
Figure 4.
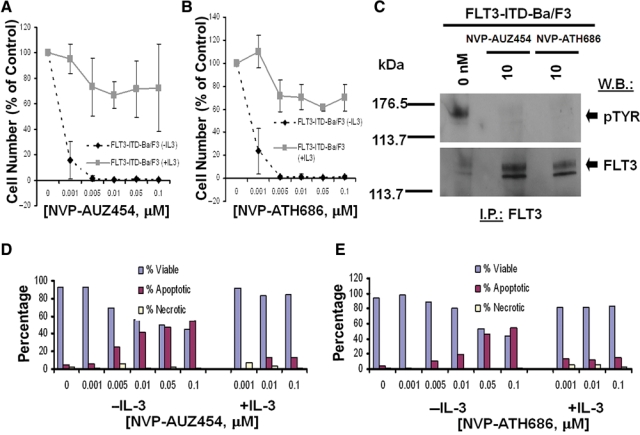
Inhibition of mutant FLT3 kinase by second-generation type II FLT3 inhibitors, AUZ454 and ATH686. (A, B) Three-day treatment of FLT3-ITD-Ba/F3 cells, in the presence and absence of IL-3, with AUZ454 (A) and ATH686 (B). (C) Treatment of FLT3-ITD-Ba/F3 cells for 15 minutes with AUZ454 or ATH686, respectively, at 10 nM. (D, E) Effects of AUZ454 (D) and ATH686 (E) on viability of FLT3-ITD-Ba/F3 cells, in the absence and presence of IL-3.
Treatment of FLT3-ITD- and D835Y-expressing cells with AUZ454 and ATH686 inhibited autophosphorylation of mutant FLT3 in these cells, with no apparent reduction in levels of the FLT3 protein (Fig. 4). In addition, cells expressing the novel point mutant, FLT3-N841I, also showed sensitivity to AUZ454 and ATH686 (Suppl. Fig. S8). These results suggest that FLT3 kinase is a target of AUZ454 and ATH686. Whole cell lysate Western analysis showed strong inhibition of pSTAT5 and pMAPK expression (with no apparent effects on total STAT5 or MAPK expression levels), following 1.5-hour treatments with 0.1 µM AUZ454 (Suppl. Fig. S3D and S3E).
Resistance of mutant FLT3-expressing cells to “type I” FLT3 inhibitor, AAE871
AAE871 is a “type I” inhibitor, like PKC412, and interacts with the same residues of the ATP site as PKC412 (Fig. 1). The synthesis of AAE871 has been previously described.25 Results of in vitro kinase studies suggested that PKC412 inhibits the tyrosine kinase activity of FLT3 with an IC50 of 0.079 µM and that AAE871 inhibits the tyrosine kinase activity of FLT3 with an IC50 of 0.034 µM. Cellular proliferation studies suggested that AAE871 potently inhibits proliferation of FLT3-ITD- and D835Y-expressing cells (IC50 < 0.01 µM) through selective inhibition of FLT3 kinase activity (Fig. 5 and Suppl. Fig. S12).
Figure 5.
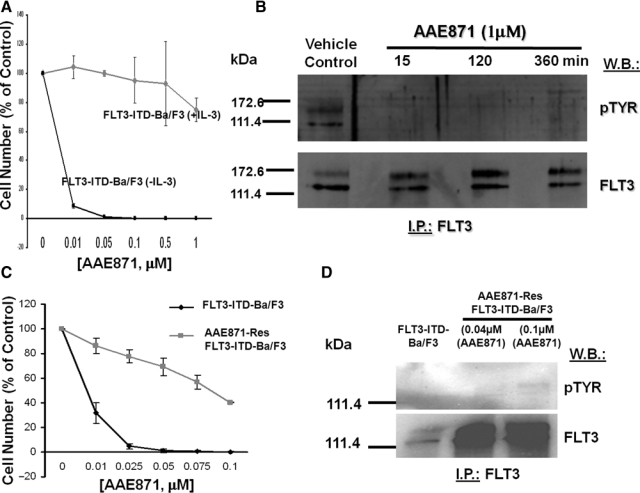
Resistance of mutant FLT3-expressing cells to “type I” FLT3 inhibitor, AAE871. (A) Approximately 3-day treatment of FLT3-ITD-Ba/F3 cells (+/− IL-3) with AAE871. (B) FLT3 I.P./Western treatment of FLT3-ITD-Ba/F3 cells for 15, 120, and 360 minutes with AAE871 at 1 µM. (C) Approximately 3-day treatment of FLT3-ITD-Ba/F3 cells and AAE871-resistant FLT3-ITD-Ba/F3 cells (made resistant to 0.04 or 0.1 µM AAE871) with AAE871. (D) FLT3 I.P./Western treatment of AAE871-sensitive or -resistant FLT3-ITD–expressing cells with AAE871.
Continuous (several months’ duration) cell culture of FLT3-ITD–expressing Ba/F3 cells in the presence of gradually increasing concentrations of AAE871 led to the development of a cell line exhibiting a drug-resistant phenotype (highest level of drug resistance achieved at 0.1 µM) (Fig. 5). AAE871-resistant cells were characterized as overexpressing FLT3-ITD (Fig. 5). The level of overexpression of FLT3-ITD in AAE871-resistant cells was comparable to levels of mutant FLT3 observed in PKC412-resistant cells (Suppl. Fig. S13A and S13B).
AAE871-resistant cells resistant to 0.1 µM AAE871 and maintained in the continuous presence of 0.1 µM AAE871 showed a modest increase in levels of phosphorylated FLT3, as compared to drug-sensitive cells (Fig. 5D). In Supplementary Figure S13A and S13B, no appreciable change in the overall levels of phosphorylated FLT3 expression was observed in AAE871-resistant cells cultured in the continuous presence of 0.04 µM. These data, which suggest that the IC50 of AAE871 against FLT3 kinase activity is >0.1 µM in drug-resistant cells, can be compared to data shown in Supplementary Figure S12A, where the IC50 of AAE871 against FLT3 kinase activity in drug-sensitive cells is >0.01 µM and <0.1 µM (which supports in vitro kinase assay results suggesting an IC50 of 0.034 µM for AAE871 against FLT3). These results combined confirm FLT3-ITD as a target of AAE871. When investigating levels of relevant signaling molecules in the AAE871-resistant cells, we did not observe a comparable increase in levels of pSTAT5 or pMAPK in AAE871-resistant Ba/F3-FLT3-ITD cells (compared to drug-sensitive Ba/F3-FLT3-ITD cells), despite overexpression of FLT3 (Suppl. Fig. S13C and S13D).
Response of “type I” FLT3 inhibitor-resistant mutant FLT3-expressing cells to “type II” first- and second-generation FLT3 inhibitors
We were interested in determining whether cellular resistance to one “type I” inhibitor would confer cross-resistance to other “type I” inhibitors. Treatment of AAE871-resistant mutant FLT3-expressing cells with PKC412 showed a significant rightward shift in the dose-response curve, as compared to treatment of drug-naive mutant FLT3-expressing cells (IC50 for PKC412 against wild-type FLT3-ITD = 0.01-0.025 µM; IC50 for PKC412 against AAE871-resistant FLT3-ITD = 0.05-0.075 µM) (Fig. 6). Similarly, treatment of PKC412-resistant mutant FLT3-expressing cells with AAE871 resulted in a rightward shift in the dose-response curve, as compared to treatment of drug-naive mutant FLT3-expressing cells (IC50 for AAE871 against wild-type FLT3-ITD <0.01 µM; IC50 for AAE871 against PKC412-resistant FLT3-ITD <0.05 µM). Treatment of PKC412-resistant mutant FLT3-expressing cells with “type II” first-generation FLT3 inhibitors, AFG206, AFG210, and AHL196, led to rightward shifts in the dose-response curves, as compared to treatment of drug-naive FLT3-expressing cells (IC50 <0.1 µM against wild-type FLT3-ITD; IC50 >0.25 µM against PKC412-resistant FLT3-ITD) (Fig. 6 and Suppl. Fig. S14). These results suggest that cross-resistance between “type I” FLT3 inhibitors can occur, as can cross-resistance between “type I” FLT3 inhibitors and “type II” FLT3 inhibitors of similar potency.
Figure 6.
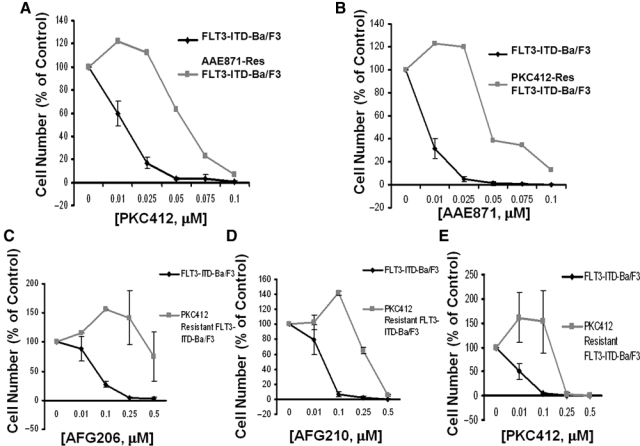
Cross-resistance of “type I” FLT3 inhibitor–resistant mutant FLT3-expressing cells to “type I” and “type II” first-generation FLT3 inhibitors. (A) Approximately 3-day treatment of FLT3-ITD-Ba/F3 cells and AAE871-resistant FLT3-ITD-Ba/F3 cells with PKC412. (B) Approximately 3-day treatment of FLT3-ITD-Ba/F3 cells and PKC412-resistant FLT3-ITD-Ba/F3 cells with AAE871. (C) Approximately 3-day treatment of FLT3-ITD-Ba/F3 and PKC412-resistant Ba/F3 cells with AFG206. (D) Approximately 3-day treatment of FLT3-ITD-Ba/F3 and PKC412-resistant Ba/F3 cells with AFG210. (E) Approximately 3-day treatment of FLT3-ITD-Ba/F3 and PKC412-resistant Ba/F3 cells with PKC412.
We were then interested in investigating the potential of the highly potent “type II” second-generation FLT3 inhibitors to override drug resistance conferred by mutant FLT3-expressing cells made resistant to “type I” FLT3 inhibitors. We observed an approximate 10-fold shift in potency of AUZ454 and ATH686 when tested against PKC412-resistant FLT3-ITD–positive cells, as compared to wild-type FLT3-ITD–expressing cells (Fig. 7). For AUZ454 and ATH686 against PKC412-resistant FLT3-ITD cells, the IC50 is approximately ≤ 0.01 µM, which, based on IL-3 rescue experiments, is a selective and physiologically relevant concentration (Fig. 7). This is in contrast to the IC50 of PKC412 against PKC412-resistant cells, which is approximately 1 µM (Fig. 7). Based on the lack of IL-3 rescue observed at this concentration of PKC412, this is not a physiologically relevant concentration, and cell death at this concentration is likely due to nonselective toxicity.
Figure 7.
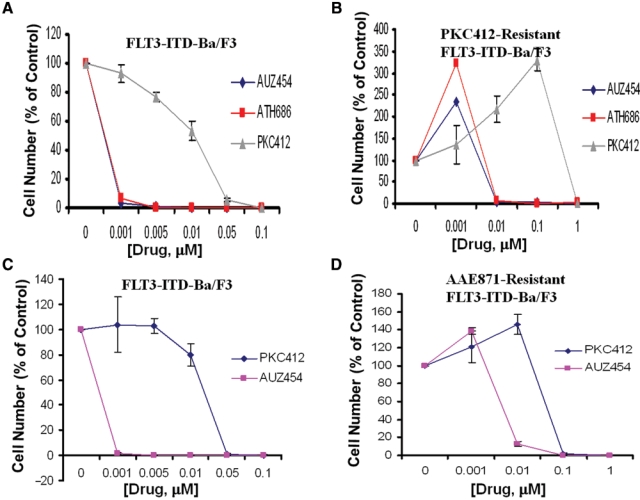
Response of “type I” FLT3 inhibitor–resistant mutant FLT3-expressing cells to “type II” second-generation FLT3 inhibitors. (A, B) Three-day treatment of FLT3-ITD-Ba/F3 cells with AUZ454, ATH686, or PKC412 (A) and 3-day treatment of PKC412-resistant FLT3-ITD-Ba/F3 cells with AUZ454, ATH686, or PKC412 (B). (C, D) Three-day treatment of FLT3-ITD-Ba/F3 cells with PKC412 or AUZ454 (C) and 3-day treatment of AAE871-resistant FLT3-ITD-Ba/F3 cells with PKC412 or AUZ454 (D).
Discussion
Clinically available therapies for AML often fail due to treatment-induced mortality or drug resistance.26 While the use of stand-alone, standard chemotherapy translates into low therapy-induced mortality, there is a high risk of relapse stemming from drug resistance.27 Conversely, a high therapy-induced mortality and a lower risk of relapse are associated with allogeneic transplantation (alloBMT), which shows more promise in younger patients and thus has a small influence on the majority of AML patients who tend to be over the age of 65 years.27,28
Constitutively activated mutant FLT3, which is expressed in a subpopulation of AML patients, is an attractive therapeutic target for AML. Presently, several FLT3 inhibitors, including the late-stage in-development “type I” inhibitor, PKC412, are undergoing clinical investigation. However, the FLT3 inhibitors clinically tested up to now generally induce only partial and transient responses in patients when used as single agents. This, coupled with the discovery of drug-resistant leukemic blasts in FLT3 inhibitor–treated AML patients, highlights a need for identification and development of novel, structurally diverse FLT3 inhibitors that could be used to override drug resistance. Such agents would ideally confer higher potency and/or less toxicity that could either be used effectively as stand-alone agents or that could be combined with other agents to effectively suppress disease progression and prolong patient survival.
We present here a structure-affinity comparison between the novel class of first-generation “type II” FLT3 inhibitors, AFG206, AFG210, and AHL196, and the novel class of second-generation “type II” derivatives and AST487 analogs, AUZ454 and ATH686. All have demonstrated the ability to potently and selectively inhibit FLT3 protein kinase activity, and each induced programmed cell death and inhibited cell cycle progression of cells expressing mutant FLT3. However, the second-generation “type II” derivatives exhibit substantially higher potency than the first-generation “type II” inhibitors. This increased potency is due to enhanced target affinity, associated with introduction of an amino group in position 2 of the pyrimidine ring, as well as introduction of a piperazine moiety. These features result in more favorable interaction with the ATP pocket of the FLT3 target. The discovery and development of novel agents, such as AUZ454 and ATH686, with unique structures conferring higher potency and selectivity toward FLT3 as a target, may allow for more complete inhibition of the FLT3 kinase protein target as compared to that of existing therapies in preclinical and clinical development.
As a way to potentially suppress the emergence of point mutations in FLT3 conferring drug resistance, 2 different FLT3 inhibitors could be used together if the mechanism whereby cells develop resistance to each is different. Dose-response curve shifts to the left were observed for the combination of AFG206 or AFG210 with PKC412, respectively, as compared to any one agent alone. CalcuSyn analysis of these data was carried out by converting the percentage of viable cells to fraction affected and carrying out standard CalcuSyn software (Biosoft, Cambridge, UK) analysis. The numerical values (combination indices) generated by CalcuSyn suggest that the combinations of AFG206 + PKC412 or AFG210 + PKC412 were generally additive to synergistic across a range of doses.
In addition to searching for novel and potent FLT3 inhibitors representative of unique structural classes, researchers are attempting to gain a better understanding of the underlying mechanisms of drug resistance in AML. Clinical trial data with tyrosine kinase inhibitors show that while there is rapid and efficient clearance of blasts in the peripheral blood of patients, a delayed or marginal decrease in bone marrow blasts has been observed. Stromal cells, which provide protective viability signals to leukemic cells, have been implicated in this mode of resistance,29 and small molecule CXCR4 inhibitors have been shown to be effective in augmenting the antileukemic effects of chemotherapy or FLT3 inhibitors.30-32 Other mechanisms of drug resistance to tyrosine kinase inhibitors include acquired point mutations in the molecular targets.14,15 For example, resistance to PKC412 in patients has been attributed to pre-existing or acquired mutations in the kinase domain of FLT3.33 Other resistance mechanisms include deregulation of signaling molecules, such as those mediating STAT signaling and apoptotic signaling,34-36 which lead to a survival advantage in leukemic cells. Also reported as mechanisms of resistance to tyrosine kinase inhibitors are gene amplification and overexpression of drug target mRNA and protein.37-39
Mutant FLT3-expressing Ba/F3-derived cells were made resistant to PKC412 as previously described,12 as well as the type I first-generation inhibitor, AAE871. Both drug-resistant cell lines were characterized as having a significantly higher expression of mutant FLT3 than their drug-sensitive counterpart, with levels of phosphorylated mutant FLT3 sustained in the drug-resistant cells, even in the continuous presence of inhibitor. However, despite overexpression of mutant FLT3 protein, levels of phosphorylated STAT5 and phosphorylated MAPK, signaling molecules associated with FLT3-mediated signal transduction, were not significantly elevated in drug-resistant cells, as compared to drug-sensitive cells. We have observed a similar lack of hyperphosphorylation of STAT5 and MAPK in MOLM13 cells made resistant to PKC412 and the type II ATP competitive inhibitor, HG-7-85-01, following long-term culture in the presence of gradually increasing concentrations of inhibitor (unpublished results). In the case of PKC412- and HG-7-85-01–resistant MOLM13 cells, a significant overexpression of mutant FLT3 was observed that was accompanied by a sharp decrease in pSTAT5 and either no increase in pMAPK (in the case of HG-7-85-01– resistant cells) or a modest increase (in the case of PKC412-resistant cells) (unpublished results). One explanation could be that the continuous presence of drug in the drug-resistant cells causes the frequently observed stabilization of target protein (previously observed for BCR-ABL in nilotinib- or imatinib-treated cells17,37 and with FLT3 in PKC412-treated cells12). The continuous presence of drug, however, which is often necessary to maintain the drug-resistant phenotype, might still inhibit downstream signaling components as it would in drug-naive cells. Just as elevated expression of BCR-ABL or FLT3 in kinase inhibitor–treated cells does not compensate for the decrease in pTYR levels observed in drug-treated, drug-sensitive cells, the same might apply here in the context of the drug-resistant phenotype. These results point toward overexpression of FLT3-ITD target protein as a possible contributor to the mechanism of drug resistance. However, the underlying complete mechanism of resistance may be more complex and suggests a need for further investigation.
Cross-resistance between “type I” FLT3 inhibitors can occur, as can cross-resistance between “type I” FLT3 inhibitors and “type II” FLT3 inhibitors of similar potency. These conclusions are supported by the observation of a significant rightward shift in the dose-response curve in PKC412-treated, AAE871-resistant mutant FLT3-expressing cells, as compared to PKC412 treatment of drug-naive mutant FLT3-expressing cells. Treatment of PKC412-resistant mutant FLT3-expressing cells with AAE871 resulted in a similar rightward shift in the dose-response curve, as compared to treatment of drug-naive mutant FLT3-expressing cells. Further, treatment of PKC412-resistant mutant FLT3-expressing cells with “type II” first-generation FLT3 inhibitors, AFG206, AFG210, and AHL196, led to rightward shifts in the dose-response curves, as compared to treatment of drug-naive FLT3-expressing cells. The high potency of the second-generation “type II” inhibitors, AUZ454 and ATH686, was sufficient to kill “type I” inhibitor-resistant mutant FLT3-expressing cells.
When considering resistance of cells to agents, the selective nature of the compound is taken into consideration as this may be predictive of how effective and nontoxic the agent might be in patients who have failed conventional therapies. Thus, when comparing 2 different agents for effectiveness against a drug-resistant cell, the comparison is relative and relevant to what can be expected in terms of efficacy and selectivity in a clinical setting. While the more potent type II inhibitors were not effective against the drug-resistant cells at 0.001 µM, they showed considerable efficacy at 0.01 µM, which is a highly selective and physiologically relevant concentration (IL-3 rescue is observed at 0.01 µM). This can be compared to the complete lack of efficacy of PKC412 against the resistant cells at concentrations (≤0.1 µM) that are selective and physiologically relevant. It can therefore be concluded that the more potent type II inhibitors can potentially override drug resistance, a conclusion that warrants testing in a clinical setting involving patients who have exhibited PKC412 intolerance or resistance.
The novel “type II” inhibitors, AUZ454 and ATH686, introduce a new class of highly potent FLT3 inhibitors able to override certain forms of drug resistance, including chemoresistance that “type I” inhibitors and first-generation “type II” FLT3 inhibitors are unable to. The structural comparisons between small molecule inhibitors representative of the different classes of agents detailed here can help to serve as a guide for development of more efficacious therapeutics designed against mutant FLT3-positive AML.
Materials and Methods
Cell lines and cell culture
The IL-3–dependent murine hematopoietic cell line Ba/F3 was transduced with either FLT3-ITD-, FLT3-D835Y-, or N841I-containing MSCV retroviruses harboring a neomycin selectable marker and selected for resistance to neomycin.8,10 Mutant FLT3- transduced cells were selected for growth in G418 (1 mg/mL). The PKC412-resistant FLT3-ITD-Ba/F3 cell line was developed as previously described.12 The AAE871-resistant FLT3-ITD-Ba/F3 cell line was similarly developed as previously described for PKC412-resistant FLT3-ITD-Ba/F3 cells.12
All cell lines were cultured with 5% CO2 at 37°C in RPMI1640 (Mediatech Inc., Herndon, VA) with 10% fetal calf serum (FCS) and supplemented with 1% L-glutamine. Parental Ba/F3 cells were similarly cultured with 15% WEHI-conditioned medium as a source of IL-3. Transfected cell lines were cultured in media supplemented with 1 mg/mL G418. PKC412- and AAE871-resistant FLT3-ITD-Ba/F3 cells were cultured in the presence of PKC412 (0.04 µM, 0.05 µM) and AAE871 (0.04 µM, 0.1 µM), respectively.
Chemical compounds and biological reagents
AFG206, AFG210, AHL196, AST487, AUZ454, ATH686, PKC412, AAE871, and imatinib were synthesized by Novartis Pharma AG. Compounds were initially dissolved in DMSO to make 10-mM stock solutions and then were serially diluted to obtain final concentrations for in vitro experiments.
Antibodies and immunoblotting
Anti-p-Tyr (clone 4G10, Upstate Biotechnology, Lake Placid, NY) was used at 1:1,000 for immunoblotting. Anti-FLT3/Flk-2 (rabbit, C-20, Santa Cruz Biotechnology Inc., Santa Cruz, CA) was used at 1:200 for immunoblotting. The monoclonal anti-β-actin antibody (clone AC-15, Sigma-Aldrich, St. Louis, MO) was used at a 1:1,000 dilution. Phospho-STAT5 Tyr694 antibody (rabbit #9351S, Cell Signaling, Danvers, MA) was used at a 1:1,000 dilution. Total STAT5 (3H7) (rabbit mAb #9358, Cell Signaling) was used at a 1:1,000 dilution. Phospho-MAPK-p44/42 (T202/Y204) antibody (rabbit, Cell Signaling) was used at a 1:1,000 dilution. Total MAPK (rabbit #9102, Cell Signaling) was used at a 1:1,000 dilution. Protein lysate preparation and immunoblotting were carried out as previously described.12
In vitro kinase assays
In vitro kinase assays were performed as previously described.17
AML patient cells
Bone marrow samples from AML patients identified as harboring mutant FLT3 were processed using Ficoll-Paque Plus (GE Healthcare, Little Chalfont, UK) for the isolation of mononuclear cells. Mononuclear cells were then tested in liquid culture (Iscove’s MDM, supplemented with 20% FCS) in the presence of different concentrations of drug. All bone marrow samples from AML patients were obtained under approval of the Dana Farber Cancer Institute Institutional Review Board.
Proliferation studies, cell cycle analysis, and apoptosis assay
Cell counts for proliferation studies were obtained using the trypan blue exclusion assay, as previously described.12 Error bars represent the standard error of the mean for each data point. Programmed cell death of inhibitor-treated cells was determined using the Annexin-V-Fluos Staining Kit (Boehringer Mannheim, Indianapolis, IN), as previously described.12 Cell cycle analysis was performed as previously described.12
Drug combination studies
For drug combination studies, compounds were added simultaneously at fixed ratios to cells, and cell viability was determined by trypan blue exclusion and expressed as the function of growth affected (FA) drug-treated versus control cells. Synergy was assessed by CalcuSyn software (Biosoft), using the Chou-Talalay method.18 The combination index = [D]1 [Dx]1 + [D]2/[Dx]2, where [D]1 and [D]2 are the concentrations required by each drug in combination to achieve the same effect as concentrations [Dx]1 and [Dx]2 of each drug alone. Values less than 1 indicate synergy, whereas values greater than 1 indicate antagonism.
Supplementary Material
Footnotes
J.D.G., J.R., P.M., G.B., P.F., G.C., and P.I. have a financial interest in Novartis Pharma AG.
This work was supported by the National Institutes of Health [grant numbers CA66996, CA36167, and DK50654] to J.D.G. and by a Specialized Center of Research Award from the Leukemia and Lymphoma Society.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. McKenzie SB. Advances in understanding the biology and genetics of acute myelocytic leukemia. Clin Lab Sci. 2005;18:28-37 [PubMed] [Google Scholar]
- 2. Stirewalt DL, Radich JP. The role of FLT3 in hematopoeitic malignancies. Nat Rev Cancer. 2003;3:650-65 [DOI] [PubMed] [Google Scholar]
- 3. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the FLT3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911-8 [PubMed] [Google Scholar]
- 4. Horiike S, Yokota S, Nakao M, et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia. 1997;11:1442-6 [DOI] [PubMed] [Google Scholar]
- 5. Kiyoi H, Towatari M, Yokota S, et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12:1333-7 [DOI] [PubMed] [Google Scholar]
- 6. Kondo M, Horibe K, Takahashi Y, et al. Prognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemia. Med Pediatr Oncol. 1999;33:525-9 [DOI] [PubMed] [Google Scholar]
- 7. Rombouts WJ, Blokland I, Lowenberg B, et al. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the FLT3 gene. Leukemia. 2000;14:675-83 [DOI] [PubMed] [Google Scholar]
- 8. Kelly LM, Liu Q, Kutok JL, et al. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310-8 [DOI] [PubMed] [Google Scholar]
- 9. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434-9 [DOI] [PubMed] [Google Scholar]
- 10. Jiang J, Paez JG, Lee JC, et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood. 2004;104:1855-8 [DOI] [PubMed] [Google Scholar]
- 11. Kindler T, Breitenbuecher F, Kasper S, et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood. 2005;105:335-40 [DOI] [PubMed] [Google Scholar]
- 12. Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433-43 [DOI] [PubMed] [Google Scholar]
- 13. Stone RM, Fischer T, Paquette R, et al. Phase 1B study of PKC412, an oral FLT3 kinase inhibitor, in sequential and simultaneous combinations with daunorubicin and cytarabine (DA) induction and high-dose cytarabine consolidation in newly diagnosed patients with AML. Blood. 2005;106:404a [Google Scholar]
- 14. Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117-25 [DOI] [PubMed] [Google Scholar]
- 15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201-14 [DOI] [PubMed] [Google Scholar]
- 16. Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;7:358-64 [DOI] [PubMed] [Google Scholar]
- 17. Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129-41 [DOI] [PubMed] [Google Scholar]
- 18. Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enz Regul. 1984;22:27-55 [DOI] [PubMed] [Google Scholar]
- 19. Dumas J. Protein kinase inhibitors from the urea class. Curr Opin Drug Discov Dev. 2002;5:718-27 [PubMed] [Google Scholar]
- 20. Mol CD, Fabbro D, Hosfield DJ. Structural insights into the conformational selectivity of STI-571 and related kinase inhibitors. Curr Opin Drug Discov Dev. 2004;7:639-48 [PubMed] [Google Scholar]
- 21. Flörsheimer A, Furet P, Manley PW, et al. Preparation of diarylurea derivatives useful for the treatment of protein kinase dependent diseases. WO. 2003;99771 [Google Scholar]
- 22. Weisberg E, Roesel J, Bold G, et al. Antileukemic effects of the novel, mutant FLT3 inhibitor AST487: effects on PKC412-sensitive and -resistant FLT3-expressing cells. Blood. 2008;112:5161-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shieh WC, McKenna J, Sclafani JA, et al. Syntheses of a triad of Flt3 kinase inhibitors: from bench to pilot plant. Org Process Res Dev. 2008;12:1146-55 [Google Scholar]
- 24. Bold G, Caravatti G, Flörsheimer A, et al. Diaryl urea derivatives, particularly N-[4-(pyrimidin-4-yloxy)phenyl]-N’-phenylureas, useful in the treatment of protein kinase dependent diseases, especially neoplasm, and their preparation. WO. 2005;51366 [Google Scholar]
- 25. Imbach P, Capraro HG, Zimmermann J, et al. Preparation of 2-amino-6-anilinopurines as inhibitors of p34cdc2/cyclin Bcdc13 kinase and protein tyrosine kinase pp60c-src. WO. 2000;49018 [Google Scholar]
- 26. Estey EH. Therapeutic options for acute myelogenous leukemia. Cancer. 2001;92:1059-73 [DOI] [PubMed] [Google Scholar]
- 27. Mathews V, DiPersio JF. Stem cell transplantation in acute myelogenous leukemia in first remission: what are the options? Curr Hematol Rep. 2004;3:235-41 [PubMed] [Google Scholar]
- 28. Witherspoon RP, Deeg HJ. Allogeneic bone marrow transplantation for secondary leukemia or myelodysplasia. Haematologica. 1999;84:1085-7 [PubMed] [Google Scholar]
- 29. Mony U, Jawad M, Seedhouse C, et al. Resistance to FLT3 inhibition in an in vitro model of primary AML cells with a stem cell phenotype in a defined microenvironment. Leukemia. 2008;22:1395-401 [DOI] [PubMed] [Google Scholar]
- 30. Zeng Z, Samudio IJ, Munsell M, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5:3113-21 [DOI] [PubMed] [Google Scholar]
- 31. Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of AML following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6045-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293-300 [DOI] [PubMed] [Google Scholar]
- 34. Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763-72 [DOI] [PubMed] [Google Scholar]
- 35. Breitenbuecher F, Markova B, Kasper S, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in acute myeloid leukemia. Blood. 2009;113:4063-73 [DOI] [PubMed] [Google Scholar]
- 36. Zhou J, Bi C, Janakakumara JV, et al. Enhanced activation of STAT pathways and overexpression of surviving confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113:3889-90 [DOI] [PubMed] [Google Scholar]
- 37. Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR-ABL-transformed hematopoietic cell lines. Blood. 2000;95:3498-505 [PubMed] [Google Scholar]
- 38. Mahon FX, Deininger MW, Schultheis B, et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070-9 [PubMed] [Google Scholar]
- 39. le Coutre P, Tassi E, Varella-Garcia M, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95:1758-66 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.