

Abstract
It has been almost a quarter century since it was first appreciated that a class of oncogenes contained in rapidly transforming avian retroviruses encoded DNA-binding transcription factors. As with other oncogenes, genetic recombination with the viral genome led to their overexpression or functional alteration. In the years that followed, alterations of numerous transcription factors were shown to be causatively involved in various cancers in human patients and model organisms. Depending on their normal cellular functions, these factors were subsequently categorized as proto-oncogenes or tumor suppressor genes. This review focuses on the role of GATA transcription factors in carcinogenesis. GATA factors are zinc finger DNA binding proteins that control the development of diverse tissues by activating or repressing transcription. GATA factors thus coordinate cellular maturation with proliferation arrest and cell survival. Therefore, a role of this family of genes in human cancers is not surprising. Prominent examples include structural mutations in GATA1 that are found in almost all megakaryoblastic leukemias in patients with Down syndrome; loss of GATA3 expression in aggressive, dedifferentiated breast cancers; and silencing of GATA4 and GATA5 expression in colorectal and lung cancers. Here, we discuss possible mechanisms of carcinogenesis vis-à-vis the normal functions of GATA factors as they pertain to human patients and mouse models of cancer.
Keywords: GATA, transcription factors, cancer
Introduction
Perturbation of normal cellular functions that lead to malignant transformation can occur at the level of extracellular molecules such as hormones and growth/survival factors, their receptors, downstream signaling molecules, and their ultimate targets in the nucleus. The latter include chromatin-modifying enzymes and DNA-binding nuclear factors. A wealth of new insights into all these layers of regulation during oncogenesis derived originally from studies of retroviruses that either carried transforming genes or activated oncogene expression by way of nearby genomic integration. One of the first oncoproteins to be recognized as a transcription factor was JUN, whose gene was contained in mutant form in the avian sarcoma virus 17.1 A remarkable intersection involving multiple areas of research (recounted vividly in a review by Vogt2) led to the realization that JUN was part of a dimeric DNA-binding transcription factor complex, called AP-1. AP-1 consists of 2 tightly growth-regulated protein subunits, JUN and FOS. Notably, FOS was already known as the oncogenic agent in the FBJ murine osteosarcoma virus.3 Following this precedent, numerous additional oncogenic transcription factors belonging to diverse families were discovered by various strategies. These included characterization of genes transduced by tumor-producing viruses, defining genes dysregulated due to nearby retroviral integration, cloning genes involved in chromosomal translocations, and screening of potential oncoproteins that produce hallmarks of transformation following their introduction into cultured cells in vitro. A comprehensive list of potentially transforming transcription factors is outside the scope of this review, and the reader is referred to other sources.4
On the other hand, nuclear factors that function as tumor suppressors were recognized as targets of transforming proteins from DNA tumor viruses. For example, the adenovirus proteins E1A and E1B interact with the cellular retinoblastoma (RB) and p53 proteins, respectively, to perturb their regulation of cell cycle progression and cell viability (see reviews by DeCaprio and Levine5,6). RB and p53 are mutated or lost in a substantial fraction of human malignancies. Both proteins function as transcriptional regulators by directly (p53) or indirectly (RB) associating with DNA. Thus, transcription factors can be dominantly acting oncogenes and tumor suppressor genes. Operationally, it is helpful to define a third class of transcription factors, whose loss contributes to malignant transformation but that are not conventional tumor suppressors. These factors, which include some GATA proteins, normally promote cellular differentiation. Impaired function or reduced expression contributes to malignant transformation due to failure of the affected cells to mature and exit the cell cycle.
Transcription factors of the GATA family are essential regulators of the specification and differentiation of numerous tissues. They all share 2 highly conserved zinc fingers of the C2H2 type that mediate not only DNA binding but also the great majority of protein interactions. GATA factors typically bind to the element A/T GATA A/G. Outside the zinc finger domains, there is relatively little conservation between distinct members of this family.
Based on initial studies of their expression, GATA1, GATA2, and GATA3 were classified as hematopoietic GATA factors, while GATA4, GATA5, and GATA6 were termed endodermal GATA factors. However, this categorization does not do justice to the much broader tissue distribution of most GATA proteins. For example, GATA3 functions in T lymphocytes but is also a critical regulator of mammary epithelial cells.7 GATA factors can function in undifferentiated progenitor cells and play a role in their expansion, or they can direct the coordinated maturation and cell cycle withdrawal in terminally differentiating cells. Thus, it is to be expected that alterations of GATA factors contribute to the development of cancer in human patients.
Here, we review our current state of knowledge about GATA factors and some relevant co-regulators in the context of human cancers and relevant mouse models of malignant diseases. Mutations, loss of expression, or overexpression of GATA factors have all been associated with a broad variety of cancers in humans, including leukemia, breast cancer, gastrointestinal cancers, and others. In addition, several proteins that associate with GATA factors are proto-oncoproteins and function in part by modulating GATA factor activities. In this review, a lot of emphasis is placed in particular on GATA1 and GATA3 since they have been very well studied in the context of human malignancies and various mouse models, and an enormous amount of past work has produced a deep understanding of their functions at the molecular level.
Transcription Factor GATA1
The first discovered member of the GATA family was GATA1.8,9 It was identified based on its ability to bind GATA elements in the regulatory regions of the erythroid-expressed globin genes. We now know that GATA1 regulates most if not all erythroid-specific genes. Moreover, it was soon recognized that GATA1 serves critical roles in additional hematopoietic lineages, including mast cells, eosinophils, and megakaryocytes (MKs).10,11 Mice with a targeted deletion of the GATA1 gene succumb to anemia in early development owing to failed erythroid maturation and apoptosis of erythroid precursor cells.12 Conditional depletion of GATA1 in the MK lineage firmly established the essential role of GATA1 in megakaryopoiesis and hence platelet formation.13 In light of the lethal phenotype of GATA1-deficient mice, it had been deemed unlikely that mutations in GATA1 would be uncovered in humans with hematological diseases. Nevertheless, in 2000, the first patients with X-linked congenital anemia and thrombocytopenia were found to have a mutation in the GATA1 gene. Specifically, a point mutation within the N-terminal zinc finger of GATA1 was identified that diminishes the interaction of GATA1 with the hematopoietic expressed transcription co-factor FOG1 (friend of GATA1).14 Subsequently, numerous additional kindreds were discovered with GATA1 point mutations in the N-terminal zinc finger. Depending on the site and type of substitutions, the severity of the anemia and thrombocytopenia varied greatly (reviewed in Ciovacco et al.15). While almost all mutations associated with congenital hematopoietic disorders are within the N-terminal zinc finger, the mechanisms by which they affect its function varied. Some substitutions reduce the ability of GATA1 to bind FOG1, while others impair DNA binding.
Although the observations in human patients confirmed the critical role for GATA1 in erythroid and MK development, they left unanswered whether alterations of GATA1 might also play a role in neoplastic diseases. Numerous mechanistic studies, mostly in murine cells, found that GATA1 not only controls the expression of genes that establish the erythroid phenotype but also genes required for cell cycle arrest. For example, GATA1 directly contributes to the silencing of genes associated with cellular proliferation such as Kit, Myc, and Myb, which are proto-oncogenes.16-18 Failure to silence these genes might be expected to result in hyperproliferation of immature erythroid cells. However, GATA1 is also essential for the viability of erythroid precursors presumably by activating the expression of the Bcl-xl gene.19 Thus, loss of GATA1 leads to apoptosis of immature erythroid precursors instead of hyperproliferation. This might explain why patients with congenital GATA1 mutations have not been noted to display a propensity towards erythroleukemias. However, it is noteworthy that in murine models, impairment of GATA1 function, in conjunction with additional changes that promote cell viability, contributes to erythroleukemia (see below). In contrast to the erythroid compartment, megakaryocytes are susceptible to transformation owing to disrupted GATA1 function in human patients, which is discussed in the following section.
Megakaryoblastic Leukemias Caused by Mutations in the Hematopoietic Transcription Factor GATA1
Only 2 years following the discovery of the first germline mutations in the zinc finger region of GATA1, acquired mutations affecting the N-terminus of the protein were found in Down syndrome patients with acute megakaryoblastic leukemia (DS-AMKL).20 Intrigued by the observation that murine GATA1-deficient MKs not only fail to mature properly but also hyperproliferate,13 it was investigated whether mutations in GATA1 might be associated with myeloid leukemias. GATA1 mutations were associated only with DS-AMKL but not any other subtypes of acute myeloid leukemia.20 DS-AMKL is characterized by an uncontrolled proliferation of immature MKs and occurs within the first 4 to 5 years of life. In the initial study, 6 of 6 patients with DS-AMKL had mutations in GATA1. Mutations were not detected in patients who were in therapy-induced remission, demonstrating that the mutations were acquired somatically. This was the first example of GATA1 mutations playing a role in human cancer. Later studies extended these findings and confirmed that almost all examined cases of DS-AMKL are associated with GATA1 mutations.10,21 Several different kinds of mutations that all fell within the N-terminus of GATA1 were catalogued in DS-AMKL patients, including insertions, deletions, splice mutations, and non-sense and mis-sense mutations. All mutations lead to the usage of a downstream start codon, producing a truncated form of GATA1 called GATA1s (GATA1 short) that lacks the N-terminal 83 amino acids.
Patients with Down syndrome are highly predisposed towards the development of leukemias, including AMKL.22 Notably, in 10% of Down syndrome patients, a transient expansion of immature MK is diagnosed during late fetal development or around the time of birth. This disorder, referred to as transient myeloproliferative disorder (TMD), displays features of leukemia but, intriguingly, resolves spontaneously. Virtually all cases of TMD contain mutations in the GATA1 gene, leading to the production of GATA1s. Approximately 20% of TMD patients progress to develop DS-AMKL. This is likely due to additional mutations that are required for full transformation. Thus, 3 entities conspire to produce DS-AMKL: trisomy 21, expression of GATA1s, and additional unknown mutation(s). Current efforts are geared towards identifying these mutations and delineating the genes on chromosome 21 that cooperate with GATA1s.
Longitudinal studies discovered that the same mutations that lead to GATA1s production in patients with TMD are found in those individuals that progress to AMKL, demonstrating that AMKL results from a clonal expansion of TMD cells.23,24 The transient nature of TMD indicates that fetal stage–specific hematopoietic precursor or stem cells, not their adult counterparts, are the cells of origin for TMD and AMKL. This is supported by studies showing that mice that express GATA1s instead of full-length GATA1 produce a transient hyperproliferative phenotype involving MKs of fetal or embryonic but not adult origin.25 Similar results were obtained when GATA1s was introduced via transgenesis into mice with low GATA1 expression.26 This suggests that a select stage-specific population of progenitor or stem cells is particularly susceptible to the effects of GATA1s. Another notable result from this study was that wild-type fetal liver progenitor cells but not postnatal bone marrow cells infected with viral vectors expressing GATA1s developed hyperproliferative MK colonies.25 This was interpreted to mean that the lack of the N-terminus of GATA1 does not simply produce a molecule with impaired function but that GATA1s might act in a dominant manner. However, since GATA1s and wild-type GATA1 are not co-expressed in human AMKL cells (one allele is lyonized), this function might not be relevant to the disease.
What Are the Mechanisms of DS-AMKL by GATA1s?
GATA1s
How does the N-terminal truncation of GATA1 contribute to the transformation of MKs? GATA1s lacks an N-terminal transcriptional activation domain as originally defined in transient transfection assays in heterologous cells.27 However, when assayed in erythroid cells, it was revealed that this domain is largely dispensable for GATA1 function, especially when the truncated form of GATA1 is overexpressed.26,28,29 When introduced into GATA1-deficient MK, GATA1s was mostly capable of restoring MK maturation but not cell cycle arrest, suggesting that the N-terminal activation domain is required for the regulation of a subset of genes involved in cell proliferation.30,31 However, the question of “quality versus quantity” of mutant GATA1 in the context of MK development and AMKL is not entirely settled. It is possible that translation initiation at the downstream start codon is less efficient in MKs, that the shorter version of GATA1 is less stable, or that the truncated molecule is functionally defective. Moreover, the levels of GATA1s expression vary between different mutations with low levels being associated with increased risk towards AMKL progression.32 Thus, the quantity of the mutant protein contributes to the clinical phenotype. In addition, a defective version of GATA1 might function in a dominant manner by competing with endogenous GATA1 in the assays described above. Notably, GATA1s failed to inhibit the expression of Myb, Myc, Gata2, and Pu.1, which are thought to be direct GATA1 target genes.25 This suggests that the N-terminus of GATA1 might also serve to repress gene expression in some contexts. How the N-terminus of GATA1 regulates transcription is unknown.
Trisomy 21 (T21)
N-terminal truncation of GATA1 is not enough to cause leukemia in humans. Notably, an inherited point mutation in exon 2 of GATA1 leading to the production of GATA1s was associated with congenital macrocytic anemia and neutropenia but normal platelet counts in human patients without Down syndrome.33 The lack of leukemia in affected individuals indicates that T21 is required in concert with GATA1s to promote malignant transformation. Most cases of childhood AMKL not associated with Down syndrome lack mutations in GATA1, which indicates the existence of other pathways leading to this disease but also highlights the specific functional synergy of T21 and GATA1 mutations. A very interesting finding supporting the T21/GATA1s synergy stems from 2 patients with GATA1s-positive AMKL without Down syndrome. Leukemic blast cells from these patients were discovered to have somatically acquired T21, again underscoring the critical role of an extra dose of chromosome 21 in conjunction with GATA1s.24 Yet, while T21 and GATA1s are hallmarks of DS-AMKL, a requirement of T21 for GATA1s-associated leukemia is not absolute. This was revealed by the observation of GATA1s expression in a rare case of AMKL in the absence of congenital or acquired T2134 and suggests that GATA1s can cooperate with other as yet unknown genetic or epigenetic events during leukemogenesis.
What is the contribution of T21 to DS-AMKL? Clearly, T21 by itself is not sufficient for AMKL development since most Down syndrome patients do not develop leukemia or TMD. However, Down syndrome fetal livers harbor elevated numbers of erythromegakaryocyte progenitor cells (MEP) that are capable of producing erythroid and MK colonies of increased size when compared to normal counterparts.35-37 Thus, T21 favors the outgrowth of 2 GATA1-dependent cell lineages (erythroid and MK) at the expense of granulocyte-monocyte progenitors. The elevated risk towards progression into TMD and AMKL might result from the increased number of cells that are susceptible to acquired mutations such as those in GATA1 or other cooperating proto-oncogenes or tumor suppressor genes. The reason why Down syndrome patients do not display a predisposition towards erythroleukemia is unknown but likely relates to an MK-specific effect of the GATA1s production. In an attempt to recapitulate the human condition in an animal model, mice were generated that bear human chromosome 21 along with transgenic GATA1s. While these animals displayed increased megakaryopoiesis, they did not develop TMD or AMKL, indicating that additional alterations are needed to produce these disorders in mice.38
A discussion of all possible candidates on the critical region of chromosome 21 is beyond the scope of this article, and the reader is referred to a recent review.22 However, it is clear that subtle changes (1.5-fold above normal) in gene expression seem to exert profound biological effects. Some of the best candidate proteins such as RUNX1, ETS2, and SON were not or only subtly overexpressed in T21 hematopoietic progenitor cells.35,37,39 Among these, RUNX1 had been deemed an especially attractive candidate due to its known involvement in leukemias, its requirement for embryonic/fetal hematopoiesis, and its ability to interact with the N-terminus of GATA1. Also, no mutations in the RUNX1 gene were found in DS-AMKL.40 Another candidate is the oncogene ERG, a member of the ETS family on human chromosome 21 that is expressed in DS-AMKL.41 ERG synergizes with GATA1s to immortalize MK progenitors.42,43 Of note, a potentially oncogenic micro-RNA miR-125b was found to be overexpressed 3-fold in DS-AMKL cells when compared to non–DS-AMKL.44 Forced expression of miR-125b-2 promotes the proliferation of MEP. Importantly, miR-125b-2 synergized with GATA1s in stimulating the growth of MKs. This established miR-125b-2 as an important candidate in DS-AMKL. Nevertheless, the hunt for additional relevant gene(s) on chromosome 21 is in full swing and will likely produce significant new insights into the multistep nature of human leukemias.
Mouse Models of Leukemias Related to GATA1 Function
Mice with targeted deletions of enhancer elements that reduce the expression of GATA1 produce disorders, including myelofibrosis and an erythroleukemia-like phenotype.45-47 It should be pointed out that there is no evidence that deletions or point mutations in the GATA1 locus contribute to these disorders in human patients. Nevertheless, relevant findings will be discussed here as they are instructive with regards to general mechanisms of leukemogenesis.
Deletion of a critical regulatory element in the murine GATA1 gene leads to reduced expression of GATA1 (GATA1low), resulting in lethal anemia.47 However, in contrast to GATA1-null mice, a small fraction of GATA1low animals survive, are thrombocytopenic, and develop myelofibrosis at around 1 year of age. Myelofibrosis is characterized by fibrotic degeneration of the bone marrow, increased numbers of immature MKs, anemia, and extramedullary hematopoiesis. In humans, myelofibrosis is a preleukemic condition. While initial studies did not detect dramatic reductions in GATA1 expression in the bone marrows of human myelofibrosis patients,48 the number of MKs lacking detectable GATA1 protein appeared elevated in bone marrow samples of such patients.49 This is consistent with the hypothesis that reduced GATA1 expression leads to expansion of immature MKs. These cells in turn are thought to increase cytokine production that contributes to the fibrotic conversion and ultimately impaired function of the bone marrow.
In a separate mouse model in which GATA1 levels amount to approximately 5% of wild-type (GATA1.05), lethal anemia was found in male but not female mice (GATA1 is on the X chromosome).46 Viable female mice exhibit a myelodysplastic syndrome–like phenotype with erythroid and MK hyperplasia. Moreover, within 5 months of age, a large proportion of GATA1.05 mice developed leukemia with erythroblastic features.45 Curiously, a fraction of GATA1.05 mice developed B lymphocytic leukemia, but the reason for this is unknown.
Together, the above studies show that structural alterations of GATA1 produce distinct phenotypes from those that are due to reduced GATA1 expression. One interesting conclusion from the study of animals with reduced GATA1 expression is that low GATA1 levels are necessary and sufficient for viability of immature precursor cells, but the full amount of GATA1 is required for their maturation and cell cycle arrest.
No review on GATA1 in leukemogenesis would be complete without mentioning that the direct impairment of GATA1 function by oncogenic proteins has been linked to leukemic transformation in murine systems. For example, the Ets family transcription factor PU.1 (also called Spi-1) is upregulated by nearby insertion of the Friend leukemia virus that causes murine erythroleukemia (MEL).50 PU.1 physically interacts with GATA1 and inhibits its activity.51 This contributes to the differentiation arrest in erythroid cells, thus aiding leukemogenesis. In support of this idea, forced expression of GATA1 in cultured MEL cells overrides the differentiation block and triggers cellular maturation and exit from the cell cycle.51 Similarly, the gene encoding the Ets factor Fli-1 (Spi-1B) is another frequent target of viral insertion-mediated activation in MEL.52 Fli-1 can interfere with GATA1 activity in certain contexts,53 perhaps through direct physical interaction,54 and might block erythroid maturation.
Finally, the AML1-ETO fusion protein, which is found in some human acute myeloid leukemias and impairs erythroid differentiation, can interact with GATA1 and inhibit its activity.55 Interestingly, both AML-ETO and PU.1 inhibit the acetylation of GATA1, a modification essential to GATA1’s ability to function in erythroid cells.55-57 These examples illustrate that GATA factors can serve as targets of binding partners with oncogenic properties. This concept can theoretically be extended to numerous other molecules with transforming potential that interact with GATA1. These include CBP and p300,58,59 components of the NuRD complex,60,61 and the SCL/TAL complex.62
Transcription Factor GATA3
GATA3 was first identified in a screen for GATA factors in the T cell lineage.63,64 GATA3 plays an essential role in early T cell development and the specification of the Th2 subset of T cells (reviewed in Ho et al.65). Later, it was discovered that GATA3 performs critical functions outside of the hematopoietic system. This includes roles in the development of the epithelial structures of the mammary gland,66,67 skin, inner ear, central nervous system, and kidney (reviewed in Chou et al.7). This section will primarily focus on the role of GATA3 in normal mammary development and breast cancer but will also briefly summarize the role of GATA3 in other cancers. For a more comprehensive review of the biology of GATA3 and its involvement in human diseases, the reader is referred to an excellent recent article by Chou et al.7
GATA3 Is Essential for the Development of Mammary Epithelium
GATA3 contributes to the development of multiple organ systems in both mice and humans. GATA3-null mice die between E11 and E12 and display diverse defects, including internal bleeding, growth retardation, brain and spinal cord abnormalities, and impaired fetal liver hematopoiesis.68 In humans, haploinsufficiency of GATA3 results in the autosomal dominant HDR (hypoparathyroidism, deafness, and renal dysplasia) syndrome, also known as Barakat syndrome.69 Notably, the same GATA3 mutation found in the germline of HDR patients was detected among diverse GATA3 mutations in breast tumors (see below).70
GATA3 has emerged as a key factor in the development of the mammary epithelium.66,67 The mammary gland is comprised of 3 mature epithelial cell types, namely ductal and alveolar luminal epithelial cells, and myoepithelial cells, which derive from common multipotent progenitors.71 GATA3 is the most abundantly expressed transcription factor in the mammary ductal epithelium, specifically in luminal epithelial cells.66,67 Conditional deletion of GATA3 in the mammary epithelium at various stages during development showed that GATA3 is required for branching morphogenesis and for the differentiation of luminal epithelial cells. Importantly, in adult mice, conditional GATA3 deficiency leads to the dedifferentiation of luminal epithelial cells with increased proliferation and diminished adhesion, ultimately resulting in apoptosis. GATA3 is also capable of promoting cellular differentiation towards specific celllineages. Overexpression of GATA3 in cell populations enriched for mammary stem cells promotes the formation of alveolar epithelial differentiation.66 The requirement of GATA3 for lineage specification and cell maturation is reminiscent of the role of GATA1 in the erythroid-MK compartment. Moreover, by constraining the proliferation of immature progenitor cells, GATA3 might function similar to a tumor suppressor, an idea further supported by studies in human cancers and mouse models. Yet, if GATA3 functions as a classic tumor suppressor in the mammary epithelium, haploinsufficiency would be expected to predispose HDR patients to breast cancer. However, to our knowledge, this has not been reported.
GATA3 in the Pathogenesis of Breast Cancer
When compared to estrogen receptor–positive (ER+) breast cancers, the ER-negative (ER−) counterparts are less well-differentiated and more malignant. Based on several microarray studies that profiled gene expression states in ER+ and ER− breast cancer, loss of GATA3 expression was found to also be a reliable indicator of poor prognosis (reviewed in Chou et al.7). In breast cancer cell lines and primary tumors, GATA3 expression was strongly correlated with ER expression.72 Moreover, low or lack of GATA3 expression is associated with shorter survival, more malignant histological features, positive lymph nodes, increased tumor mass, lack of progesterone receptor expression, and overexpression of the epidermal growth factor receptor 2 (HER2/Neu), which in its own right is associated with aggressive forms of breast cancer.73 Strikingly, even in cases of ER+ breast cancers, failure to respond to hormonal therapy and poor prognosis are associated with lack of GATA3 expression.73,74 At present, it remains unresolved whether GATA3 expression has prognostic status independent of ER.73,75 In addition, a small fraction of human breast cancers contained acquired mutations in GATA3 that are predicted to alter GATA3 function.70 However, these mutations occurred in a heterozygous state. Moreover, the GATA3 mutation-bearing cancers were of the luminal ER+ subtype and expressed GATA3 from the wild-type allele. Thus, the contribution of GATA3 haploinsufficiency to the pathogenesis of these cancers, if any, is uncertain.
Mouse Models Implicate GATA3 in Breast Cancer Progression and Metastasis
GATA3 was implicated in the malignant progression of luminal breast cancer in a mouse model in which an oncoprotein, the polyoma middle T antigen (PyMT), is expressed from a transgene under the control of the mammary epithelial-specific MMTV promoter.76 The progression of tumors from hyperplasia to well-differentiated adenomas and, subsequently, metastatic adenocarcinomas in the MMTV-PyMT model recapitulates the development of the disease in humans.77-79 Transcriptome analysis comparing adenomas with late carcinomas revealed a loss in the latter of differentiation markers, including luminal differentiation genes.76 Notably, GATA3 expression was also downregulated. Thus, tumor cells assumed features and surface markers similar to mammary epithelial stem cells. In addition, loss of GATA3 was associated with the ability of tumor cells to disseminate. Retroviral delivery of GATA3 into cells from early tumors led to luminal cell differentiation and decreased tumor spreading.76 In late carcinomas, the GATA3 gene was hypermethylated consistent with its lack of expression. One possible scenario suggests that GATA3 is silenced epigenetically in early adenomas, triggering conversion into more malignant cells, which then outgrow the other cells. However, this idea was tested elegantly and found to be unlikely. Specifically, conditional deletion of GATA3 in early tumors led to apoptosis and detachment of adenoma cells from their basement membrane,76 indicating that deletion of GATA3 in differentiated cells is not sufficient for tumor development. This suggests an alternative scenario in which transforming events occur in a cell population that is a priori GATA3 negative, that is, early progenitors or stem cells, which then outpaces the other cells in the lesion.76 Thus, GATA3 does not appear to function as a conventional tumor suppressor but as a differentiation agent. This might help to explain why predisposition to breast cancer has not been reported in HDR patients. Moreover, this raises the question as to what extent somatic mutations in GATA3 found in human breast cancers are bone fide drivers of tumor progression. Finally, it becomes important to consider that “silencing” of GATA factors in other cancers, such as GATA4 and GATA5 in colorectal cancers (see below), might similarly reflect an outgrowth of a population of cells that never expressed these factors in the first place.
In a separate mouse model of breast cancer, GATA3 was found to inhibit the metastatic seeding of breast cancer cells.80 When overexpressed in a cell line selected for high metastatic potential to the lung, GATA3 was capable of reducing tumor burden and metastases. While generally in agreement with the study above, it was additionally noted that, in this particular model, GATA3’s ability to suppress metastasis could be uncoupled from its ability to promote differentiation of malignant mammary epithelial cells. This conclusion was reached based on the downregulation by GATA3 of prometastatic genes and the upregulation of genes inhibitory to metastasis while luminal differentiation markers were unchanged.80
Epithelial mesenchymal transition (EMT) is one of the mechanisms by which tumors can develop invasiveness and the ability to detach from the tissue of origin and seed distant organs. One study found that GATA3, when expressed in a GATA3-deficient breast cancer cell line, reverses EMT in part by promoting the expression of E-cadherin and repressing mesenchymal proteins N-cadherin and vimentin.81 This was associated with reduced tumor burden and diminished dissemination into the lungs. Conversely, knockdown of GATA3 in a GATA3-positive breast cancer cell line increased tumor volume and lung metastasis compared to controls. One mechanism by which GATA3 can reverse EMT is potentially through its activation of the cell-cell adhesion protein E-cadherin, which is normally downregulated in EMT.81
GATA3 in Other Cancers
While GATA3 is inhibitory to tumor formation in the breast, it seems capable of promoting carcinogenesis in lymphoid precursor cells. Mice in which GATA3 is overexpressed under the control of the human CD2 locus control region develop CD4+ CD8+ double-positive (DP) T cell lymphoma.82 GATA3 converts DP thymocytes into a premalignant state, which is marked by increased cell size and increased Myc expression.83 The stimulatory effects on Myc expression appear to be indirect and contrast with the inhibition of Myc expression by GATA1 in erythroid cells. This is yet another example demonstrating that the function of GATA factors is highly context dependent. Whether GATA3 plays a role in human T cell malignancies is unknown. However, we speculate that overexpression of SCL/Tal1, which occurs frequently as a result of chromosomal translocations in human T cell acute lymphoblastic leukemias (T-ALL), might affect GATA3 function in immature T cells. SCL/Tal1 via an intermediary protein can physically interact with GATA factors and modulate their activities. In support of this model, in human T-ALL cell lines, SCL/Tal1 was recruited via GATA3 to the promoter of the NKX3.1 homeobox gene to upregulate its expression.84 Nkx3.1 in turn is required for T-ALL cell proliferation and leukemia development in mice.84 This provides another example of how a GATA factor might serve as a conduit for the action of an oncogenic protein.
In this context, it is worth mentioning that there are several studies assessing the expression of GATA3 and other GATA factors in diverse cancers. For example, GATA3 is overexpressed in pancreatic cancer cell lines and primary pancreatic cancers.85 GATA2, GATA3, GATA4, and FOG2 are expressed in human neuroblastoma, with overexpression of GATA4 correlating with less favorable subtypes and overexpression of GATA2, GATA3, and FOG2 with more favorable subtypes.86 In neuroblastoma cell lines, GATA3 was found to positively regulate cyclin D1, which maintains cells in an undifferentiated state,87,88 suggesting that GATA3 has oncogenic potential in neuroblastoma.
GATA4, GATA5, and GATA6 in Human Cancers
These 3 GATA factors will be discussed here together mostly because our knowledge of their function in human cancers is not yet as rich as that about GATA1 and GATA3. Altered expression of GATA4, GATA5, and GATA6 is associated with a broad range of tumors emerging from the gastrointestinal tract, lungs, ovaries, and even the brain. However, it should be stated right at the outset that, to date, published reports are mostly correlative and, in contrast to studies on GATA1 and GATA3, are not yet fully complemented by functional experiments and mouse models of these cancers.
GATA4, GATA5, and GATA6 are expressed predominantly in endoderm- and mesoderm-derived tissues (for review, see Molkentin89). They all harbor a highly conserved double zinc finger domain and even share a significant degree of similarity among their activation domains. All 3 are expressed in distinct but overlapping patterns. For example, they are all expressed in the heart and gut epithelium, although GATA4 is detected in the proximal parts of the gastrointestinal tract, whereas GATA6 is expressed throughout the small and large intestines.90,91 As to the intestinal cell types of expression, the literature is quite complex and not always in agreement, which might be due to the study of distinct organisms and limitations of the antibodies used. Nevertheless, it has been suggested that GATA4 and GATA5 tend to mark fully differentiated epithelial cells, while GATA6 is expressed in the immature proliferating cells in the intestinal crypts. This would implicate GATA4 and GATA5 as potential tumor suppressors and GATA6 as a potential oncogene.91-93
Point mutations in GATA4 cause congenital cardial septal defects in human patients, highlighting the importance of GATA4 for normal heart development.94 Mice null for GATA4 die in utero due to failed heart morphogenesis, but animals heterozygous for GATA4 deletion (GATA4 +/−) appear normal.95,96 A role of GATA4 as a tumor suppressor might be expected to manifest itself in increased susceptibility to cancer in GATA4 +/− mice, which has not been described. The augmentation in cancer rates due to loss of heterozygosity at the GATA4 locus might require the cooperation of additional genetic events. Likewise, no increased rates of cancer have been described in mice nullizygous for GATA597 or heterozygous for loss of GATA6 (homozygous loss of GATA6 is an early embryonic lethal).98
While, to date, no mutations or deletions of the GATA4 and GATA5 genes have been discovered in human cancers, silencing of their expression seems to be widespread in colorectal and gastric cancers. Expression of GATA4 and GATA5 but not GATA6 was extinguished in the majority of cell lines from colorectal (CRC) and gastric (GC) cancers as well as in primary tumors.92 Silencing was associated with hypermethylation of the GATA4 and GATA5 promoter sequences. Moreover, candidate target genes of these GATA factors were commensurately decreased in their expression and also displayed methylated promoters. Treatment with a DNA methyltransferase inhibitor or genetic disruption of DNA methyltransferases led to reactivation of GATA4, GATA5, and their target genes in CRC cell lines. Moreover, forced expression of GATA5 in CRC cells also led to reactivation of target gene expression. A more recent study confirmed these findings by showing GATA4 and GATA5 promoter methylation in over two thirds of CRC.99 Methylation was not significantly correlated with stage, histological subtype, or grade of differentiation of the disease, suggesting that GATA4 and GATA5 silencing might be an early event during carcinogenesis. Forced expression of GATA4 or GATA5 in CRC cell lines impaired their proliferation and migration, suggesting that loss of GATA5 expression in cancer cells is functionally important.
As mentioned above, GATA4 is expressed only in the proximal parts of the intestinal tract in humans and mice. The question then is if GATA4 is not expressed in colorectal epithelial cells in the first place, how can lack of GATA4 expression be relevant to the disease? It remains possible that expression is too low to be detectable or that it is restricted to only a subpopulation of cells that give rise to CRC such as the stem/progenitor cells in the colonic crypts. Alternatively, even if GATA4 were already silenced in colorectal epithelial cells prior to their transformation and therefore be irrelevant to the process, overexpression of GATA4 in CRC cells might generate the same effects as GATA5 due to overlapping activities.
One interesting aspect of these studies is that the high rate of promoter methylation could be exploited as a marker for early detection. Indeed, stool samples from CRC patients frequently revealed methylated GATA4 promoter DNA, but the sensitivity of the assay requires further improvement to be clinically useful.99
In conceptually related studies, GATA4 and GATA5 were found to be extinguished in a large fraction of lung and esophageal cancers. GATA4 and GATA5 promoter methylation and loss of expression were detected in 67% and 41%, respectively, in primary human lung cancers, while GATA6 was continuously expressed.100 Promoter methylation was unrelated to the stage of the disease. Remarkably similar results were obtained in studies of esophageal cancer. While normal esophageal mucosa expresses GATA4 and GATA5, squamous cell carcinomas displayed GATA4 and GATA5 promoter methylation in 61% and 32%, respectively. The numbers in adenocarcinomas amounted to 71% (GATA4) and 55% (GATA5). GATA6 expression was maintained in all samples. Together, these studies support the idea that loss of GATA5 and GATA4 by epigenetic silencing might contribute to malignant transformation and are consistent with these factors functioning as tumor suppressors. It remains an open question as to what causes these silencing events. It is possible that genetic insults or even stochastic events lead to repression of the GATA4 and GATA5 genes that is subsequently maintained by epigenetic mechanisms such as DNA methylation.
The function of GATA factors depends on cell and promoter context. While GATA4 might promote differentiation in one cell type and thus function as a tumor suppressor, its role in other cell types might be distinct. For example, elevated GATA4 levels are associated with poor prognosis in ovarian granulosa cell tumors.101 It has been suggested that GATA4 promotes the expression of the antiapoptotic factor Bcl2 and cyclin D2.102 Thus, in granulosa cells, GATA4 might function as an oncoprotein.
Seemingly opposing functions during carcinogenesis have also been described for GATA6. In a very elegant study, GATA6 was discovered as a tumor suppressor of astrocytoma in a gene trapping screen.103 The great majority of human glioblastomas (which can arise from low-grade astrocytomas) but not low-grade astrocytomas displayed loss of GATA6 expression, mutations in GATA6, and loss of heterozygosity. Re-expression of GATA6 in human malignant astrocytoma cells inhibited their growth. This establishes GATA6 as a bona fide tumor suppressor in this disease, marking the progression from low-grade astrocytoma to malignant glioblastoma. Conversely, overexpression of GATA6, in part due to gene amplification, was observed in approximately half of pancreatic carcinomas.104,105 Knockdown or overexpression of GATA6 in pancreatic carcinoma cell lines modestly increased and decreased, respectively, cell proliferation rates, supporting a role for GATA6 during tumorigenesis. GATA6 was also found to be highly expressed in human colon cancer samples.106 Thus, not unlike other transcription factors, GATA6 has a split personality with regard to its oncogenicity, depending on cell type.
If select GATA factors can function as tumor suppressors, it might be expected that impairment of co-factors might also predispose to cancer. FOG1 and FOG2 are multitype zinc finger proteins that associate with the N-terminal zinc finger of GATA proteins107; FOG proteins are expressed in a tissue-specific manner and are required for most but not all GATA factor functions. To our knowledge, mutations in the FOG genes have not been described in human cancers. However, a chromosomal translocation fusing the AML1 gene to FOG2 has been observed in a case of lethal myelodysplastic syndrome.108 The reciprocal translocation (FOG2-AML1) is also expressed. It is possible that the resulting fusion proteins interfere with the functions of AML1 (also known as RUNX1) and/or the hematopoietic GATA factors GATA1 and GATA2. In this regard, it is worth mentioning preliminary data showing that aged mice homozygous for targeted point mutations in FOG1 that disrupt binding to the NuRD chromatin remodeling complex109 have a propensity to develop liver cancer at a higher rate than controls (R.Z. and G.A.B., unpublished observation). Since GATA4 and GATA6 are expressed in the liver, this suggests that the GATA/FOG1/NuRD axis might function as a tumor suppressor pathway in the liver.
Finally, evidence for a direct role of another GATA factor, GATA2, in human malignancies is sparse. This is somewhat surprising since GATA2 is associated with early progenitor and stem cells in the hematopoietic compartment and marks proliferative cell populations. However, there have been hints at its involvement in leukemia. For example, it was shown that the PML-RARa translocation fusion protein characteristic of acute promyelocytic leukemia (APL) interacts with GATA2 and modulates its activity,110 suggesting that part of PML-RAR’s leukemic action is mediated by GATA2. Moreover, 2 distinct types of mutations in the zinc finger domain of GATA2 were found in a subset of human chronic myelogenous leukemias (CML) but not other types of leukemias such as AML or ALL.111 Although these mutations had opposite effects on the transactivation properties of GATA2 in transfection assays, they both appeared capable of inhibiting myeloid differentiation of leukemic cell lines in vitro. These studies suggest that further searches for changes in the structure or expression of GATA2 might uncover a broader role of this factor in human diseases.
Summary and Perspective
All the above considerations implicate GATA factors and their co-factors as participants in pathways, leading to cancer in diverse tissues. GATA factor activity can be altered by mutation, overexpression, loss of expression, or functional interference by interacting proteins (Fig. 1). This multitude of mechanisms capable of subverting normal transcription factor function likely applies to other proto-oncoproteins and tumor suppressor proteins as well.
Figure 1.
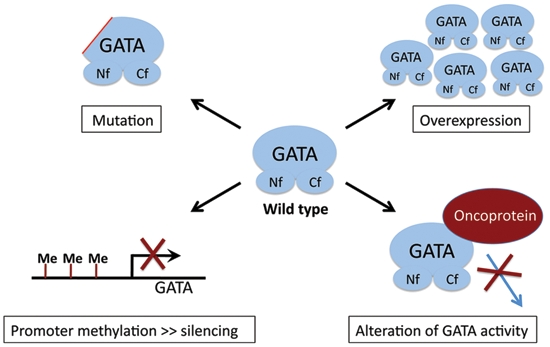
Mechanisms by which GATA factor activity can be altered during carcinogenesis include mutation, overexpression, silencing by promoter methylation, or interference by proteins that bind GATA factors. Please see text for details. Nf = N-terminal zinc finger; Cf = C-terminal zinc finger.
It has been argued perhaps somewhat cynically that most transcription factors when overexpressed have the potential to either promote or inhibit cell proliferation or viability. Whether this turns out to be true remains to be seen. Nevertheless, most transcription factors are restricted in their expression patterns and display a degree of selectivity towards a limited set of genes. Importantly, as with all eukaryotic transcription factors, GATA factors work in combination with other DNA binding proteins and co-regulator molecules to establish specific patterns of gene expression. This degree of specificity could be exploited to manipulate gene expression in a targeted manner as an approach to treat diseases involving GATA factors. DNA binding proteins are generally considered relatively poor drug targets. However, GATA factors, very much like most transcription regulators, seed higher order molecular complexes consisting of diverse co-regulators with enzymatic activity. This includes chromatin-modifying enzymes such as acetyl-transferases, de-acetylases, methyl-transferases, and ATPases. Although these enzymes are typically expressed in many cell types, they certainly do not all function in a ubiquitous manner and might be limiting in select gene contexts. To identify these vulnerabilities and to exploit them through the use of pharmacological compounds is a challenge that is likely going to be rewarding.
Acknowledgments
The authors apologize to those whose work could not be mentioned due to space restrictions. They thank Dr. John Crispino for helpful suggestions on the article. This review is dedicated to the memory of Saburo and Teruko Hanafusa as a small token of gratitude for their steady guidance, kindness, and generosity and for welcoming one of the authors (G.A.B.) to the Hanafusa laboratory family.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the National Institutes of Health [grant numbers DK054937, DK058044].
References
- 1. Maki Y, Bos TJ, Davis C, Starbuck M, Vogt PK. Avian sarcoma virus 17 carries the jun oncogene. Proc Natl Acad Sci U S A. 1987;84:2848-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogt PK. Fortuitous convergences: the beginnings of JUN. Nat Rev Cancer. 2002;2:465-9 [DOI] [PubMed] [Google Scholar]
- 3. Curran T, Peters G, Van Beveren C, Teich NM, Verma IM. FBJ murine osteosarcoma virus: identification and molecular cloning of biologically active proviral DNA. J Virol. 1982;44:674-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cooper G, editor. Oncogenes. 2nd ed. London, UK: Jones and Bartlett Learning; 1995 [Google Scholar]
- 5. DeCaprio JA. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology. 2009;384:274-84 [DOI] [PubMed] [Google Scholar]
- 6. Levine AJ. The common mechanisms of transformation by the small DNA tumor viruses. The inactivation of tumor suppressor gene products: p53. Virology. 2009;384:285-93 [DOI] [PubMed] [Google Scholar]
- 7. Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it! J Cell Physiol. 2010;222:42-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsai SF, Martin DI, Zon LI, D’Andrea AD, Wong GG, Orkin SH. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature. 1989;339:446-51 [DOI] [PubMed] [Google Scholar]
- 9. Evans T, Felsenfeld G. The erythroid-specific transcription factor Eryf1: a new finger protein. Cell. 1989;58:877-85 [DOI] [PubMed] [Google Scholar]
- 10. Crispino JD. GATA1 in normal and malignant hematopoiesis. Semin Cell Dev Biol. 2005;16:137-47 [DOI] [PubMed] [Google Scholar]
- 11. Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol. 2005;25:1215-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pevny L, Simon MC, Robertson E, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature. 1991;349:257-60 [DOI] [PubMed] [Google Scholar]
- 13. Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16:3965-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nichols KE, Crispino JD, Poncz M, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24:266-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ciovacco WA, Raskind WH, Kacena MA. Human phenotypes associated with GATA-1 mutations. Gene. 2008;427:1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Munugalavadla V, Dore LC, Tan BL, et al. Repression of c-kit and its downstream substrates by GATA-1 inhibits cell proliferation during erythroid maturation. Mol Cell Biol. 2005;25:6747-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rylski M, Welch JJ, Chen Y-Y, et al. GATA-1-mediated proliferation arrest during erythroid maturation. Mol Cell Biol. 2003;23:5031-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bartůnek P, Králová J, Blendinger G, Dvorák M, Zenke M. GATA-1 and c-myb crosstalk during red blood cell differentiation through GATA-1 binding sites in the c-myb promoter. Oncogene. 2003;22:1927-35 [DOI] [PubMed] [Google Scholar]
- 19. Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood. 1999;94:87-96 [PubMed] [Google Scholar]
- 20. Wechsler J, Greene M, Mcdevitt MA, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148-52 [DOI] [PubMed] [Google Scholar]
- 21. Vyas P, Crispino JD. Molecular insights into Down syndrome-associated leukemia. Curr Opin Pediatr. 2007;19:9-14 [DOI] [PubMed] [Google Scholar]
- 22. Roy A, Roberts I, Norton A, Vyas P. Acute megakaryoblastic leukaemia (AMKL) and transient myeloproliferative disorder (TMD) in Down syndrome: a multi-step model of myeloid leukaemogenesis. Br J Haematol. 2009;147:3-12 [DOI] [PubMed] [Google Scholar]
- 23. Hitzler JK, Cheung J, Li Y, Scherer SW, Zipursky A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood. 2003;101:4301-4 [DOI] [PubMed] [Google Scholar]
- 24. Rainis L, Bercovich D, Strehl S, et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003;102:981-6 [DOI] [PubMed] [Google Scholar]
- 25. Li Z, Godinho FJ, Klusmann J-H, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37:613-9 [DOI] [PubMed] [Google Scholar]
- 26. Shimizu R, Kobayashi E, Engel JD, Yamamoto M. Induction of hyperproliferative fetal megakaryopoiesis by an N-terminally truncated GATA1 mutant. Genes Cells. 2009;14:1119-31 [DOI] [PubMed] [Google Scholar]
- 27. Martin DI, Orkin SH. Transcriptional activation and DNA binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes Dev. 1990;4:1886-98 [DOI] [PubMed] [Google Scholar]
- 28. Blobel GA, Simon MC, Orkin SH. Rescue of GATA-1-deficient embryonic stem cells by heterologous GATA-binding proteins. Mol Cell Biol. 1995;15:626-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weiss MJ, Yu C, Orkin SH. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol Cell Biol. 1997;17:1642-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuhl C, Atzberger A, Iborra F, Nieswandt B, Porcher C, Vyas P. GATA1-mediated megakaryocyte differentiation and growth control can be uncoupled and mapped to different domains in GATA1. Mol Cell Biol. 2005;25:8592-606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muntean AG, Crispino JD. Differential requirements for the activation domain and FOG-interaction surface of GATA-1 in megakaryocyte gene expression and development. Blood. 2005;106:1223-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanezaki R, Toki T, Terui K, et al. Down syndrome and GATA1 mutations in transient abnormal myeloproliferative disorder: mutation classes correlate with progression to myeloid leukemia. Blood. 2010;116:4631-8 [DOI] [PubMed] [Google Scholar]
- 33. Hollanda LM, Lima CSP, Cunha AF, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38:807-12 [DOI] [PubMed] [Google Scholar]
- 34. Harigae H, Xu G, Sugawara T, Ishikawa I, Toki T, Ito E. The GATA1 mutation in an adult patient with acute megakaryoblastic leukemia not accompanying Down syndrome. Blood. 2004;103:3242-3 [DOI] [PubMed] [Google Scholar]
- 35. Chou ST, Opalinska JB, Yao Y, et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112:4503-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Vita S, Devoy A, Groet J, Kruslin B, Kuzmić-Prusac I, Nizetić D. Megakaryocyte hyperproliferation without GATA1 mutation in foetal liver of a case of Down syndrome with hydrops foetalis. Br J Haematol. 2008;143:300-3 [DOI] [PubMed] [Google Scholar]
- 37. Tunstall-Pedoe O, Roy A, Karadimitris A, et al. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112:4507-11 [DOI] [PubMed] [Google Scholar]
- 38. Alford KA, Slender A, Vanes L, et al. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010;115:2928-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bourquin J-P, Subramanian A, Langebrake C, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci U S A. 2006;103:3339-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu G, Nagano M, Kanezaki R, et al. Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood. 2003;102:2960-8 [DOI] [PubMed] [Google Scholar]
- 41. Rainis L, Toki T, Pimanda JE, et al. The proto-oncogene ERG in megakaryoblastic leukemias. Cancer Res. 2005;65:7596-602 [DOI] [PubMed] [Google Scholar]
- 42. Salek-Ardakani S, Smooha G, de Boer J, et al. ERG is a megakaryocytic oncogene. Cancer Res. 2009;69:4665-73 [DOI] [PubMed] [Google Scholar]
- 43. Stankiewicz MJ, Crispino JD. ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood. 2009;113:3337-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klusmann J-H, Li Z, Böhmer K, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. 2010;24:478-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimizu R, Kuroha T, Ohneda O, et al. Leukemogenesis caused by incapacitated GATA-1 function. Mol Cell Biol. 2004;24:10814-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takahashi S, Komeno T, Suwabe N, et al. Role of GATA-1 in proliferation and differentiation of definitive erythroid and megakaryocytic cells in vivo. Blood. 1998;92:434-42 [PubMed] [Google Scholar]
- 47. Vannucchi AM, Bianchi L, Cellai C, et al. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood. 2002;100:1123-32 [DOI] [PubMed] [Google Scholar]
- 48. Martyré M-C, Steunou V, LeBousse-Kerdilès M-C, Wietzerbin J. Lack of alteration in GATA-1 expression in CD34+ hematopoietic progenitors from patients with idiopathic myelofibrosis. Blood. 2003;101:5087-8, author reply 8-9 [DOI] [PubMed] [Google Scholar]
- 49. Vannucchi AM, Pancrazzi A, Guglielmelli P, et al. Abnormalities of GATA-1 in megakaryocytes from patients with idiopathic myelofibrosis. Am J Pathol. 2005;167:849-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moreau-Gachelin F. Spi-1/PU.1: an oncogene of the Ets family. Biochim Biophys Acta. 1994;1198:149-63 [DOI] [PubMed] [Google Scholar]
- 51. Rekhtman N, Radparvar F, Evans T, Skoultchi AI. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999;13:1398-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ben-David Y, Giddens EB, Letwin K, Bernstein A. Erythroleukemia induction by Friend murine leukemia virus: insertional activation of a new member of the ets gene family, Fli-1, closely linked to c-ets-1. Genes Dev. 1991;5:908-18 [DOI] [PubMed] [Google Scholar]
- 53. Athanasiou M, Mavrothalassitis G, Sun-Hoffman L, Blair DG. FLI-1 is a suppressor of erythroid differentiation in human hematopoietic cells. Leukemia. 2000;14:439-45 [DOI] [PubMed] [Google Scholar]
- 54. Eisbacher M, Holmes ML, Newton A, et al. Protein-protein interaction between Fli-1 and GATA-1 mediates synergistic expression of megakaryocyte-specific genes through cooperative DNA binding. Mol Cell Biol. 2003;23:3427-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Choi Y, Elagib KE, Delehanty LL, Goldfarb AN. Erythroid inhibition by the leukemic fusion AML1-ETO is associated with impaired acetylation of the major erythroid transcription factor GATA-1. Cancer Res. 2006;66:2990-6 [DOI] [PubMed] [Google Scholar]
- 56. Hong W, Kim AY, Ky S, et al. Inhibition of CBP-mediated protein acetylation by the Ets family oncoprotein PU.1. Mol Cell Biol. 2002;22:3729-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hung HL, Lau J, Kim AY, Weiss MJ, Blobel GA. CREB-Binding protein acetylates hematopoietic transcription factor GATA-1 at functionally important sites. Mol Cell Biol. 1999;19:3496-505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Blobel GA. CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood. 2000;95:745-55 [PubMed] [Google Scholar]
- 59. Blobel GA, Nakajima T, Eckner R, Montminy M, Orkin SH. CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci U S A. 1998;95:2061-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433-8 [DOI] [PubMed] [Google Scholar]
- 61. Hong W, Nakazawa M, Chen Y-Y, et al. FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J. 2005;24:2367-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lécuyer E, Hoang T. SCL: from the origin of hematopoiesis to stem cells and leukemia. Exp Hematol. 2004;32:11-24 [DOI] [PubMed] [Google Scholar]
- 63. Ho IC, Vorhees P, Marin N, et al. Human GATA-3: a lineage-restricted transcription factor that regulates the expression of the T cell receptor alpha gene. EMBO J. 1991;10:1187-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ko LJ, Yamamoto M, Leonard MW, George KM, Ting P, Engel JD. Murine and human T-lymphocyte GATA-3 factors mediate transcription through a cis-regulatory element within the human T-cell receptor delta gene enhancer. Mol Cell Biol. 1991;11:2778-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ho I-C, Tai T-S, Pai S-Y. GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat Rev Immunol. 2009;9:125-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Asselin-Labat M-L, Sutherland KD, Barker H, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9:201-9 [DOI] [PubMed] [Google Scholar]
- 67. Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127:1041-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pandolfi PP, Roth ME, Karis A, et al. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. 1995;11:40-4 [DOI] [PubMed] [Google Scholar]
- 69. Van Esch H, Groenen P, Nesbit MA, et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419-22 [DOI] [PubMed] [Google Scholar]
- 70. Usary J, Llaca V, Karaca G, et al. Mutation of GATA3 in human breast tumors. Oncogene. 2004;23:7669-78 [DOI] [PubMed] [Google Scholar]
- 71. Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84-8 [DOI] [PubMed] [Google Scholar]
- 72. Hoch RV, Thompson DA, Baker RJ, Weigel RJ. GATA-3 is expressed in association with estrogen receptor in breast cancer. Int J Cancer. 1999;84:122-8 [DOI] [PubMed] [Google Scholar]
- 73. Mehra R, Varambally S, Ding L, et al. Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 2005;65:11259-64 [DOI] [PubMed] [Google Scholar]
- 74. Parikh P, Palazzo JP, Rose LJ, Daskalakis C, Weigel RJ. GATA-3 expression as a predictor of hormone response in breast cancer. J Am Coll Surg. 2005;200:705-10 [DOI] [PubMed] [Google Scholar]
- 75. Voduc D, Cheang M, Nielsen T. GATA-3 expression in breast cancer has a strong association with estrogen receptor but lacks independent prognostic value. Cancer Epidemiol Biomarkers Prev. 2008;17:365-73 [DOI] [PubMed] [Google Scholar]
- 76. Kouros-Mehr H, Bechis SK, Slorach EM, et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dydensborg AB, Rose AAN, Wilson BJ, et al. GATA3 inhibits breast cancer growth and pulmonary breast cancer metastasis. Oncogene. 2009;28:2634-42 [DOI] [PubMed] [Google Scholar]
- 81. Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J Biol Chem. 2010;285:14042-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nawijn MC, Ferreira R, Dingjan GM, et al. Enforced expression of GATA-3 during T cell development inhibits maturation of CD8 single-positive cells and induces thymic lymphoma in transgenic mice. J Immunol. 2001;167:715-23 [DOI] [PubMed] [Google Scholar]
- 83. van Hamburg JP, de Bruijn MJW, Dingjan GM, et al. Cooperation of Gata3, c-Myc and Notch in malignant transformation of double positive thymocytes. Mol Immunol. 2008;45:3085-95 [DOI] [PubMed] [Google Scholar]
- 84. Kusy S, Gerby B, Goardon N, et al. NKX3.1 is a direct TAL1 target gene that mediates proliferation of TAL1-expressing human T cell acute lymphoblastic leukemia. J Exp Med. 2010;207:2141-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gulbinas A, Berberat PO, Dambrauskas Z, et al. Aberrant gata-3 expression in human pancreatic cancer. J Histochem Cytochem. 2006;54:161-9 [DOI] [PubMed] [Google Scholar]
- 86. Hoene V, Fischer M, Ivanova A, Wallach T, Berthold F, Dame C. GATA factors in human neuroblastoma: distinctive expression patterns in clinical subtypes. Br J Cancer. 2009;101:1481-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Molenaar JJ, Ebus ME, Koster J, et al. Cyclin D1 is a direct transcriptional target of GATA3 in neuroblastoma tumor cells. Oncogene. 2010;29:2739-45 [DOI] [PubMed] [Google Scholar]
- 88. Molenaar JJ, Ebus ME, Koster J, et al. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008;68:2599-609 [DOI] [PubMed] [Google Scholar]
- 89. Molkentin JD. The zinc finger-containing transcription factors GATA-4, -5, and -6: ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949-52 [DOI] [PubMed] [Google Scholar]
- 90. Bosse T, Piaseckyj CM, Burghard E, et al. Gata4 is essential for the maintenance of jejunal-ileal identities in the adult mouse small intestine. Mol Cell Biol. 2006;26:9060-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Haveri H, Westerholm-Ormio M, Lindfors K, et al. Transcription factors GATA-4 and GATA-6 in normal and neoplastic human gastrointestinal mucosa. BMC Gastroenterol. 2008;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Akiyama Y, Watkins N, Suzuki H, et al. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 2003;23:8429-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gao X, Sedgwick T, Shi YB, Evans T. Distinct functions are implicated for the GATA-4, -5, and -6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol. 1998;18:2901-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443-7 [DOI] [PubMed] [Google Scholar]
- 95. Kuo CT, Morrisey EE, Anandappa R, et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048-60 [DOI] [PubMed] [Google Scholar]
- 96. Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061-72 [DOI] [PubMed] [Google Scholar]
- 97. Molkentin JD, Tymitz KM, Richardson JA, Olson EN. Abnormalities of the genitourinary tract in female mice lacking GATA5. Mol Cell Biol. 2000;20:5256-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Morrisey EE, Tang Z, Sigrist K, et al. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12:3579-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hellebrekers DMEI, Lentjes MHFM, van den Bosch SM, et al. GATA4 and GATA5 are potential tumor suppressors and biomarkers in colorectal cancer. Clin Cancer Res. 2009;15:3990-7 [DOI] [PubMed] [Google Scholar]
- 100. Guo M, Akiyama Y, House MG, et al. Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res. 2004;10:7917-24 [DOI] [PubMed] [Google Scholar]
- 101. Anttonen M, Unkila-Kallio L, Leminen A, Butzow R, Heikinheimo M. High GATA-4 expression associates with aggressive behavior, whereas low anti-Müllerian hormone expression associates with growth potential of ovarian granulosa cell tumors. J Clin Endocrinol Metab. 2005;90:6529-35 [DOI] [PubMed] [Google Scholar]
- 102. Kyronlahti A, Ramo M, Tamminen M, et al. GATA-4 regulates Bcl-2 expression in ovarian granulosa cell tumors. Endocrinology. 2008;149:5635-42 [DOI] [PubMed] [Google Scholar]
- 103. Kamnasaran D, Qian B, Hawkins C, Stanford WL, Guha A. GATA6 is an astrocytoma tumor suppressor gene identified by gene trapping of mouse glioma model. Proc Natl Acad Sci U S A. 2007;104:8053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fu B, Luo M, Lakkur S, Lucito R, Iacobuzio-Donahue CA. Frequent genomic copy number gain and overexpression of GATA-6 in pancreatic carcinoma. Cancer Biol Ther. 2008;7:1593-601 [DOI] [PubMed] [Google Scholar]
- 105. Kwei KA, Bashyam MD, Kao J, et al. Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genetics. 2008;4:e1000081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shureiqi I, Zuo X, Broaddus R, et al. The transcription factor GATA-6 is overexpressed in vivo and contributes to silencing 15-LOX-1 in vitro in human colon cancer. FASEB J. 2007;21:743-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tsang AP, Visvader JE, Turner CA, et al. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell. 1997;90:109-19 [DOI] [PubMed] [Google Scholar]
- 108. Chan EM, Comer EM, Brown FC, et al. AML1-FOG2 fusion protein in myelodysplasia. Blood. 2005;105:4523-6 [DOI] [PubMed] [Google Scholar]
- 109. Miccio A, Wang Y, Hong W, et al. NuRD mediates activating and repressive functions of GATA-1 and FOG-1 during blood development. EMBO J. 2010;29:442-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tsuzuki S, Towatari M, Saito H, Enver T. Potentiation of GATA-2 activity through interactions with the promyelocytic leukemia protein (PML) and the t(15;17)-generated PML-retinoic acid receptor alpha oncoprotein. Mol Cell Biol. 2000;20:6276-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang S-J, Ma L-Y, Huang Q-H, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2008;105:2076-81 [DOI] [PMC free article] [PubMed] [Google Scholar]