

Abstract
Our understanding of cell cycle control has been based largely upon studies of synchronized cultures, often focused upon the early stages of the cell cycle following stimulation of quiescent cultures. These studies showed that cyclin D1 and p27Kip1 (p27) each respond to the growth environment of the cell and together control entry into the cell cycle. In contrast, all cell cycle phases were considered in these studies of actively growing cultures, including events leading to withdrawal from the cell cycle. This approach relies upon the techniques of microinjection, quantitative image analysis, and time-lapse microscopy. The results provide critical new detail to our understanding of the roles of cyclin D1 and p27 in cell cycle regulation. Three critical observations resulting from this work will be described here to demonstrate that 1) cyclin D1 levels oscillate through the normal cell cycle, 2) checkpoint kinases are able to suppress cyclin D1 during S phase, and 3) the level of p27 is determined by a dynamic interaction between cyclin D1 and p27 so as to determine the rate of cell cycle progression. Based upon these observations, a model of cell cycle control is presented in which ras activity stimulates cyclin D1 during G2 phase, resulting in commitment of the cell to continued cell cycle progression. During G1 phase, ras activity suppresses the level of p27 protein, most of which is bound to cyclin D1, resulting in regulation of the rate of proliferation. This model predicted the involvement of checkpoint kinases in regulating cyclin D1 and the role of checkpoint kinases in the protection of neural cells against reactive oxygen. The substantiation of these 2 predictions serves as general validation of the model.
Keywords: cell cycle, ras, cyclin D1, p27, G1 phase
Introduction
Control of cell cycle progression is vital in many, if not most, physiological processes, including development, repair, tumor formation, and the function of various tissues. Our understanding of this fundamental biological process has been based in large part upon studies with traditional biochemical techniques performed upon synchronized cells.1,2 The identity and biochemical properties of the molecules involved in cell cycle control were revealed by these studies. For example, growth factor signaling induces the elevation of cyclin D1 and suppresses p27Kip1 (p27) expression.3 Cyclin D1 then associates with cdk4 to form the kinase for the retinoblastoma protein, whose phosphorylation activates E2F transcription factors, resulting in the expression of cell cycle control proteins during G1 phase. Entry into S phase requires suppression of the cyclin inhibitory p27 protein, leading to activation of the critical cyclin E/cdk2 kinase.4 Thus, proliferative signaling was shown to promote expression of cyclin D1 and to block expression of p27, both of which are required for entry into the cell cycle.
Past biochemical analyses in synchronized cultures, however, provided information primarily of the early stages of the cell cycle, required disruption of the actual events under investigation, and seldom addressed the question of what happened to cells later in the cell cycle as they ultimately faced the decision to stop proliferating. For this purpose, a set of quantitative cytometric techniques were developed to make it possible to follow individual cells throughout the cell cycle in a variety of growth conditions. These techniques—microinjection, quantitative image analysis, and time-lapse microscopy—yielded 3 observations to be described here that together provide an entirely new perspective in our understanding of cell cycle control.5 The results demonstrate the necessity for cyclin D1 levels to oscillate through the normal cell cycle, the need for checkpoint kinases in this oscillation, and the role of p27 in regulating the rate of cell cycle progression.
Observation 1: Cyclin D1 Levels Fall during S Phase
Ras Is Required Only during G2 Phase
Based upon traditional studies, primarily upon quiescent cells, the notion developed that cyclin D1 levels are induced by growth factor action and stay elevated so long as the cells continue active cell cycle progression. It followed that when growth conditions no longer allowed active proliferation, the levels of the short-lived cyclin D1 protein would fall, and cells would stop dividing. Our evidence that this simple model is incomplete began not with a study of cyclin D1 but with cell cycle analyses of cellular ras activity. Ras is known to be a central mediator of proliferative signaling and is essential for cell cycle progression.6 Our initial goal was to determine when during the cell cycle cellular ras activity is required for continued cell cycle progression. These studies were aided by the anti-ras monoclonal antibody, which had been shown previously to block all ras activity immediately upon introduction into the cell by physical microinjection.7 The inhibition of ras activity was complete within a few minutes following injection, and such injections were found to have no other identifiable long-term effects upon the cell. Anti-ras injections were performed into actively proliferating cultures with cells in all cell cycle phases. In order to determine the cell cycle phase of individual cells at the time of injection, the culture was followed in time lapse prior to injection. Immediately after the injection was complete, the cells were once again followed in time lapse to determine which of the cells were able to continue cell cycle progression. The results were totally unexpected: each injected cell divided once and only once following anti-ras injection.
If ras activity were required during any specific cell cycle period, it would be expected to interrupt cell cycle progression immediately if introduced during or prior to that point, resulting in failure to pass through mitosis. The fact that each and every cell passed through mitosis exactly once indicated that this simple interpretation could not be applied. Rather, this result could only be explained if cellular ras activity were required during G2 phase and if neutralization of ras activity during G2 phase allowed progression through mitosis prior to final cell cycle blockage in the subsequent G1 phase (Fig. 1).8 As a control, anti–cyclin D1 antibody in the same type of study blocked cell cycle progression immediately whenever present during G1 phase, without passage through mitosis.
Figure 1.
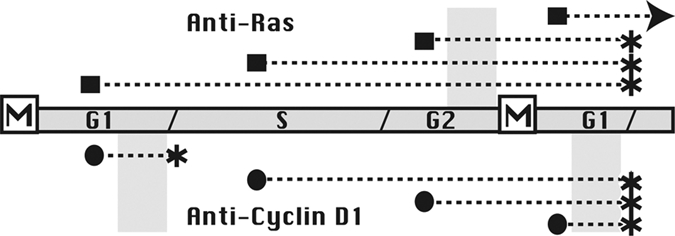
Ras activity is required during G2 phase. A timeline representing each cell cycle period is depicted. Above this timeline, injections with anti-ras antibody are represented, with a closed square indicating the cell cycle period in which injection took place, with a dashed line representing the progress of the cell through subsequent cell cycle stages, and with an asterisk indicating the point at which the cell stops proliferating. A similar depiction with closed circles below the timeline represents injections with anti–cyclin D1 antibody. Note that each cell injected with anti-ras antibody must pass through G2 phase (indicated with a shaded area above the timeline), which then results in blockage of cell cycle progression in the subsequent G1 phase. On the other hand, anti–cyclin D1–injected cells are inhibited immediately once they enter G1 phase (shaded areas below the timeline). In this way, each cell injected with anti-ras antibody passes through mitosis exactly once following injection.
The suggestion that ras activity is required only during G2 phase required a re-evaluation of traditional models of cell cycle control. For comparison, in quiescent cells stimulated to re-enter the cell cycle, ras activity was shown to be required up until the cells passed the restriction point, several hours prior to DNA synthesis.9 Once a cell had passed the restriction point, the neutralization of ras, removal of growth factors, and even removal of energy sources failed to block entry into S phase. The above results suggest that in the proliferating cells tested, the restriction point precedes mitosis. While blockage of ras activity has the same effect as removal of serum in most situations, we and others have found that the removal of serum early in G1 phase does block cell cycle progression.10 While further study will be needed to explain the difference between anti-ras injection and serum removal, it is clear that ras is required only during G2 phase of cultured NIH3T3 cells.
Cyclin D1 Levels Fall during S Phase
The above result was difficult to explain until the cell cycle expression characteristics of cyclin D1 were determined with the use of quantitative image analysis. This technique was developed to carefully quantitate the levels of cellular markers identified by fluorescence staining. Extensive comparison demonstrated the results with quantitative image analysis to be directly proportional to determinations with traditional biochemical analyses.11 Since these studies were conducted on microscopic images of monolayer cells, precise analysis of each cell compartment was possible with limited interference from light scatter or overlay of different cellular compartments. The results clearly revealed that cyclin D1 levels are high during G1 and G2 phases but are low during S phase in actively cycling cells from a variety of lineages (Fig. 2).11
Figure 2.
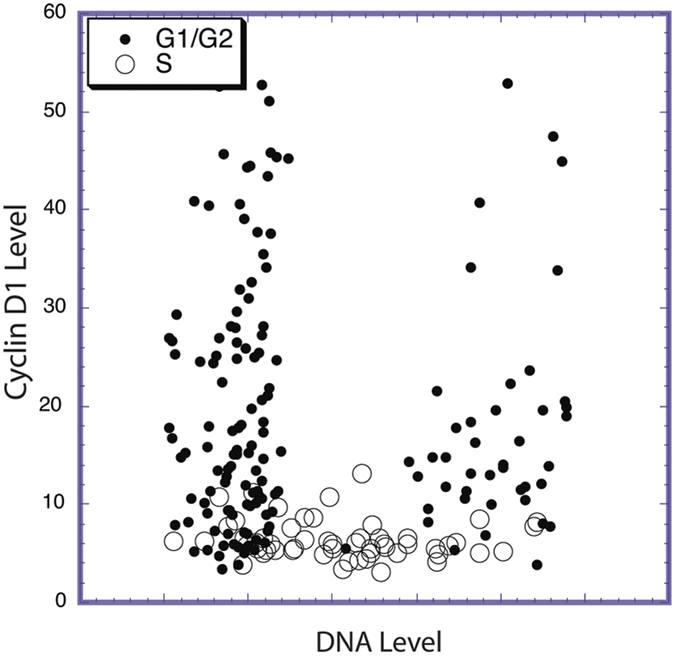
Cyclin D1 levels fall during S phase. Actively cycling Mrc5, diploid human fibroblast cells, were pulsed with BrdU, fixed, and stained for BrdU, cyclin D1, and DNA (with DAPI). Images of each fluorochrome were then subjected to analysis to determine the levels of each marker in each cell. In this figure, the level of cyclin D1 is plotted versus its DNA content on a cell-by-cell basis, with BrdU positive (S phase cells) shown as large open circles. Cyclin D1 levels are high in G1 and G2 phase cells and nearly undetectable in S phase cells. This pattern has been observed in a variety of actively cycling cells of different types and lineages.
The decline of cyclin D1 during S phase was somewhat controversial when first reported, despite the fact that cyclin D1 had long been known to be absent from the nucleus of S phase cells as revealed by fluorescent staining.12 Moreover, it had been shown that cyclin D1 binds PCNA to block DNA synthesis, as an explanation for why cyclin D1 must be low during S phase.13,14 The observation that total cyclin D1 levels fall during S phase, therefore, should not have been surprising. Nevertheless, experiments were performed to validate this result biochemically and to verify that cyclin D1 had not simply left the nucleus and entered the cytoplasm during S phase.15-17 In support of this conclusion, results from other laboratories demonstrate the decline in total cyclin D1 levels during S phase with the use of FACS analysis,17,18 following separation of cells into separate cell cycle phases by elutriation,19 following mitosis selection,20 or by serum stimulation of quiescent cells.21-23 At this time, there is little basis to question that cyclin D1 levels fall during S phase during normal cell cycle progression. Because of the central importance of cyclin D1 in cell cycle control, it is logical to assume that the regulation of cyclin D1 through the cell cycle must hold a critical key in understanding this control. The next goal, therefore, was to determine the molecular mechanism(s) involved in regulating cyclin D1 levels through the cell cycle and to determine the biological importance of this process.
Before moving on to a molecular analysis of cyclin D1 regulation, it is important to reconsider the results with anti-ras injection. Recall that ras is required only during G2 phase to promote cell cycle progression and that cyclin D1 is specifically elevated during G2 phase. Since it is well known that ras stimulates cyclin D1 expression, it was possible that ras had to be active during G2 specifically to promote elevation of cyclin D1. To test this possibility, anti-ras antibody was injected into cycling cells, and cyclin D1 expression was analyzed at various times thereafter. In these injected cells, cyclin D1 levels declined rapidly during G2 phase but remained high in G1 phase. The level of cyclin D1 observed in G1 phase cells declined only after sufficient time had passed following anti-ras injection to allow cells originally in G2 phase to pass through mitosis and enter G1 phase with reduced cyclin D1 levels and for cells originally in G1 phase with high cyclin D1 levels to enter S phase.11 This result suggests that ras activity induces cyclin D1 elevation specifically during G2 phase and that, once elevated in G2 phase, cyclin D1 remains high through the subsequent G1 phase. To test this hypothesis, constitutively active oncogenic ras protein was injected into actively cycling cells and the levels of cyclin D1 expression determined 3 hours thereafter. Cyclin D1 levels were rapidly induced by the injected ras protein, but only in G2 phase cells.24 Once again, as these cells divided and moved into G1 phase 6 to 9 hours following oncogenic ras injection, the levels of cyclin D1 increased in G1 phase.
The above results show that growth factor signaling leading to ras activation promotes cyclin D1 elevation specifically during G2 phase and that, once so induced, cyclin D1 levels remain high through mitosis and the subsequent G1 phase. On the other hand, without ras or proliferative signaling activity, the low levels of cyclin D1 established during S phase fail to increase upon entry into G2 phase and remain low through mitosis and the subsequent G1 phase. Thus, the level of cyclin D1 established during G2 phase by ras signaling determines the proliferative capacity of the cell once it passes through mitosis and enters the next cell cycle. In this way, the decision to continue proliferating is made in cycling cultured cells during G2 phase when proliferative signaling induces an increase in cyclin D1 levels. If cyclin D1 levels fail to increase during G2 phase, the cell passes through mitosis into quiescence (Fig. 3). These studies in individual cells, therefore, suggest the molecular means by which the cell determines its continuing proliferative fate.
Figure 3.
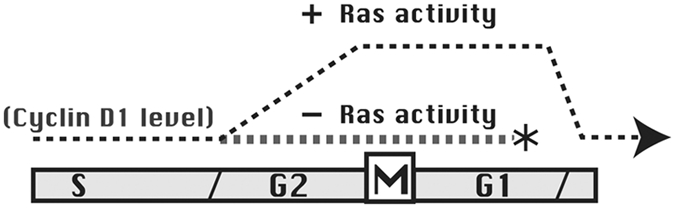
Cyclin D1 elevation during G2 phase determines proliferative fate. This diagram represents progressive stages of the cell cycle, with broken lines indicating cyclin D1 levels. As the cells enter G2 phase in the presence of cellular ras activity, cyclin D1 levels increase and remain at high levels throughout G1 phase. This promotes entry into the subsequent S phase and a commitment to complete the entire upcoming cell cycle. If ras activity is low upon entry into G2 phase, cyclin D1 levels remain low, and the cell enters quiescence following mitosis.
Phosphorylation of Cyclin D1 Thr 286
The activity of cyclin D1 is strongly linked to its expression level, which has been postulated to be controlled in part by phosphorylation on Thr 286, leading to proteasomal degradation.25 It was possible that the decline in cyclin D1 during S phase might be similarly controlled. Amino acid labeling studies, analysis of cyclin D1 mRNA levels at the biochemical and cellular level, nuclear run-on analysis of the mRNA synthetic rate, and image analysis in the presence and absence of proteasomal inhibitors all confirmed that reduced protein stability, rather than reduced protein production or altered mRNA stability, was responsible for the decline in cyclin D1 levels during S phase.26,27 To confirm this conclusion, cyclin D1 mRNA was shown to be quite stable through the cell cycle so long as growth conditions remained positive, but declined rapidly with the removal of serum. Moreover, even in the presence the RNA polymerase II inhibitor α-amanitin, the levels of cyclin D1 were observed to increase when cells entered G2 phase in the presence of positive growth conditions,26 an observation that strongly argues against the involvement of an increase in cyclin D1 mRNA during G2 phase. These observations confirm that the decline in cyclin D1 levels during S phase is determined by protein stability.
To determine if protein stability is regulated by phosphorylation as previously described, mutations were introduced into several potential phosphorylation sites of cyclin D1. These mutant proteins were then linked to green fluorescent protein and expressed in living cells. The expression level of each mutant protein was then determined from the fluorescence level of the associated green fluorescent protein in a time-lapse, fluorescence analysis. The cyclin D1–associated fluorescence was determined each hour for each cell and related to the time the cell passed through mitosis. As expected, with the wild-type protein, cyclin D1–associated fluorescence declined within a few hours after the cell was observed to pass through mitosis, only to increase again just prior to mitosis. No accumulation of fluorescence was observed in the cytoplasm of cells in any cell cycle phase. On the other hand, a mutation at position 286 produced a protein that was expressed uniformly throughout the cell cycle and failed to decline upon entry into S phase.28 Other mutants tested behaved as wild-type protein. Moreover, with an antibody specific for phosphate on Thr 286 of cyclin D1 (from Michelle Garrett, Institute of Cancer Research, Sutton, UK), the 286 phosphorylation was shown to increase specifically during S phase. The decline in cyclin D1 levels during S phase was thus shown to result from the phosphorylation of Thr 286, leading to proteasomal degradation. We then initiated experiments to demonstrate if the kinase responsible was glycogen synthase kinase 3 (GSK3) as previously reported.29
GSK3 Is Not Involved in S Phase Suppression of Cyclin D1
GSK3 is a tightly regulated enzyme with a variety of functions, including the regulation of glycogen synthesis as the name implies. It is inactivated by phosphorylation by the AKT kinase, which is in turn regulated by phosphatidylinositol-3 kinase (PI3K), a target of ras activity and a general mediator of proliferative signaling.30,31 If GSK3 promotes phosphorylation of cyclin D1, leading to its degradation during S phase, the activity of GSK3 would be most prominent during S phase. Similarly, AKT and PI3K would be most active during G1 and G2 phases so they could phosphorylate and inactivate GSK3. The activities of these 3 kinases were, therefore, analyzed biochemically in synchronized cultures, and by fluorescence analysis of individual cells of actively cycling cultures, but in no instance was there any evidence for cell cycle–specific alterations in the activity of any of them.32 To extend this analysis, the activities of AKT and PI3K were blocked with siRNA and specific inhibitors, respectively, without alteration in cyclin D1 phosphorylation or expression levels through the cell cycle.32 These observations raised the likelihood that GSK3 is not responsible for regulation of cyclin D1 levels during S phase.
To gain additional evidence for the above conclusion, the activity of GSK3 itself was artificially elevated by the introduction of a nonphosphorylated, constitutively active GSK3 plasmid (from J.R. Woodgett , Samuel Lunenfeld Research Institute, Toronto, Canada), without any affect upon cyclin D1 levels. Similarly, no effect upon cyclin D1 expression was observed in cells following GSK3 ablation with siRNA. Finally, GSK3 was inhibited in cultures with the potent, specific inhibitor LiCl. The phosphorylation of β-catenin, a known target of GSK3, was efficiently blocked following such treatments, but the phosphorylation of cyclin D1 was not altered in the same cultures.32 These experiments clearly demonstrated that GSK3 is not responsible for the phosphorylation of cyclin D1 during S phase, but it remained possible that another proliferative signaling kinase might be involved. However, even when serum was totally removed from cultures, or anti-ras antibody injected to disrupt proliferative signaling, cyclin D1 fell to low levels specifically during S phase. It was clear, therefore, that no kinase regulated by the mitogenic signaling environment of the cell could be responsible for the S phase suppression of cyclin D1. Rather, it was necessary to conclude that some event specifically controlled by S phase or DNA synthesis promotes the phosphorylation of Thr 286 and the resulting decline in cyclin D1 levels.
Observation 2: ATR and ATM Regulate Cyclin D1 Phosphorylation
When it became clear that neither GSK3 nor any kinase controlled by proliferative signaling regulates cyclin D1 phosphorylation and degradation during Sphase, consideration focused upon kinases that might be activated by a process specific to S phase itself. The checkpoint kinase ATR was a likely candidate. It was identified in consequence of its activation by single-stranded DNA, such as produced following ultraviolet (UV) irradiation. Recent studies have shown that ATR activating structures are also produced during normal DNA synthesis.33,34 Indeed, it has been shown that ATR serves as a normal modulator of the rate of DNA synthesis.35
ATR Promotes Thr 286 Phosphorylation
The ability of ATR to promote phosphorylation of cyclin D1 on Thr 286 was tested by artificially inducing its activity by UV irradiation. The phosphorylation of cyclin D1 was then analyzed with specific antibodies in individual cells. Phosphorylation of both cyclin D1 Thr 286 and histone H2AX were induced by UV irradiation. This phosphorylation was inhibited by pretreatment of the cells with siRNA against ATR and was interestingly restricted to S phase.36 To extend this observation, the activity of ATR was artificially activated by its molecular inducer, the TopBP1 protein. A plasmid expressing the active region of this protein linked to green fluorescence protein (from A. Kumagai, Division of Biology, California Institute of Technology) was injected into living cells, which were then tested for cyclin D1 levels and phosphorylation. As a control, an inactive mutant of the TopBP1 protein similarly expressed was also tested. The active peptide, but not the inactive mutant TopBP1 peptide, strongly induced cyclin D1 phosphorylation and reduced total cyclin D1 levels within injected cells.36
Both of the above experiments clearly indicated the ability of ATR to promote cyclin D1 phosphorylation, leading to its degradation. These studies in individual cells were confirmed biochemically in bulk cultures following ablation of ATR by siRNA, where it was shown that cyclin D1 Thr 286 phosphorylation following UV irradiation was reduced by siRNA ablation of ATR.36 The fact that ATR is able to promote cyclin D1 phosphorylation and degradation supported the original assumption that it might normally function during S phase to suppress cyclin D1 levels. To test this idea, cells treated with siRNA against ATR were analyzed without any other treatment. In these normally proliferating cells, ablation of ATR reduced the normal levels of Thr 286 phosphorylation during S phase. These studies indicate not only that ATR is able to promote phosphorylation and degradation of cyclin D1 but also that it does so in the course or normal S phase, exactly as predicted above. The confirmation of this seemingly unlikely prediction serves as direct support for the results and hypotheses discussed above (Fig. 4). While it is clear that ATR is at least in part responsible for the suppression of cyclin D1 during S phase, it is not clear if other kinases might also be involved.
Figure 4.
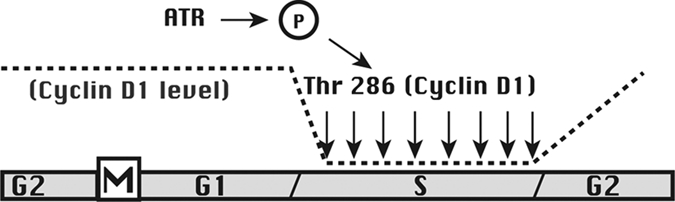
The mechanism of cyclin D1 suppression during S phase. This diagram represents progression through successive cell cycle phases, with the level of cyclin D1 indicated with a broken line. High levels of cyclin D1 during G2 and G1 phases allow the cell to enter S phase. The ATR checkpoint kinase is activated by DNA synthesis and promotes (through an unknown signaling pathway) the phosphorylation of Thr 286 on cyclin D1. This phosphorylation leads to rapid proteasomal degradation of cyclin D1 so long as DNA synthesis continues. At the end of S phase, however, the active phosphorylation of cyclin D1 ceases, and the levels of cyclin D1 are again allowed to increase so long as the growth environment of the cell permits.
ATM Also Promotes Cyclin D1 Thr 286 Phosphorylation
The ATM kinase is closely related to ATR, but is induced by double-stranded breaks, and is normally involved in blockage of cell cycle progression following DNA damage.33,37 To determine if ATM is also able to promote cyclin D1 phosphorylation, its activity was induced by treatment with a drug able to induce double-stranded DNA breaks. This treatment was shown to induce phosphorylation of cyclin D1 Thr 286 as expected. In this case, however, phosphorylation of cyclin D1 and histone H2AX were observed equally in all cell cycle phases (while ATR promoted phosphorylation of these 2 proteins only during S phase, as described above). As direct evidence that ATM was responsible, the Thr 286 phosphorylation of cyclin D1 was inhibited by pretreatment of cells with KU55933, a specific ATM inhibitor, prior to the introduction of double-stranded breaks.36
ATM is a checkpoint kinase, normally involved in cell cycle blockage following DNA damage.38 Experiments were, therefore, designed to determine if the phosphorylation of cyclin D1 plays any role in this ATM-induced checkpoint response. Time-lapse and quantitative image analysis demonstrated that double-stranded breakage of DNA blocks cell cycle progression in all 3 cell cycle phases.36 The pathway leading from checkpoint kinases to blockage of the critical S phase kinase cyclin A/cdk2 is well known.39 It is not as clear, however, how cell cycle progression might be interrupted during G1 phase. However, when the nonphosphorylated Thr 286 mutant cyclin D1 was expressed near endogenous levels prior to the induction of double-stranded breakage, the cell cycle inhibition profile was altered. The nonphosphorylatable cyclin D1 had promoted passage from G1 to S phase of over half of the cells that would have otherwise been blocked in G1 phase by the DNA damage.36 This is consistent with the idea that activated ATM promoted phosphorylation and degradation of cyclin D1 during G1 phase, leading to G1 phase blockage. The mutant cyclin D1, however, could not be phosphorylated by active ATM and therefore remained stable and was able to promote passage from G1 to S phase even in the presence of double-stranded DNA breakage. This confirms not only that ATM promotes cyclin D1 phosphorylation and degradation but also that this phosphorylation plays an important role in the ability of ATM to block cell cycle progression. It is important to emphasize that these results with ATM and ATR were predicted by the cytometric studies of cyclin D1 described above and serve as important validation of those studies.
ATM and Phosphorylation of Cyclin D1 in Neural Cells
The role of cyclin D1 in the checkpoint response of ATM made a potentially critical biological prediction. Mutation of ATM produces ataxia telangiectasia, a disease characterized by increased sensitivity to ionizing radiation and ataxia (a neural dysfunction).40 Recent studies indicate that unscheduled expression of cyclin D1, leading to inappropriate initiation of DNA synthesis, is in part responsible for neuronal degeneration in ataxia telangiectasia and other neural diseases.41-43 It is possible that since ATM functions normally to suppress cyclin D1 levels following DNA damage, its loss might allow elevated cyclin D1 levels under conditions of stress, potentially explaining the inappropriate cell cycle entry and neural cell death in ataxia telangiectasia. This possibility was tested in fully differentiated neurons in culture. These neurons were exposed to hydrogen peroxide in the presence or absence of the ATM inhibitor Ku55933. The expression of cyclin D1 was then tested in cells shown by marker expression to be mature neurons, which would never express cyclin D1 under normal circumstances. However, in these cultures, cyclin D1 was expressed in some cases to high levels, but only in cultures in which ATM activity had been inhibited by a highly specific inhibitor. Moreover, the overall numbers of neurons surviving in such cultures were reduced, exactly as predicted.44 Further study will be needed to determine if the cell death seen is linked to elevated cyclin D1 in these terminally differentiated cells and if these observations have any relationship to neural dysfunction in any disease. This result, however, provides compelling substantiation for conclusions reached above in the studies of cyclin D1 regulation through the cell cycle that predicted them.
Observation 3: The Rate of Cell Proliferation Depends upon a Dynamic Interaction between Cyclin D1 and p27
To this point, the analysis has focused upon cyclin D1, a positive regulator of cell cycle progression whose expression is stimulated by growth factor signaling during G2 phase and required throughout G1 phase. The p27 molecule is a negative regulator of cell cycle progression whose levels are suppressed by proliferative signaling. p27 inhibits cell cycle progression following binding to the critical S phase kinase cdk2 associated with cyclins A or E45 and must be reduced to low levels during the initiation and progression of S phase. The p27 molecule also binds the cyclin D/cdk4 kinase during G1 phase, but this binding is not inhibitory so long as proliferative signaling promotes p27 phosphorylation on tyrosine.46 Interestingly, it is known that p27 serves a vital role in cell cycle progression by promoting the association between cyclin D1 and cdk4.47,48 It has been suggested that the binding between p27 and cyclin D/cdk4 is able to neutralize p27 and thus “titrate” it away from binding and inhibiting cdk2. The studies of p27 in single cells to be described below reveal an interaction between p27 and cyclin D1 that is much more dynamic than simple neutralization. This interaction regulates the rate of progression through the cell cycle.
Ras Activity Regulates p27 Levels
When serum is added to quiescent cells, p27 levels decline gradually to very low levels prior to S phase entry. In actively cycling cells, p27 levels remain relatively low but are observed to increase gradually throughout G1 phase, only to decline rapidly upon the initiation of DNA synthesis.49 This suppression was shown to depend upon proliferative signaling and upon ras activity. The role of ras in p27 regulation was demonstrated when anti-ras antibody was injected into actively cycling cells, resulting in increased p27 expression in all cell cycle phases soon thereafter. Alternatively, the introduction of constitutively active oncogenic ras protein into cycling cells by microinjection resulted in suppression of p27 in each cell cycle phase.49 Interestingly, detailed analyses indicated that the suppression of p27 levels by proliferative signaling involved a separate mechanism for each cell cycle period. For example, p27 expression during G1 phase was elevated when the signaling kinases MEK and PI3K were specifically inhibited. Expression of p27 was elevated during S phase when AKT was inhibited, while it is well known that late in the cell cycle, the Skp2 ubiquitin ligase promotes p27 degradation following phosphorylation by cdk2.49 Interestingly, the level of Skp2 is also regulated by ras activity. Of particular note for the purposes of this discussion is the fact that ras controls the levels of p27 during G1 phase through a defined signaling pathway. This consideration became important in studies to determine the rate of cell cycle progression described below.
Cyclin D1 Is Not Involved in the Rate of Passage through the Cell Cycle
The overall rate of cell cycle progression is normally determined by the length of G1 phase since the lengths of other cell cycle phases tend to be relatively uniform. It is reasonable to expect that this rate is determined by growth conditions of the cell and, therefore, related to ras activity. Since both cyclin D1 and p27 levels are determined by ras activity, it follows that one or both of them could be involved in determining the length of G1 phase and, therefore, the overall rate of cell cycle progression. To determine the potential role of cyclin D1 in regulating G1 phase length, cyclin D1 levels were artificially elevated slightly above endogenous levels following introduction of an expression vector into cells. As has been reported with other cyclins,50 the length of G1 phase was decreased by the elevated level of cyclin D1, while the rate of passage through S phase was increased, resulting in an overall increase in cell cycle length. The increase in cell cycle length was proportional to the additional expression of cyclin D1. In addition, when cyclin D1 levels were reduced following microinjection of siRNA, there was no apparent alteration in cell cycle length.51 These results make it apparent that cyclin D1 is not involved in the normal regulation of the rate of cell cycle progression, despite the fact that it plays a vital role in the overall regulation of the proliferative state of the cell, as demonstrated above. In support of this conclusion, evidence presented above indicates that ras regulates cyclin D1 levels only during G2 phase and, therefore, would be unlikely to target cyclin D1 to regulate the rate of passage through G1 phase. Moreover, a variety of studies have demonstrated that the cyclin D/cdk4 retinoblastoma kinase is active early in G1 phase, well before the critical decision to initiate S phase is made. For example, cyclin D/cdk4 is required to inactivate the retinoblastoma protein and release E2F to promote synthesis of cyclin E, the synthesis of which must be accomplished well before the G1/S phase transition. Therefore, while cyclin D1 is clearly required for passage through G1 phase, it is apparently not normally a central player in determining the length of G1 phase or of the cell cycle.
p27 Determines the Length of G1 Phase
While cyclin D1 is apparently required early during G1 phase, the inhibitory activity of p27 is particularly important late in G1 phase. It blocks the activity of cyclin E/cdk2, which is essential for the initiation of DNA synthesis. The fact that p27 levels are controlled by ras activity during G1 phase further supports the idea that it might be involved in regulating the timing of G1/S phase transition. In an effort to determine if p27 can be involved in modulating the length of G1 phase, a plasmid expressing the p27 sequence linked to green fluorescent protein was microinjected into cycling cells. Even the slightest exogenous expression of p27 as indicated by the associated fluorescence completely inhibited cell cycle progression, as an indication of its profound inhibitory activity.51 This observation created an important concern. Cyclin D1 is apparently not involved in regulating G1 phase length, and p27 is so inhibitory it is difficult to imagine how it could effect the delicate modulation required for sensitive control over the rate of cell growth. Nevertheless, an siRNA able to reduce p27 expression levels to varying degrees in individual cells was microinjected into actively cycling cells, which were then incubated for 40 hours prior to a time-lapse analysis of the cell cycle. The length of G1 phase was determined. In those cells with reduced p27 levels, G1 phase lasted between 3 and 5 hours in the NIH3T3 cultures analyzed. Uninjected cells with normal p27 levels passed through S phase in 3 to 9 hours. The reduction in G1 phase length was inversely correlated with p27 level, especially in cells with the longest G1 phase lengths.51 It should be noted that even cells with extremely low levels of p27 required at least 3 to 5 hours for G1 phase. Apparently, this minimal length allows completion of molecular events required during G1 phase, while the levels of p27 serve to determine any additional time spent in this cell cycle period. It is clear that p27 determines the length of G1 phase, but how this is achieved with such a profoundly inhibitory protein remains to be determined.
Cyclin D1 and p27 Are Mutually Regulated
As a means to determine how p27 inhibitory activity might be regulated, previous studies reporting the titration of p27 by cyclin D1 were considered. A plasmid expressing the p27 protein linked to the green fluorescent protein was again microinjected. In a parallel injection, however, the p27 plasmid was injected together with a 3-fold excess of plasmid expressing unmodified cyclin D1. As above, the p27 alone blocked entry into S phase at the lowest detectable levels of expression. In cells receiving the combined p27 and cyclin D1 injections, however, entry into S phase was observed in cells with considerably higher levels of p27, levels that alone would have been completely inhibitory. This confirms the ability of cyclin D1 to modulate the inhibitory potential of p27. Unexpectedly, however, it was also found that in those cells receiving the combined injection, the overall level of p27 expression had been increased above that seen in cells receiving p27 alone.51 Thus, cyclin D1 had not only inhibited p27 activity, but it also had actually increased p27 expression. If cyclin D1 functions simply to block p27 activity, it would not be expected to promote expression of p27. Clearly, the interaction between these 2 molecules with opposite proliferative activity is more involved than a simple, static inhibitory effect.
The unexpected interaction between cyclin D1 and p27 was further explored by microinjection of either p27 or cyclin D1 alone, followed by determination of the expression levels of both proteins in recipient cells. In cells with elevated expression of exogenous p27, the endogenous levels of cyclin D1 were coordinately elevated. Similarly, exogenous cyclin D1 expression promoted increased expression of endogenous p27.51 Clearly, the expression of these 2 proteins is coordinately regulated. To determine if this is the case in normal cells, the p27 and cyclin D1 expression levels were each determined in the same individual cells of an actively cycling culture. While the absolute levels of each of these proteins varied dramatically throughout the culture, the ratio between them was highly consistent in each of a variety of cell lines tested. High levels of p27 were tolerated in cycling cells so long as cyclin D1 was proportionally elevated. When serum was removed or the culture became crowded, the ratio of p27 to cyclin D1 increased, and the cells stopped proliferating.51 It is apparent that cell cycle progression is regulated by the interaction between these 2 regulatory molecules. It follows that cyclin D1 functions in a complex way to modulate the inhibitory activity of p27 and allow it to promote the marginal alterations in G1 phase length required for the sensitive control of the rate of cellular proliferation described above.
p27 Plays a Central Role in Determining the Restriction Point in Quiescent Cells
For some time, the actual mechanism by which a quiescent cell becomes committed to enter S phase has been uncertain. This restriction point, which commits a cell to enter S phase several hours before DNA synthesis begins, has been postulated to involve the retinoblastoma protein, or cyclin D1.9,52 While these molecules are certainly involved, the above experiments reveal the central role played by p27 in regulating progression through G1 phase and suggest its potential role in the restriction point as well. To test this possibility, NIH3T3 cells were rendered quiescent by serum deprivation and restimulated with serum. Approximately 50% of the cells passed the restriction point 7 hours later, while DNA synthesis in these cultures began at 12 hours following serum addition. Thus, when serum is removed after only 7 hours of serum stimulation, half the cells go on to complete the cell cycle, and the other half remain quiescent. Consequently, p27 levels were analyzed in these cultures 7 hours after serum addition. It was found that the levels of p27 varied dramatically among the cells of such cultures. When the cells were then further cultured in the absence of serum, those cells with low p27 levels were the ones that proceeded through the cell cycle and initiated DNA synthesis. Cells with the higher p27 levels at 7 hours of serum stimulation failed to progress in the cell cycle.53
The above results suggest that the reduction in p27 is closely correlated with passage through the restriction point. The possibility that this reduction in p27 is actually necessary was shown by microinjection of a p27 expression plasmid at 9 hours after serum stimulation. This elevation of p27 levels in cells, which had passed the restriction point, blocked entry into S phase.53 It should be recalled that the injection of anti-ras or anti–cyclin D1 antibody, and even the removal of an energy source from the medium, failed to block entry into S phase following passage through the restriction point. The reduction in p27 levels, therefore, plays a central role in establishing and maintaining the restriction point. This conclusion was strengthened when siRNA was microinjected into quiescent cells prior to serum stimulation. At 7 hours following serum addition under normal conditions, half the cells would have passed the restriction point and would enter S phase even if serum were removed, as described above. In those cultures in which p27 levels had been artificially reduced by siRNA prior to serum stimulation, however, the proportion of cells committed to S phase entry after 7 hours’ serum stimulation was dramatically increased.53 Thus, the reduction in p27 levels is both necessary and sufficient for determination and maintenance of the restriction point. These observations are consistent with the critical role played by p27 in regulating the rate of passage through the cell cycle54 and provide independent support for this conclusion.
Model of Cell Cycle Control Based upon Studies of Cyclin D1 and p27
In the past, proliferative signaling was shown to promote cell cycle entry by suppressing the cyclin inhibitory protein p27 and elevating the positive growth regulatory cyclin D1 protein. When proliferative signaling ceased, it was presumed that these events would be reversed, leading to exit from the cell cycle. Those past studies, however, were not designed to study cell cycle progression after it had begun, or its rate, both of which are vital considerations in cell and organ physiology. The studies described here focused upon individual cells in actively cycling cultures, where no cell cycle blockage was involved and where all cell cycle stages and growth conditions could be analyzed. Microinjection targeted individual molecules in specific cell cycle stages, while quantitative image analysis determined with precision the expression or phosphorylation of individual proteins in each cell. This approach revealed 3 novel observations, which together refine our understanding of cell cycle control.
First, cyclin D1 levels invariably decline during S phase. This decline not only eliminates the inhibitory effect of cyclin D1 upon DNA synthesis, but it also effectively eliminates any cellular memory of past growth regulatory events and requires the reassessment of current growth conditions prior to making a decision to continue active proliferation. The decline of cyclin D1 during S phase makes its elevation during G1 phase all the more important as it commits the cell to continued cell cycle progression.
The second critical observation reported here links checkpoint kinases with the phosphorylation and suppression of cyclin D1 during S phase. The fact that the checkpoint kinase ATR performs this function in normal cells has important implications. For example, evidence is accumulating to support the possibility that checkpoint kinases function to suppress cyclin D1 elevation and cell death among cultured neurons, as a possible explanation for the neural effects of ATM mutation in ataxia telangiectasia.
Finally, these studies indicate that cyclin D1 could not regulate G1 phase length, while the profound inhibitory activity of p27 also made it an unlikely candidate. The observation that cyclin D1 has a dynamic relationship with p27 explained how the 2 can interact together to adjust the rate of G1 phase progression. These observations lead to the experiments that demonstrated the central role played by p27 in the restriction point.
Combined Model for Cell Cycle Control
Based upon the above observations, the following model is proposed to explain cell cycle control (Fig. 5). It is known that once the cell enters S phase, it is normally committed to complete DNA synthesis. Both cyclin D1 and p27 are suppressed during this cell cycle period regardless of the growth environment. Upon completion of DNA synthesis, the cell must reassess its growth state prior to making a commitment to continue proliferating or enter quiescence. If growth conditions remain conducive for continued proliferation upon entry into G2 phase, active ras stabilizes cyclin D1 mRNA to promote elevation of cyclin D1 (as soon as the S phase–regulated kinase is no longer able to promote cyclin D1 degradation). Once elevated during G2 phase, cyclin D1 levels remain high through the mitosis and G1 phase to allow entry into the subsequent S phase and completion of the next cell cycle. Thus, during G2 phase, ras activity signals for the cell to continue active proliferation by promoting high cyclin D1 levels. If growth conditions fail to promote positive proliferative signaling prior to entry into G2 phase, cyclin D1 mRNA becomes unstable, cyclin D1 levels are unable to increase, and the cell enters quiescence following mitosis (Fig. 5).
Figure 5.
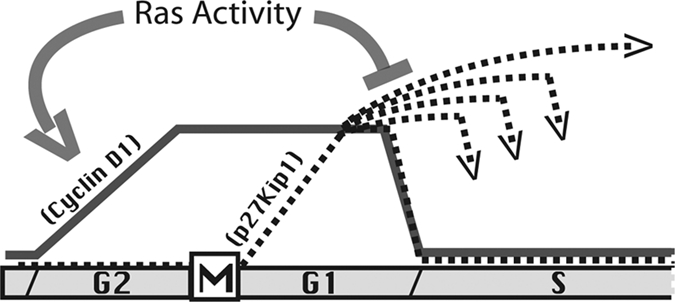
Model outlining the postulated roles of cyclin D1 and p27 in cell cycle regulation. This diagram represents progression through the cell cycle, with the levels of cyclin D1 indicated by a solid line and those of p27 indicated with broken lines. In positive growth conditions, ras activity promotes the increase of cyclin D1 during G2 phase and then into G1 phase. This event commits the cell to continued proliferation. The levels of p27, however, are suppressed by Skp2 during G2 phase and allowed to increase only upon entry into G1 phase. As p27 accumulates through G1 phase, it binds cyclin D1, which modulates its inhibitory activity. p27 levels, however, are likely to increase above the levels of cyclin D1 unless ras signals through MEK to promote its degradation. If p27 levels remain within the dynamic range of cyclin D1, the cell enters S phase as soon as metabolic requirements for cell cycle passage are met. If ras signaling fails to completely eliminate p27, the increased levels, in combination with cyclin D1, are able to promote a graded increase in the length of G1 phase. This increase is proportional to the increased level of p27 over cyclin D1, so that the combination of cyclin D1 levels, ras activity, and p27 expression determines the overall length of G1 phase. In this way, ras activity regulates progression through the cell cycle. In G2 phase, it stimulates increased cyclin D1 levels to signal continued proliferation. In G1 phase, it suppresses p27 levels to determine the rate of cell cycle progression.
Once a cell passes through mitosis, p27 begins to accumulate and bind cyclin D1. As this is occurring, events required for entry into S phase are complete, including formation of the replication origins and synthesis of cyclin E. Once these basic processes are complete, the cell is competent to enter S phase and will do so unless inhibited by p27. The inhibitory activity of p27 during G1 phase is regulated by its binding to cyclin D1, and its degradation as the result of ras and MEK signaling. If ras is active, p27 levels are suppressed, and the cell enters S phase as soon as basal metabolic functions are complete. If p27 suppression is incomplete, it interacts with cyclin D1 to promote a gradual increase in G1 phase length. Thus, it is during G1 phase that sensitive variations in ras and MEK activities set the levels of free and cyclin D1–bound p27 to regulate the overall rate of cell cycle progression. Once DNA synthesis is initiated, the active cdk2 kinases phosphorylate p27, leading to proteasomal degradation modulated by the SKP2 ubiquitin ligase to ensure that p27 is unable to interfere with the completion of the cell cycle.
It is worth noting that this model eliminates the potential problem resulting from inappropriate expression of these 2 critical growth regulatory molecules. For example, if cyclin D1 were mutated to allow constitutive high levels, the cell might experience unregulated progression through G2, mitosis, and G1 phase. But according to the considerations discussed above, the high level of cyclin D1 would inhibit progression through S phase and block overall cell growth. Likewise, it is known that p27 serves a vital function by promoting the association of cyclin D1 with cdk4 to generate an active retinoblastoma protein kinase. Thus, if p27 expression were blocked, the cell would lose a powerful growth inhibitor until the requirement for cyclin D1/cdk4 became apparent, at which time the cell would once again stop proliferating. The fact that these critical regulatory molecules have both positive as well as negative growth promoting properties ensures that their regulation is properly maintained for active cell cycle progression.55
Unanswered Questions
The above model raises several questions that must be addressed in future studies. In a broad range of cultured cells studied, cyclin D1 levels always increase during G2 phase, indicating the requirement for ras activity during this cell cycle period. The removal of serum, however, has the ability to block cell cycle progression during the early part of G1 phase. It is not possible, at present, to explain why removal of serum and neutralization of ras would have different cell cycle effects; or if in normal tissues, where G1 phase might be much longer, the same cell cycle pattern exists. It is also not clear if other kinases in addition to ATR are involved in phosphorylation of cyclin D1 during S phase or the signaling pathway involved downstream of ATR. In addition, the detailed nature of the interaction between cyclin D1 and p27 is yet to be explained, along with the signaling pathway, leading from MEK to p27 degradation specifically during G1 phase. Finally, the mechanism by which proliferative signaling stabilizes cyclin D1 mRNA is yet to be described.
Acknowledgments
This work is a direct consequence of the training I received from Hidesaburo Hanafusa. It follows in a direct sequence from the studies I began in his laboratory. That work was the result of his careful training in the formulation of an experiment, the interpretation of results, and an understanding of where the conclusions might lead. These lessons have been my guide ever since. My goal has ever been to do something he would appreciate. For all he taught me, however, I am most grateful for the example of his excitement for science and his dedication to accuracy. I count the time I spent under his mentorship as a priceless opportunity. May his memory and influence live with those of us who shared this gift.
I also acknowledge the central role played by Masahiro Hitomi throughout these studies, together with his review of this article. In addition, I acknowledge the tireless work of Jyoti Harwalkar, Yang Guo, Ke Yang, and Gaurisankar Sa. Together, we worked as a team to produce these results. It is difficult to imagine how this work might have progressed without the contributions of any one of these co-investigators.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the Lerner Research Institute of the Cleveland Clinic.
References
- 1. Sherr CJ. Mammalian G1 cyclins and cell cycle progression. Proc Assoc Am Physicians. 1995;107(2):181-6 [PubMed] [Google Scholar]
- 2. Musgrove EA. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Factors. 2006;24(1):13-9 [DOI] [PubMed] [Google Scholar]
- 3. Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol. 1997;17(7):3850-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672-7 [DOI] [PubMed] [Google Scholar]
- 5. Stacey DW, Hitomi M. Cell cycle studies based upon quantitative image analysis. Cytometry A. 2008;73(4):270-8 [DOI] [PubMed] [Google Scholar]
- 6. Smith MR, DeGudicibus SJ, Stacey DW. Requirement for c-ras proteins during viral oncogene transformation. Nature. 1986;320(6062):540-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mulcahy LS, Smith MR, Stacey DW. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature. 1985;313(5999):241-3 [DOI] [PubMed] [Google Scholar]
- 8. Hitomi M, Stacey DW. Cellular ras and cyclin D1 are required during different cell cycle periods in cycling NIH 3T3 cells. Mol Cell Biol. 1999;19(7):4623-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1(2):103-10 [PubMed] [Google Scholar]
- 10. Zetterberg A, Larsson O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescnec of Swiss 3T3 cells. Proc Natl Acad Sci U S A. 1985;82:5365-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr Biol. 1999;9(19):1075-84 [DOI] [PubMed] [Google Scholar]
- 12. Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17:7362-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pagano M, Theodoras AM, Tam SW, Draetta GF. Cycling D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblassts. Genes Dev. 1994;8:1627-39 [DOI] [PubMed] [Google Scholar]
- 14. Fukami-Kobayashi J, Mitsui Y. Cyclin D1 inhibits cell proliferation through binding to PCNA and cdk2. Exp Cell Res. 1999;246(2):338-47 [DOI] [PubMed] [Google Scholar]
- 15. Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr Biol. 1999;9(19):1075-84 [DOI] [PubMed] [Google Scholar]
- 16. Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14(24):3102-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scovassi AI, Stivala LA, Rossi L, Bianchi L, Prosperi E. Nuclear association of cyclin D1 in human fibroblasts: tight binding to nuclear structures and modulation by protein kinase inhibitors. Exp Cell Res. 1997;237(1):127-34 [DOI] [PubMed] [Google Scholar]
- 18. Collecchi P, Santoni T, Gnesi E, et al. Cyclins of phases G1, S and G2/M are overexpressed in aneuploid mammary carcinomas. Cytometry. 2000;42(4):254-60 [DOI] [PubMed] [Google Scholar]
- 19. Sclafani RA. Cyclin dependent kinase activating kinases. Curr Opin Cell Biol. 1996;8:788-94 [DOI] [PubMed] [Google Scholar]
- 20. Lukas J, Pagano M, Staskova Z, Draetta G, Bartek J. Cyclin D1 protein oscillates and is essential for cell cycle progression in human tumour cell lines. Oncogene. 1994;9(3):707-18 [PubMed] [Google Scholar]
- 21. Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7(5):812-21 [DOI] [PubMed] [Google Scholar]
- 22. Sewing A, Bürger C, Brüsselbach S, Schalk C, Lucibello FC, Müller R. Human cyclin D1 encodes a labile nuclear protein whose synthesis is directly induced by growth factors and suppressed by cyclic AMP. J Cell Sci. 1993;104:545-54 [DOI] [PubMed] [Google Scholar]
- 23. Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701-13 [DOI] [PubMed] [Google Scholar]
- 24. Sa G, Hitomi M, Harwalker J, Stacey A, Chen G, Stacey DW. Ras is active throughout the cell cycle, but is able to induce cyclin D1 only during G2 phase. Cell Cycle. 2002;1:50-8 [PubMed] [Google Scholar]
- 25. Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957-72 [DOI] [PubMed] [Google Scholar]
- 26. Guo Y, Harwalkar J, Stacey DW, Hitomi M. Destabilization of cyclin D1 message plays a critical role in cell cycle exit upon mitogen withdrawal. Oncogene. 2005;24(6):1032-42 [DOI] [PubMed] [Google Scholar]
- 27. Guo Y, Stacey DW, Hitomi M. Post-transcriptional regulation of cyclin D1 expression during G2 phase. Oncogene. 2002;21(49):7545-56 [DOI] [PubMed] [Google Scholar]
- 28. Guo Y, Yang K, Harwalkar J, et al. Phosphorylation of cyclin D1 at Thr 286 during S phase leads to its proteasomal degradation and allows efficient DNA synthesis. Oncogene. 2005;24(16):2599-612 [DOI] [PubMed] [Google Scholar]
- 29. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodgett JR, Plyte SE, Pulverer BJ, Mitchell JA, Hughes K. Roles of glycogen synthase kinase-3 in signal transduction. Biochem Soc Transactions. 1993;21:905-7 [DOI] [PubMed] [Google Scholar]
- 31. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785-9 [DOI] [PubMed] [Google Scholar]
- 32. Yang K, Guo Y, Stacey WC, et al. Glycogen synthase kinase 3 has a limited role in cell cycle regulation of cyclin D1 levels. BMC Cell Biol. 2006;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol. 2004;6:648-55 [DOI] [PubMed] [Google Scholar]
- 34. Jirmanova L, Bulavin DV, Fornace AJ. Inhibition of the ATR/Chk1 pathway induces a p38-dependent S-phase delay in mouse embryonic stem cells. Cell Cycle. 2005;4(10):1428-34 [DOI] [PubMed] [Google Scholar]
- 35. Sorensen CS, Syljuasen RG, Lukas J, Bartek J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle. 2004;3(7):941-5 [PubMed] [Google Scholar]
- 36. Hitomi M, Yang K, Stacey AW, Stacey DW. Phosphorylation of cyclin D1 regulated by ATM or ATR controls cell cycle progression. Mol Cell Biol. 2008;28(17):5478-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15(17):2177-96 [DOI] [PubMed] [Google Scholar]
- 38. Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31(7):402-10 [DOI] [PubMed] [Google Scholar]
- 39. Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30(3):290-4 [DOI] [PubMed] [Google Scholar]
- 40. McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5(8):772-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 1996;132(3):413-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24(42):9232-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hitomi M, Stacey DW. The checkpoint kinase ATM protects against stress-induced elevation of cyclin D1 and potential cell death in neurons. Cytometry A. 2010;77(6):524-33 [DOI] [PubMed] [Google Scholar]
- 45. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501-12 [DOI] [PubMed] [Google Scholar]
- 46. Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7(7):892-8 [DOI] [PubMed] [Google Scholar]
- 47. Cheng M, Olivier P, Diehl JA, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18(6):1571-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bagui TK, Mohapatra S, Haura E, Pledger WJ. P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol Cell Biol. 2003;23(20):7285-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sa G, Stacey DW. P27 expression is regulated by separate signaling pathways, downstream of Ras, in each cell cycle phase. Exp Cell Res. 2004;300(2):427-39 [DOI] [PubMed] [Google Scholar]
- 50. Han EK, Sgambato A, Jiang W, et al. Stable overexpression of cyclin D1 in a human mammary epithelial cell line prolongs the S-phase and inhibits growth. Oncogene. 1995;10(5):953-61 [PubMed] [Google Scholar]
- 51. Sa G, Guo Y, Stacey DW. The regulation of S phase initiation by p27Kip1 in NIH3T3 cells. Cell Cycle. 2005;4:618-27 [PubMed] [Google Scholar]
- 52. Zetterberg A, Larsson O, Wiman KG. What is the restriction point? Curr Opin Cell Biol. 1995;7(6):835-42 [DOI] [PubMed] [Google Scholar]
- 53. Hitomi M, Yang K, Guo Y, Fretthold J, Harwalkar J, Stacey DW. p27Kip1 and cyclin dependent kinase 2 regulate passage through the restriction point. Cell Cycle. 2006;5(19):2281-9 [DOI] [PubMed] [Google Scholar]
- 54. Coats S, Flanagan M, Nourse J, Roberts JM. Requirement of p27kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877-80 [DOI] [PubMed] [Google Scholar]
- 55. Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15(2):158-63 [DOI] [PubMed] [Google Scholar]