

Abstract
The discovery of the Src oncogene was the first step on a long journey toward improved cancer chemotherapy. In this review, we explore Src and BCR-ABL, signal transduction, and recent advances in oncogene addiction and celebrate Hidesaboro Hanafusa and the many researchers who ushered in the age of target-directed therapy against tyrosine kinase oncoproteins.
Keywords: Hanafusa, Src, Abl
Introduction
In 1911, Peyton Rous discovered that chickens could transmit sarcomas to one another, thereby implying that tumors could be caused by transmissible agents. Subsequently, he showed that cell-free filtered tumor extracts could transfer sarcomas through the avian sarcoma virus.1 The significance of this finding was hotly contested because of the lack of obvious infectious tumors in humans. Nevertheless, the mystery over this cause of cancer was lifted after several decades when the nonreceptor tyrosine kinase src became the first oncogene identified, thereby ushering in a new paradigm in basic and clinical research.
v-src and c-src
The tumor retrovirus genetics field flourished in the mid-20th century when several groups searched for the component of the Rous sarcoma virus (RSV) responsible for tumor propagation (for detailed reviews of Src, see Martin2,3 and Vogt4). In the 1960s, working in Harry Rubin’s laboratory, Hanafusa discovered that the RSV preparation contained an additional associated virus.5 He purified the RSV-associated virus and was able to uncouple the ability of RSV to infect and transform cells.6 Hanafusa’s ability to segregate defective viruses from replication-competent viruses was a technical tour de force, testimony to his experimental prowess, and one of his most seminal contributions. In the early 1970s, the Duesberg and Martin laboratories used temperature-sensitive mutants of RSV to map the transforming region of the virus, allowing for the identification of v-src.7-9 In 1976, Varmus and Bishop identified these sequences in cells using a complementary library of deletions of Src as probes.10 Around the same time, Hanafusa’s laboratory infected chickens with a transformation-defective virus containing large deletions in the v-src region. The transformation was rescued by a homologous recombination event from within the cell.11 The discovery and characterization of the recovered viruses suggested appropriation of cellular oncogenes through viral recombination events.12 Finally, in the 1980s, viral and cellular src was sequenced, proving that the drivers of cancer could be mutated or overexpressed genes.13,14 Moreover, these findings pointed to the cellular origin for cancer and the coining of the term “proto-oncogene.”
More genes containing similar sequences were cloned, including v-fps and v-crk from Hanafusa’s laboratory.15-17 Tony Pawson and Hanafusa compared the sequences and identified similarities between the oncogenes they cloned that contributed to characterization of the Src homology domains (SH1-SH4).15,18-20 Src was identified to be a tyrosine kinase (SH1) that also contained a region that enables binding to phosphorylated tyrosines (SH2).15,21,22 Another domain binds to proline-rich regions located in other proteins (SH3). Subsequently, 8 more Src family members (Fgr, Blk, Fyn, Lck, Hck, Yrk, Yes, and Lyn) were identified as they share the same structural format with Src, besides their unique domain (SH4).23,24 These and other modular domains became critical in classifying a function to newly identified genes.
CML and ABL
The groundbreaking work on Src and fundamental advances in molecular biology were paralleled by the discovery of another oncogene, v-abl, in the Abelson murine leukemia virus. In 1970, v-abl was found to cause lymphosarcoma of a nonthymic origin in mice.25 This virus model ultimately contributed to an understanding of the oncogenic driver of chronic myelogenous leukemia (CML), a clonal disorder marked by an increase in mature myeloid cells in the blood. This myeloproliferative disorder can progress toward an acute or blast phase, which is characterized by a sudden increase in immature cells in the bone marrow and the blood.
Presaging the discovery of the altered ABL oncogene, in 1960, Nowell and Hungerford identified a recurring chromosomal aberration in patients with CML.26 Over a decade later, Janet Rowley identified this reciprocal chromosomal translocation as t(9;22)(q34.1;q11.23).27 The c-ABL1 gene, the normal cellular counterpart to v-abl, was cloned and identified fused to a gene in a region in which chromosomal break points were tightly clustered, hence thereafter called the break-point cluster region gene, or BCR.28-30 The bcr-abl fusion was found in 95% of patients with CML and in 10% to 30% of patients with acute lymphoblastic leukemia (ALL), a disorder more similar to the leukemia that develops from the Abelson murine leukemia virus26,31 than to CML. Moreover, the BCR-ABL fusion was shown in a mouse model that it could solely drive CML32 or ALL.33 The discovery of the BCR-ABL translocation became the first of many mutations or chromosomal aberrations found in leukemia.34,35
The Structure and Activation of Src and Abl
Besides their parallel history, Src and Abl share a similar structure and function. Src and Abl both have conserved SH3, SH2, and tyrosine kinase domains. Both proteins maintain their tightly regulated kinase activity through similar principles of autoinhibition and autoregulation36 but with slightly differing features. For example, the phosphorylated C-terminal domain of Src self-interacts with its SH2 domain as a “latch” mechanism, constraining an inactive conformation, which is further stabilized by a “clamp” that is mediated by a tight interaction between the SH2 and SH3 domains.37,38 In comparison, ABL can be inactivated by its N-terminal myristoylation cap locking to the C-lobe of the kinase domain.39,40 In contrast, when disrupted by chromosomal translocation, the c-ABL N-terminal region is replaced by BCR in the BCR-ABL fusion, and a coiled-coil domain of BCR mediates oligomerization, thus pushing the kinase toward an activated state.37,41 Besides structural characteristics that modify kinase activity, the C-terminal region of ABL mediates signaling interactions with the cytoskeleton and other signaling adaptors.42-45
Src and BCR-ABL Signaling
Signaling via the Src family kinases and BCR-ABL are closely related; a more detailed description can be found within these reviews.46,47 BCR-ABL and Src family members Hck and Lyn physically interact with each other and play a key role in leukemogenesis.48-50 The SH2, SH3, and C-terminal domain of BCR-ABL can interact with Hck,51 which can in return phosphorylate the SH2, SH3, and Grb2 binding site.49 BCR-ABL can activate Hck, leading to phosphorylation of STAT5.52 When infected with a BCR-ABL retrovirus, mice lacking Lyn, Hck, and Fgr develop a CML-like disease but not a lymphoid disease.48 These and other data implicate the Src family of kinases in progression to blast crisis,53-55 and increased src activity has been observed in a subset of patients resistant to inhibitors.54 A mutation in BCR-ABL within a target residue of the src family kinases can lead to resistance to therapy.46,56
Signaling Complexes
Historically, many groups have focused on the signaling cascades downstream of BCR-ABL and receptor tyrosine kinases to elucidate mechanisms for oncogenic transformation. Adaptor molecules such as Grb2/Gab2/Shc, Dok1/Dok2, and CrkI complexes orchestrate the activation of key signaling nodes.57,58 Hanafusa’s laboratory contributed many critical observations to this enormous compendium of data, such as elucidating the role of v-crk in activating the PI3K/Akt signaling pathway.59,60 CrkI was initially considered dispensable for lymphoid ABL transformation but proved in recent studies to be required for myeloid leukemia.61,62 A complex interplay of adaptors with kinases contributes to BCR-ABL activation of a variety of signaling nodes, including the c-MYC, PI3K/AKT, RAS, and JAK/STAT pathways.63-67 These pathways together drive proliferation and maintain survival.
Molecular Targeted Therapy
The discovery of Src and a host of related tyrosine kinase oncoproteins not only enhanced our understanding of cancer’s origins but also triggered a revolution in cancer therapy. Prior to the 1990s, CML had a poor prognosis, with allogenic stem transplantation as the only available curative therapy.68 Cytotoxic drugs such as hydroxyurea were administrated to control symptoms and leukocytosis but did nothing to change the inexorable progression of leukemia to a fatal blast crisis.69 Although interferon therapy could alter the course of disease and prolong survival for a subset of responders, the majority of patients ultimately succumbed to CML.70
Because of the highly conserved nature of the ATP-binding catalytic pocket of tyrosine kinases, conventional wisdom considered it impossible to develop a specific inhibitor against a given kinase without inducing intolerable toxicity. In the late 1980s, scientists at Ciba-Geigy (now Novartis), in collaboration with Dana Farber, performed a screen for inhibitors against a panel of kinases, developing selective inhibitors of PDGFR that later proved to also inhibit BCR-ABL.71 Being the first molecular lesion consistently associated with and also causative of human CML,32 the BCR-ABL kinase was considered an attractive drug target. The potent ABL inhibitor known as STI-571 was identified and tested in human cell lines and patient cells in Brian Druker’s laboratory72 and shown to induce death in human cancer cell lines.72-74 Serendipitously, this inhibitor also exhibited favorable pharmacokinetic properties and was readily translated to the clinic. Despite the structural similarities between ABL and Src, STI-571 had no effect on tumors driven by v-src.75 The drug, later dubbed imatinib, was subsequently shown to bind tightly within the ATP-binding cleft of ABL, trapping the kinase in an inactive conformation.76 Structural differences between ABL and Src determined that imatinib was specific to the former but not the latter.76 Druker, Charles Sawyers, Moshe Talpaz, and Novartis, which now owned the compound, conducted a phase I study to test its efficacy. The drug showed impressively low toxicity and a remarkable hematological response rate of >90%.77 Over the past decade, imatinib (Gleevec, Novartis, Basel, Switzerland) has evolved into the standard of care, transforming CML from an inevitably fatal malignancy to a manageable chronic condition for most patients.78 Moreover, it has been shown to be effective in treating other malignancies such as gastrointestinal stromal tumors driven by mutated c-kit and other translocations containing PDGF fusions.79,80
Resistance to Targeted Therapy
Even though imatinib has yielded dramatic results, some patients nonetheless develop resistance or convert to blast crisis.78 Investigation of the mechanisms of resistance has revealed mutations within BCR-ABL that reactivate the kinase and abrogate drug binding.81 The reactivation of kinase activity, as opposed to activation of alternative pathways, highlights the essential function of ABL kinase activity as the main driver of disease. Despite the success of imatinib, the many mutations identified in patients compelled the development of next-generation inhibitors that are more potent and active against imatinib-resistant mutations. Nilotinib (Novartis) and Dasatinib (Bristol-Myers Squibb, New York, NY), a dual SRC/ABL inhibitor, have proven their efficacy as second-line therapies in CML.82-84 Of note, dual SRC/ABL inhibitors have also been considered in solid malignancies.85 But one challenge remains—the T315I mutation,86 which places a bulky isoleucine residue in the kinase domain, blocking the access of inhibitors to the active site while also creating conformational changes that activate the kinase itself.86 Inhibitors like ponatinib (AP24534), currently in phase II clinical trials, and HG-7-85-01, currently preclinical, are aimed at targeting the T315I mutation.83,87,88 Moreover, ABL inhibitors that act allosterically, like GNF-2, are being combined with Nilotinib (Novartis) and show activity against T315I.89 In addition to the search for more potent inhibitors that mitigate resistance, alternative strategies are being developed to target the quiescent BCR-ABL stem cell population that appears insensitive to kinase inhibition and thus acts as a reservoir from which relapse can occur.90,91
Oncogene Addiction
Early work on viral oncogenes suggested that a single gene could hijack multiple pathways, leaving the cell dependent upon the oncogene for growth and survival signals. Indeed, another aspect of the legacy of Src biology is the concept of oncogene addiction: cancer cells become “addicted” to signals emanating from a particular oncoprotein, such that abrogation of that pathway via therapeutic targeting could potentially affect only cancer cells while sparing normal cells.92,93
A particularly good validation of the concept of oncogene addiction stems from the remarkable activity of imatinib in the context of CML.93-96 The exquisite sensitivity of leukemia cells to imatinib, and their death in response to drug treatment, suggested that the cells had indeed become entirely dependent on oncogenic signaling and had somehow lost the natural controls on cell proliferation and survival. Given that the BCR-ABL oncoprotein represents a “gain of function,” one might have supposed that drug inhibition would merely revert cells to their normal routine. Instead, in many experimental models, cell death is a consequence of drug treatment. Nevertheless, whereas oncogene addiction might describe the effectiveness of imatinib against rapidly amplifying progenitor pools in CML patients, the more quiescent hematopoietic stem cell pool harboring the BCR-ABL translocation is not eliminated by imatinib, arguing that the stem cell compartment is not suffering addiction, which helps explain the challenge in eradicating leukemic clones.97 In addition to BCR-ABL, oncogene addiction has also been reported in other cancer signaling pathways such as Myc or Ras.98,99 Alternatively, “oncogenic shock” has been invoked to explain tumor cells’ sensitivity upon the loss of oncogenic signals,93 whereby acute removal of the oncogene can lead to rapid apoptosis and cell cycle arrest.93,96 Whether addiction or shock, the effectiveness of target-directed chemotherapy owes to epigenetic changes in the cellular landscape that renders cells dependent upon a given oncogenic signal, thereby allowing for therapeutic targeting and the sparing of normal tissues.
The paradigm of inhibiting kinases remains a dominant focus of today’s therapeutic cancer treatments. To expand the current collection of targets, several groups have surveyed the entire kinome in cancer cell lines.100-103 Interestingly, addiction does not necessarily depend on overexpression or mutation of one particular gene.104 For example, when STK33 is knocked down in K-RAS–dependent tumors, rapid apoptosis occurs specifically in cells addicted to K-RAS but not N-RAS.105 These findings suggest that a specific kinase or a particular checkpoint within a pathway could be required uniquely within the cellular context for survival.
Novel Targets
Besides the activation of kinases, hematopoietic transformation results in massive perturbations of the transcriptional and translational networks. Modifications of myeloid-specific transcription factors such as CCAAT/enhancer binding protein (CEBP) and PU1 are known contributors to leukemia.106,107 Besides these transcription factors, RNA binding proteins can also regulate leukemia progression as shown in a recent study that identified the RNA binding protein MUSASHI-2 (MSI2) to regulate translation in myeloid leukemias. Moreover, MSI2 can be utilized as a diagnostic marker for aggressive AML and CML-BC.108,109 MSI2 blocks the translation of Numb and regulates myeloid differentiation and proliferation. Human myeloid leukemic cells are addicted to high MSI2 levels, based on the results of differentiation and apoptosis in cell lines that have been depleted for MSI2. Admittedly, while transcription factors and RNA binding proteins provide a novel class of therapeutic targets, it is still currently difficult to develop inhibitors against them. Potential strategies include therapeutic inhibitory RNAs and/or small molecules targeting interactions between DNA or RNA and proteins.
Conclusion
It has been a century since avian viruses were first identified in the sarcomas of chickens. Hanafusa was one of the titans of cancer research in that last century, during which he and many others through the study of Src pioneered the field of oncogenic signaling, which ultimately resulted in molecularly targeted cancer therapy (Fig. 1). One of us (G.Q.D.) was privileged to have worked for a brief period as a young medical student in the Hanafusa laboratory and to have witnessed both this great scientist’s command of experimental detail and deep appreciation for the complexities of biology. His legacy will long be respected.
Figure 1.
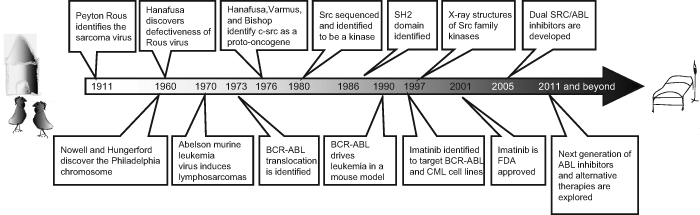
Timeline: the legacy of Hanafusa.
Acknowledgments
The authors thank Harith Rajagopalan, Hao Zhu, and Tingting Zhang for critically reviewing the article.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the US National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Career Development Award (M.G.K.) and grants from the NIH and the Leukemia and Lymphoma Society (G.Q.D.). G.Q.D. is an investigator of the Howard Hughes Medical Institute.
References
- 1. Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med. 1911;13:397-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martin G. The hunting of the src. Nat Rev Mol Cell Biol. 2001;2:467-75 [DOI] [PubMed] [Google Scholar]
- 3. Martin GS. The road to src. Oncogene. 2004;23:7910-7 [DOI] [PubMed] [Google Scholar]
- 4. Vogt PK. Oncogenes and the revolution in cancer research: homage to Hidesaburo Hanafusa (1929-2009). Genes Cancer. 2010;1:6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rubin H, Vogt PK. An avian leukosis virus associated with stocks of Rous sarcoma virus. Virology. 1962;17:184-94 [DOI] [PubMed] [Google Scholar]
- 6. Hanafusa H, Hanafusa T, Rubin H. The defectiveness of Rous sarcoma virus. Proc Natl Acad Sci U S A. 1963;49:572-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang LH, Duesberg PH, Kawai S, Hanafusa H. Location of envelope-specific and sarcoma-specific oligonucleotides on RNA of Schmidt-Ruppin Rous sarcoma virus. Proc Natl Acad Sci U S A. 1976;73:447-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duesberg PH, Vogt PK. Differences between the ribonucleic acids of transforming and nontransforming avian tumor viruses. Proc Natl Acad Sci U S A. 1970;67:1673-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin GS. Rous sarcoma virus: a function required for the maintenance of the transformed state. Nature. 1970;227:1021-3 [DOI] [PubMed] [Google Scholar]
- 10. Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170-3 [DOI] [PubMed] [Google Scholar]
- 11. Hanafusa H, Halpern CC, Buchhagen DL, Kawai S. Recovery of avian sarcoma virus from tumors induced by transformation-defective mutants. J Exp Med. 1977;146:1735-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang LH, Halpern CC, Nadel M, Hanafusa H. Recombination between viral and cellular sequences generates transforming sarcoma virus. Proc Natl Acad Sci U S A. 1978;75:5812-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeya T, Hanafusa H. Structure and sequence of the cellular gene homologous to the RSV src gene and the mechanism for generating the transforming virus. Cell. 1983;32:881-90 [DOI] [PubMed] [Google Scholar]
- 14. Shalloway D, Zelenetz AD, Cooper GM. Molecular cloning and characterization of the chicken gene homologous to the transforming gene of Rous sarcoma virus. Cell. 1981;24:531-41 [DOI] [PubMed] [Google Scholar]
- 15. Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272-5 [DOI] [PubMed] [Google Scholar]
- 16. Shibuya M, Hanafusa H, Balduzzi PC. Cellular sequences related to three new onc genes of avian sarcoma virus (fps, yes, and ros) and their expression in normal and transformed cells. J Virol. 1982;42:143-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Groffen J, Heisterkamp N, Shibuya M, Hanafusa H, Stephenson JR. Transforming genes of avian (v-fps) and mammalian (v-fes) retroviruses correspond to a common cellular locus. Virology. 1983;125:480-6 [DOI] [PubMed] [Google Scholar]
- 18. Foster DA, Levy JB, Daley GQ, Simon MC, Hanafusa H. Isolation of chicken cellular DNA sequences with homology to the region of viral oncogenes that encodes the tyrosine kinase domain. Mol Cell Biol. 1986;6:325-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sadowski I, Stone JC, Pawson T. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus p130gag-fps. Mol Cell Biol. 1986;6:4396-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stone JC, Atkinson T, Smith M, Pawson T. Identification of functional regions in the transforming protein of Fujinami sarcoma virus by in-phase insertion mutagenesis. Cell. 1984;37:549-58 [DOI] [PubMed] [Google Scholar]
- 21. Eckhart W, Hutchinson MA, Hunter T. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell. 1979;18:925-33 [DOI] [PubMed] [Google Scholar]
- 22. Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259:1157-61 [DOI] [PubMed] [Google Scholar]
- 23. Lowell CA, Soriano P. Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 1996;10:1845-57 [DOI] [PubMed] [Google Scholar]
- 24. Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121-49 [DOI] [PubMed] [Google Scholar]
- 25. Abelson HT, Rabstein LS. Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Res. 1970;30:2213-22 [PubMed] [Google Scholar]
- 26. Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497-501 [Google Scholar]
- 27. Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290-3 [DOI] [PubMed] [Google Scholar]
- 28. Shields A, Goff S, Paskind M, Otto G, Baltimore D. Structure of the Abelson murine leukemia virus genome. Cell. 1979;18:955-62 [DOI] [PubMed] [Google Scholar]
- 29. Goff SP, Gilboa E, Witte ON, Baltimore D. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: studies with cloned viral DNA. Cell. 1980;22:777-85 [DOI] [PubMed] [Google Scholar]
- 30. Reddy EP, Smith MJ, Srinivasan A. Nucleotide sequence of Abelson murine leukemia virus genome: structural similarity of its transforming gene product to other onc gene products with tyrosine-specific kinase activity. Proc Natl Acad Sci U S A. 1983;80:3623-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Voncken JW, Kaartinen V, Pattengale PK, Germeraad WT, Groffen J, Heisterkamp N. BCR/ABL P210 and P190 cause distinct leukemia in transgenic mice. Blood. 1995;86:4603-11 [PubMed] [Google Scholar]
- 32. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824-30 [DOI] [PubMed] [Google Scholar]
- 33. Heisterkamp N, Jenster G, Ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251-3 [DOI] [PubMed] [Google Scholar]
- 34. Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179-98 [DOI] [PubMed] [Google Scholar]
- 35. Fröhling S, Döhner H. Chromosomal abnormalities in cancer. N Engl J Med. 2008;359:722-34 [DOI] [PubMed] [Google Scholar]
- 36. Harrison SC. Variation on an src-like theme. Cell. 2003;112:737-40 [DOI] [PubMed] [Google Scholar]
- 37. Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-src. Nature. 1997;385:595-602 [DOI] [PubMed] [Google Scholar]
- 38. Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell. 1993;72:779-90 [DOI] [PubMed] [Google Scholar]
- 39. Wang J. Controlling Abl: auto-inhibition and co-inhibition? Nat Cell Biol. 2004;6:3-7 [DOI] [PubMed] [Google Scholar]
- 40. Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33-44 [DOI] [PubMed] [Google Scholar]
- 41. Zhang X, Subrahmanyam R, Wong R, Gross AW, Ren R. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol Cell Biol. 2001;21:840-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pendergast A. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51-100 [DOI] [PubMed] [Google Scholar]
- 43. Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol. 1994;124:325-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Etten RA, Jackson P, Baltimore D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell. 1989;58:669-78 [DOI] [PubMed] [Google Scholar]
- 45. Mcwhirter JR, Wang JY. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. EMBO J. 1993;12:1533-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li S. Src-family kinases in the development and therapy of Philadelphia chromosome-positive chronic myeloid leukemia and acute lymphoblastic leukemia. Leuk Lymphoma. 2008;49:19-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li S. Src kinase signaling in leukaemia. Int J Biochem Cell Biol. 2007;39:1483-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu Y, Liu Y, Pelletier S, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453-61 [DOI] [PubMed] [Google Scholar]
- 49. Warmuth M, Bergmann M, Priess A, Häuslmann K, Emmerich B, Hallek M. The Src family kinase Hck interacts with Bcr-Abl by a kinase-independent mechanism and phosphorylates the Grb2-binding site of Bcr. J Biol Chem. 1997;272:33260-70 [DOI] [PubMed] [Google Scholar]
- 50. Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56:3589-96 [PubMed] [Google Scholar]
- 51. Lionberger JM, Wilson MB, Smithgall TE. Transformation of myeloid leukemia cells to cytokine independence by Bcr-Abl is suppressed by kinase-defective Hck. J Biol Chem. 2000;275:18581-5 [DOI] [PubMed] [Google Scholar]
- 52. Klejman A, Schreiner SJ, Nieborowska-Skorska M, et al. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002;21:5766-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat Med. 2004;10:1187-9 [DOI] [PubMed] [Google Scholar]
- 54. Donato NJ, Wu JY, Stapley J, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690-8 [DOI] [PubMed] [Google Scholar]
- 55. Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J Biol Chem. 2004;279:34227-39 [DOI] [PubMed] [Google Scholar]
- 56. Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831-43 [DOI] [PubMed] [Google Scholar]
- 57. Titz B, Low T, Komisopoulou E, Chen SS, Rubbi L, Graeber TG. The proximal signaling network of the BCR-ABL1 oncogene shows a modular organization. Oncogene. 2010;29:5895-910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pawson T. Dynamic control of signaling by modular adaptor proteins. Curr Opin Cell Biol. 2007;19:112-6 [DOI] [PubMed] [Google Scholar]
- 59. Akagi T, Murata K, Shishido T, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway by utilizing focal adhesion kinase and H-Ras. Mol Cell Biol. 2002;22:7015-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Akagi T, Shishido T, Murata K, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci U S A. 2000;97:7290-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seo J-H, Wood LJ, Agarwal A, et al. A specific need for CRKL in p210BCR-ABL-induced transformation of mouse hematopoietic progenitors. Cancer Res. 2010;70:7325-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hemmeryckx B, Reichert A, Watanabe M, et al. BCR/ABL P190 transgenic mice develop leukemia in the absence of Crkl. Oncogene. 2002;21:3225-31 [DOI] [PubMed] [Google Scholar]
- 63. Goga A, Mclaughlin J, Afar DE, Saffran DC, Witte ON. Alternative signals to RAS for hematopoietic transformation by the BCR-ABL oncogene. Cell. 1995;82:981-8 [DOI] [PubMed] [Google Scholar]
- 64. Hoover RR, Gerlach MJ, Koh EY, Daley GQ. Cooperative and redundant effects of STAT5 and Ras signaling in BCR/ABL transformed hematopoietic cells. Oncogene. 2001;20:5826-35 [DOI] [PubMed] [Google Scholar]
- 65. Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kharas MG. Abl oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005;65:2047-53 [DOI] [PubMed] [Google Scholar]
- 67. Sonoyama J, Matsumura I, Ezoe S, et al. Functional cooperation among Ras, STAT5, and phosphatidylinositol 3-kinase is required for full oncogenic activities of BCR/ABL in K562 cells. J Biol Chem. 2002;277:8076-82 [DOI] [PubMed] [Google Scholar]
- 68. Geary CG. The story of chronic myeloid leukaemia. Br J Haematol. 2000;110:2-11 [DOI] [PubMed] [Google Scholar]
- 69. Wintrobe MM, Huguley CM. Nitrogen mustard as a therapeutic agent for Hodgkin’s disease, lymphosarcoma and leukemia. Ann Intern Med. 1947;27:529-40 [DOI] [PubMed] [Google Scholar]
- 70. Talpaz M, McCredie KB, Mavligit GM, Gutterman JU. Leukocyte interferon-induced myeloid cytoreduction in chronic myelogenous leukemia. Blood. 1983;62:689-92 [PubMed] [Google Scholar]
- 71. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808-17 [DOI] [PubMed] [Google Scholar]
- 72. Druker BJ. Perspectives on the development of imatinib and the future of cancer research. Nat Med. 2009;15:1149-52 [DOI] [PubMed] [Google Scholar]
- 73. Gambacorti-Passerini C, Le Coutre P, Mologni L, et al. Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood Cells Mol Dis. 1997;23:380-94 [DOI] [PubMed] [Google Scholar]
- 74. Deininger MW, Goldman JM, Lydon N, Melo JV. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood. 1997;90:3691-8 [PubMed] [Google Scholar]
- 75. Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561-6 [DOI] [PubMed] [Google Scholar]
- 76. Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938-42 [DOI] [PubMed] [Google Scholar]
- 77. Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-17 [DOI] [PubMed] [Google Scholar]
- 78. Hochhaus A, O’Brien SG, Guilhot F, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23:1054-61 [DOI] [PubMed] [Google Scholar]
- 79. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-32 [DOI] [PubMed] [Google Scholar]
- 80. Cools J, Deangelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201-14 [DOI] [PubMed] [Google Scholar]
- 81. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876-80 [DOI] [PubMed] [Google Scholar]
- 82. Kantarjian HM, Giles FJ, Bhalla KN, et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase after imatinib resistance or intolerance: 24-month follow-up results. Blood. 2011;117:1141-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. O’Hare T, Deininger MW, Eide CA, Clackson T, Druker BJ. Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin Cancer Res. 2011;17:212-21 [DOI] [PubMed] [Google Scholar]
- 84. O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500-5 [DOI] [PubMed] [Google Scholar]
- 85. Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010;16:3526-32 [DOI] [PubMed] [Google Scholar]
- 86. Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Weisberg E, Choi HG, Ray A, et al. Discovery of a small-molecule type II inhibitor of wild-type and gatekeeper mutants of BCR-ABL, PDGFRalpha, Kit, and Src kinases: novel type II inhibitor of gatekeeper mutants. Blood. 2010;115:4206-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang J, Adrián FJ, Jahnke W, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Testa U. Leukemia stem cells. Ann Hematol. 2011;90:245-71 [DOI] [PubMed] [Google Scholar]
- 91. Helgason GV, Young GAR, Holyoake TL. Targeting chronic myeloid leukemia stem cells. Curr Hematol Malig Rep. 2010;5:81-7 [DOI] [PubMed] [Google Scholar]
- 92. Weinstein IB. Cancer. Addiction to oncogenes: the Achilles heal of cancer. Science. 2002;297:63-4 [DOI] [PubMed] [Google Scholar]
- 93. Sharma SV. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12:4392S-5S [DOI] [PubMed] [Google Scholar]
- 94. Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009;15:1158-61 [DOI] [PubMed] [Google Scholar]
- 95. Cooper S, Giles FJ, Savona MR. Overcoming resistance in chronic myelogenous leukemia. Leuk Lymphoma. 2009;50:1785-93 [DOI] [PubMed] [Google Scholar]
- 96. Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214-31 [DOI] [PubMed] [Google Scholar]
- 97. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396-409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. D’Cruz CM, Gunther EJ, Boxer RB, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235-9 [DOI] [PubMed] [Google Scholar]
- 99. Jonkers J, Berns A. Oncogene addiction: sometimes a temporary slavery. Cancer Cell. 2004;6:535-8 [DOI] [PubMed] [Google Scholar]
- 100. Schlabach MR, Luo J, Solimini NL, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319:620-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Luo B, Cheung HW, Subramanian A, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105:20380-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986-8 [DOI] [PubMed] [Google Scholar]
- 105. Scholl C, Fröhling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821-34 [DOI] [PubMed] [Google Scholar]
- 106. Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007;7:105-17 [DOI] [PubMed] [Google Scholar]
- 107. Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89-101 [DOI] [PubMed] [Google Scholar]
- 108. Ito T, Kwon HY, Zimdahl B, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010;466:765-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kharas MG, Lengner CJ, Al-Shahrour F, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16:903-8 [DOI] [PMC free article] [PubMed] [Google Scholar]