

Abstract
The C-28 methyl ester of the oleane triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO-Me) induces apoptosis of human cancer cells by disrupting redox balance and is in clinical trials. CDDO-Me contains α,β-unsaturated carbonyl groups that form reversible adducts with thiol nucleophiles. The present studies show that CDDO-Me blocks interleukin-6 (IL-6)–induced and constitutive activation of the Janus-activated kinase 1 (JAK1) in cells. In support of a direct mechanism, CDDO-Me forms adducts with JAK1 at Cys1077 in the kinase domain and inhibits JAK1 activity. In concert with these results, CDDO-Me blocked IL-6–induced and constitutive activation of signal transducer and activator of transcription 3 (STAT3). Moreover, we show that CDDO-Me (a) binds directly to STAT3 by a mechanism dependent on the alkylation of Cys259 and (b) inhibits the formation of STAT3 dimers. These findings indicate that CDDO-Me inhibits activation of the JAK1→STAT3 pathway by forming adducts with both JAK1 and STAT3.
Introduction
The synthetic oleanane triterpenoids are a new class of agents that have antiproliferative and proapoptotic activity (1). One of the synthetic oleanane triterpenoids, C-28 methyl ester of 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO-Me), is under study in phase I and II clinical trials for the treatment of patients with hematologic malignancies and solid tumors. In this regard, CDDO-Me and other derivatives with modifications at the C-28 position induce apoptosis of human myeloid leukemia (2–6), multiple myeloma (7–9), osteosarcoma (10), lung cancer (11, 12), breast cancer (13, 14), and pancreatic cancer (15). The synthetic oleanane triterpenoids activate the phase II response of cells at low nanomolar concentrations and thereby protect against oxidative stress by decreasing levels of reactive oxygen species (1). By contrast, at low micromolar concentrations, CDDO-Me and related derivatives induce apoptosis in vitro by increasing reactive oxygen species and decreasing intracellular glutathione (5, 7, 15, 16). How the synthetic oleanane triterpenoids disrupt redox balance and induce apoptosis at low micromolar concentrations is not known. However, structure-activity analysis has shown that α,β-unsaturated carbonyl groups on rings A and C confer Michael addition with a nucleophilic target (17, 18). In this context, nuclear factor κB activates the transcription of diverse genes that regulate cell survival (19), and CDDO directly inhibits the IκB kinase β and thereby the nuclear factor-κB pathway by interacting with Cys179 in the IκB kinase β activation loop (20, 21). These findings have indicated that CDDO-Me induces apoptosis, in part, by alkylating critical cysteines in proteins that regulate survival.
Members of the signal transducer and activator of transcription (STAT) family of transcription factors have, like nuclear factor κB, been implicated in transformation, tumor cell survival, invasion, and metastasis (22). Activation of STAT3, in particular, has been detected in diverse hematologic malignancies and solid tumors (22–24) and induces transformation (25). STAT3 is also constitutively activated in immune cells in the tumor microenvironment (26). STAT3 is activated by Janus-activated kinase (JAK)-1 phosphorylation of the interleukin-6 (IL-6) receptor, recruitment of STAT3, and, in turn, phosphorylation of STAT3 (27). STAT3 then forms dimers that translocate to the nucleus and activate STAT3 target genes, which encode regulators of cell cycle progression (cyclin D1 and c-Myc) and inhibitors of apoptosis (survivin and Bcl-xL; refs. 28, 29). The pattern of STAT3-induced gene expression is regulated by diverse signals that converge on JAK1 and STAT3 (27), indicating that both proteins are targets for inhibiting this pathway. Importantly, small-molecule inhibitors of the STAT3 pathway are effective as anticancer agents in vitro and in animal models (30, 31). Other studies have shown that the triterpenoids decrease pSTAT3 levels (9, 32, 33); however, the mechanistic basis for this response is not known.
The present results show that CDDO-Me directly inhibits IL-6–induced and constitutive JAK1 activity. The results also show that CDDO-Me directly blocks STAT3 signaling and the induction of growth and survival genes.
Materials and Methods
Cell culture
Human HeLa cervical cancer and MDA-MB-468 breast cancer cells were cultured in DMEM containing 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine. Cells were treated with CDDO-Me (provided by Reata Pharmaceuticals) and human IL-6 (20 ng/mL; R&D Systems).
Subcellular fractionation
Nuclear fractions were prepared as described (34).
Immunoblot analysis
Lysates were analyzed by immunoblotting with antibodies against phospho-JAK1, phospho-JAK2 (Millipore), JAK1, JAK2, STAT3, IκBα, cyclin D1, survivin (Santa Cruz Biotechnology), phospho-STAT3 (Cell Signaling Technology), STAT3 monoclonal antibody (BD Transduction Laboratory), lamin B (Calbiochem), and β-actin (Sigma). The immune complexes were detected with horseradish peroxidase–conjugated second antibodies and enhanced chemiluminescence (Amersham Biosciences).
JAK1 kinase assays
Soluble proteins were incubated with anti-JAK1 or anti–green fluorescent protein (GFP; Abcam) and precipitated with protein A/G beads. The immune complexes were incubated in kinase buffer [20 mmol/L Tris-HCl (pH 7.5), 10 mmol/L MgCl2, and 100 μmol/L ATP] with 1 μg of recombinant glutathione S-transferase (GST)-STAT3 and [γ-32P]ATP (Perkin-Elmer Life Sciences), as described (35), in the absence and presence of 1 μmol/L CDDO-Me for 30 min at 30°C. The reaction products were analyzed by SDS-PAGE and autoradiography.
Binding of CDDO-Me-biotin to JAK1 and STAT3
CDDO-Me and CDDO were biotinylated as described (36). Lysates were incubated with 1 μmol/L biotin (Calbiochem) or CDDO-Me-biotin for 2 h, followed by addition of streptavidin-Sepharose beads (GE Healthcare) for 1 h. Alternatively, intact cells were pretreated with CDDO-Me for 2 h and then incubated with 1 μmol/L CDDO-Me-biotin. Complexes were isolated with streptavidin-Sepharose beads. The beads were washed and then subjected to immunoblotting with anti-JAK1 or anti-STAT3. In other studies, recombinant GST-JAK1 kinase domain (KD; amino acids 870–1,152), GST-STAT3 or GST-JAK1-KD(C1077A), and GST-STAT3(C259A), generated by site-directed mutagenesis, were cleaved with thrombin to remove the GST moiety. The purified JAK1-KD and STAT3 proteins were incubated with 1 μmol/L biotin, CDDO-Me-biotin, or CDDO-biotin for 30 min. Complexes were isolated with streptavidin-Sepharose beads and analyzed by immunoblotting with anti-JAK1 and anti-STAT3.
Luciferase assays
Cells were transfected with pSTAT3-Luc (Panomics) and SV40-Renilla-Luc (Promega) in the presence of Lipofectamine 2000 (Invitrogen). At 24 h after transfection, lysates were analyzed with the Dual Luciferase Assay Kit (Promega).
Analysis of STAT3 dimerization
Cell lysates were incubated with anti-STAT3 (Santa Cruz Biotechnology) and precipitated with protein A/G beads. The precipitates were immunoblotted with anti-STAT3 (BD Transduction Laboratories). In other studies, MDA-MB-468 cells were cotransfected with pCMV-STAT3-Flag and pCMV-STAT3-HA (30) in the presence of Lipofectamine 2000 (Invitrogen). At 48 h after transfection, lysates were incubated with anti-hemagglutinin (HA; Sigma) and the precipitates analyzed by immunoblotting with anti-Flag (Sigma).
Results
CDDO-Me inhibits JAK1 activity
To determine whether CDDO-Me affects JAK signaling, we first studied HeLa cells that were stimulated with IL-6. Phospho-JAK1 levels were low to undetectable in control HeLa cells and were increased with IL-6 stimulation (Fig. 1A). Notably, treatment of the HeLa cells with CDDO-Me suppressed IL-6–induced JAK1 activation (Fig. 1A). IL-6 stimulation was also associated with increases in phospho-JAK2 levels (Fig. 1B). However, in contrast to JAK1, CDDO-Me treatment had no apparent effect on JAK2 activation (Fig. 1B). There was no detectable activation of JAK3 in the IL-6–stimulated HeLa cells (data not shown). In contrast to HeLa cells, JAK1 is constitutively activated in human MDA-MB-468 cells (Fig. 1C). Treatment of MDA-MB-468 cells with CDDO-Me was associated with down-regulation of phospho-JAK1 levels (Fig. 1C). Moreover, the effect of CDDO-Me on phospho-JAK1 was detectable within 2 hours of treatment (Fig. 1D). These findings indicate that CDDO-Me inhibits JAK1, but not JAK2, activation.
Figure 1.
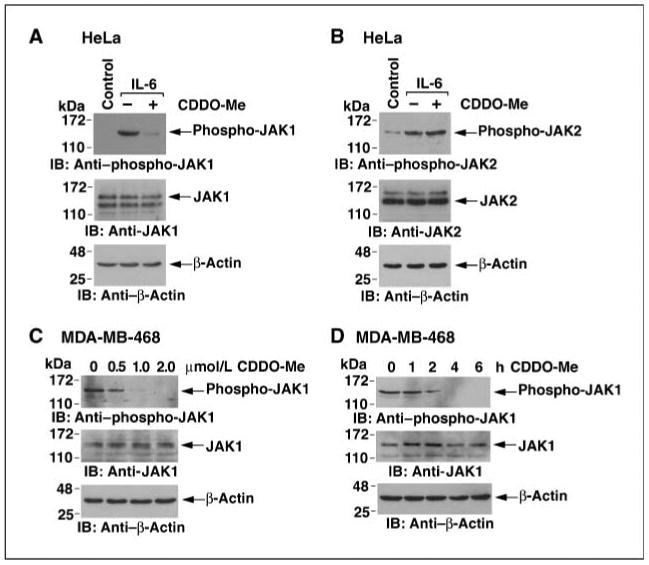
CDDO-Me inhibits IL-6–induced and constitutive JAK1 activity. A and B, HeLa cells were pretreated with 1 μmol/L CDDO-Me for 6 h and then left unstimulated (Control) or stimulated with IL-6 for 15 min. Whole-cell lysates were subjected to immunoblotting with the indicated antibodies. C, MDA-MB-468 cells were treated with the indicated concentrations of CDDO-Me for 6 h. Lysates were immunoblotted with the indicated antibodies. D, MDA-MB-468 cells were treated with 1 μmol/L CDDO-Me for the indicated times. Lysates were immunoblotted with the indicated antibodies.
CDDO-Me interacts directly with JAK1
To further assess the effects of CDDO-Me, we immunoprecipitated JAK1 from control and IL-6–stimulated HeLa cells and performed in vitro kinase assays with GST-STAT3 as substrate. IL-6 stimulation was associated with increases in JAK1-mediated phosphorylation of STAT3 (Fig. 2A, left). However, addition of CDDO-Me to the reactions blocked STAT3 phosphorylation, indicating that CDDO-Me directly blocks JAK1 activity (Fig. 2A, left). In vitro kinase assays with constitutively activated JAK1 immunoprecipitated from MDA-MB-468 breast cancer cells also showed that addition of CDDO-Me to the reactions blocks JAK1-mediated phosphorylation of STAT3 (Fig. 2A, right). To confirm these findings, we expressed GFP-tagged JAK1 in MDA-MB-468 cells. Analysis of anti-GFP precipitates showed that phosphorylation of GST-STAT3 is inhibited by CDDO-Me (Fig. 2B). To determine whether CDDO-Me interacts with JAK1, lysates from HeLa cells were incubated with CDDO-Me conjugated to biotin. Analysis of the adsorbates by immunoblotting with anti-JAK1 showed binding to CDDO-Me and not biotin (Fig. 2C, left). By contrast, there was no detectable binding of CDDO-Me to JAK2 (Fig. 2C, left). Incubation of intact MDA-MB-468 cells showed binding of JAK1 to CDDO-Me-biotin and not biotin (Fig. 2C, right). In addition, pretreatment of intact cells with CDDO-Me competitively blocked binding of JAK1 to CDDO-Me-biotin (Fig. 2C, right). JAK1 contains a cysteine residue at position 1077 in the kinase domain that is not present in JAK2. Binding of CDDO-Me-biotin was detectable with purified recombinant JAK1-KD (Fig. 2D). By contrast, the formation of adducts was substantially decreased when recombinant JAK1-KD mutated at Cys1077 was incubated with CDDO-Me-biotin (Fig. 2D). These findings indicate that CDDO-Me interacts directly with JAK1 on Cys1077 and inhibits JAK1 activity.
Figure 2.
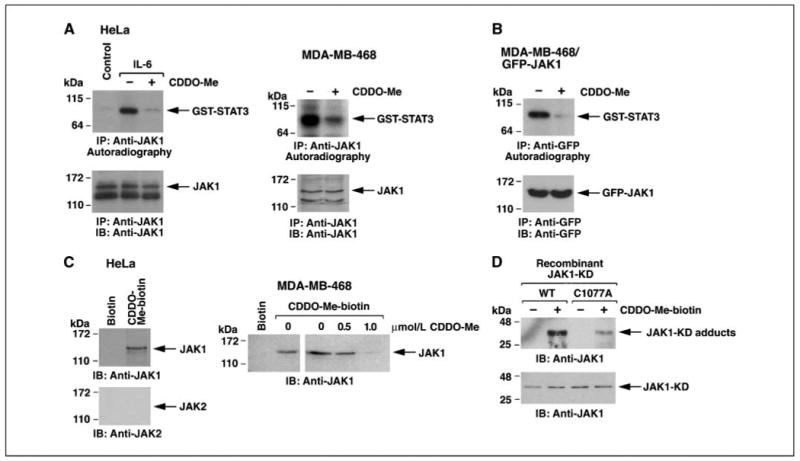
CDDO-Me directly inhibits JAK1 activity. A, HeLa cells were left untreated as the control or stimulated with IL-6 for 15 min (left). MDA-MD-468 cells were analyzed without stimulation (right). Anti-JAK1 precipitates were incubated with GST-STAT3 and [γ-32P]ATP in the absence and presence of 1 μmol/L CDDO-Me. The reaction products were analyzed by SDS-PAGE and autoradiography (top). The precipitates were also analyzed by immunoblotting with anti-JAK1 (bottom). B, MDA-MB-468 cells were transfected with GFP-JAK1. At 48 h after transfection, lysates were precipitated with anti-GFP. The precipitates were incubated with GST-STAT3 and [γ-32P]ATP in the absence and presence of 1 μmol/L CDDO-Me. The reaction products were analyzed by SDS-PAGE and autoradiography (top). The precipitates were also analyzed by immunoblotting with anti-GFP (bottom). C, lysates from HeLa cells were incubated with biotin or CDDO-Me-biotin and then precipitated with streptavidin-Sepharose beads. The adsorbates were immunoblotted with anti-JAK1 and anti-JAK2 (left). Intact MDA-MB-468 cells were pretreated with the indicated concentrations of CDDO-Me for 2 h and then incubated with 1 μmol/L CDDO-Me-biotin. Lysates were precipitated with streptavidin-Sepharose beads and the adsorbates were immunoblotted with anti-JAK1 (right). D, recombinant JAK1-KD or JAK1-KD(C1077A) was incubated with 1 μmol/L CDDO-Me-biotin for 30 min. The complexes were isolated with streptavidin-Sepharose beads and the precipitates were subjected to immunoblotting with anti-JAK1 (top). Input of the proteins was determined by immunoblotting with anti-JAK1 (bottom).
CDDO-Me inhibits the IL-6–induced STAT3 pathway
JAK1 phosphorylates and thereby activates STAT3 for targeting to the nucleus (22, 27). In this context, IL-6 stimulation of HeLa cells was associated with increases in phospho-STAT3 levels (Fig. 3A). Treatment of the HeLa cells with CDDO-Me blocked IL-6–induced phospho-STAT3 (Fig. 3A). In addition, CDDO-Me blocked IL-6–induced targeting of STAT3 to the nucleus (Fig. 3B). Transfection of HeLa cells with a pSTAT3-luciferase (Luc) reporter showed that IL-6 stimulates STAT3-mediated transcription and that CDDO-Me inhibits this response (Fig. 3C). STAT3 activates transcription of the cyclin D1 and survivin genes. Consistent with inhibition of STAT3-mediated transcription, IL-6–induced increases in cyclin D and survivin expression were inhibited by CDDO-Me treatment (Fig. 3D). These results and our findings with JAK1 indicate that CDDO-Me blocks IL-6–induced activation of the JAK1→STAT3 pathway.
Figure 3.
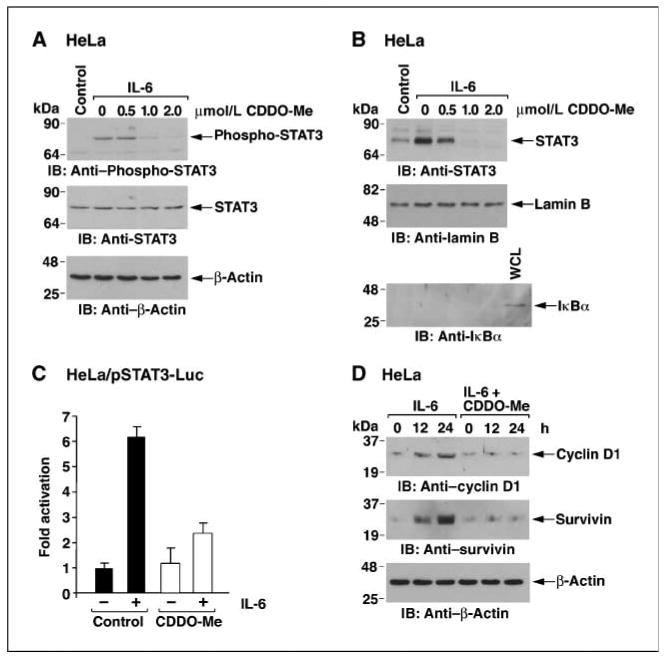
CDDO-Me inhibits IL-6–induced STAT3 activation. A and B, HeLa cells were pretreated with the indicated concentrations of CDDO-Me for 6 h and then stimulated with IL-6 for 15 min. Whole-cell lysates were immunoblotted with the indicated antibodies (A). Nuclear lysates were immunoblotted with anti-STAT3 and, as controls for equal loading and purity, with antibodies against nuclear lamin B and cytosolic IκBα (B). WCL, whole-cell lysate. C, HeLa cells were transfected with pSTAT3-Luc and SV40-Renilla-Luc. At 24 h after transfection, cells were pretreated with 1 μmol/L CDDO-Me for 6 h, stimulated with IL-6 for 15 min, and then assayed for luciferase activity. Columns, mean fold activation relative to that obtained for the control (assigned a value of 1) from three separate experiments; bars, SD. D, HeLa cells were treated with 1 μmol/L CDDO-Me for 2 h and then IL-6 was added for the indicated times. Lysates were immunoblotted with the indicated antibodies.
CDDO-Me blocks constitutive activation of the JAK1→STAT3 pathway
The JAK1→STAT3 pathway is constitutively activated in MDA-MB-468 breast cancer cells (37). Consistent with the results obtained in IL-6–stimulated HeLa cells, treatment of MDA-MB-468 cells with CDDO-Me down-regulated constitutive phosphorylation of STAT3 (Fig. 4A, left). Analysis of the kinetics of this response showed decreases in phospho-STAT3 levels within 2 hours of CDDO-Me treatment (Fig. 4A, right). Furthermore, CDDO-Me inhibited the constitutive nuclear localization of STAT3 in MDA-MB-468 cells (Fig. 4B). Constitutive activation of the pSTAT3-Luc reporter was inhibited by CDDO-Me (Fig. 4C). In addition, CDDO-Me treatment was associated with down-regulation of cyclin D1 and survivin expression (Fig. 4D). These results indicate that CDDO-Me blocks constitutive activation of the JAK1→STAT3 pathway in MDA-MB-468 cells.
Figure 4.
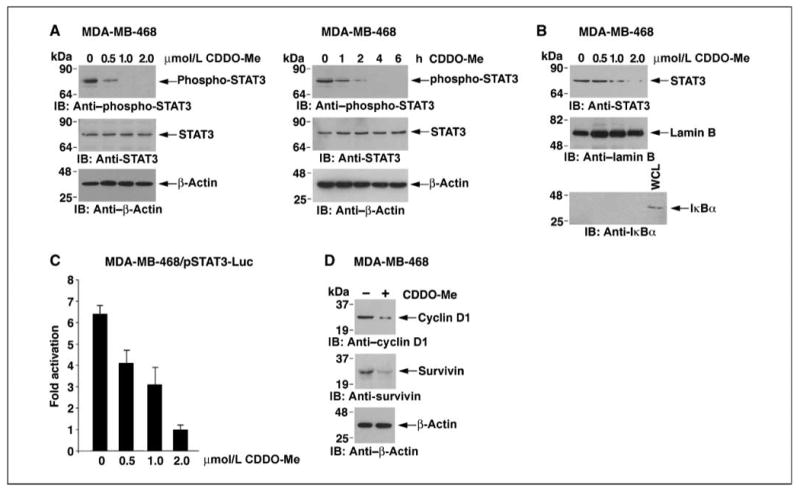
CDDO-Me inhibits constitutive activation of the STAT3 pathway. A and B, MDA-MB-468 cells were treated with the indicated concentrations of CDDO-Me for 6 h or 1 μmol/L CDDO-Me for the indicated times. Whole-cell lysates were immunoblotted with the indicated antibodies (A). Nuclear lysates were immunoblotted with anti-STAT3 and, as controls for equal loading and purity, with antibodies against nuclear lamin B and cytosolic IκBα (B). C, MDA-MB-468 cells were transfected with pSTAT3-Luc and SV40-Renilla-Luc. At 24 h after transfection, cells were treated with the indicated concentrations of CDDO-Me for 6 h and then assayed for luciferase activity. Columns, mean fold activation relative to that obtained for cells treated with 2 μmol/L CDDO-Me (assigned a value of 1) from three separate experiments; bars, SD. D, MDA-MB-468 cells were left untreated or treated with 1 μmol/L CDDO-Me for 6 h. Lysates were immunoblotted with the indicated antibodies.
CDDO-Me blocks STAT3 dimerization
JAK1-mediated phosphorylation of STAT3 is associated with the targeting of phospho-STAT3 dimers to the nucleus for activation of STAT3-dependent genes. Thus, IL-6 stimulation of HeLa cells results in the formation of STAT3 dimers (Fig. 5A). To determine if CDDO-Me affects STAT3 dimerization, lysates from IL-6–stimulated HeLa cells were incubated with CDDO-Me. The results show that CDDO-Me disrupts the STAT3 dimers (Fig. 5A). Constitutive STAT3 dimerization in MDA-MB-468 cells was also disrupted by CDDO-Me (Fig. 5B). To confirm these results, we transfected MDA-MB-468 cells with HA-tagged STAT3 (HA-STAT3) and Flag-tagged STAT3 (Flag-STAT3). Immunoblot analysis of anti-HA precipitates with anti-Flag showed the formation of Flag-STAT3-HA-STAT3 dimers (Fig. 5C). Moreover, incubation of the anti-HA precipitates with CDDO-Me resulted in disruption of the Flag-STAT-HA-STAT3 dimers (Fig. 5C). These results indicate that CDDO-Me blocks STAT3 dimerization.
Figure 5.

CDDO-Me blocks dimerization of STAT3. A and B, HeLa cells were stimulated with IL-6 for 15 min (A). MDA-MB-468 cells were untreated (B). Lysates were immunoprecipitated with anti-STAT3 (polyclonal, anti–COOH-terminal). The precipitates were incubated with 0 or 1 μmol/L CDDO-Me for 2 h, washed, suspended in nonreducing sample buffer, boiled for 5 min, and immunoblotted with anti-STAT3 (monoclonal, anti–NH2-terminal). C, MDA-MB-468 cells were transfected with the empty pCMV vector or cotransfected with Flag-STAT3 and HA-STAT3. At 48 h after transfection, lysates were precipitated with anti-HA. The precipitates were incubated with 0 or 1 μmol/L CDDO-Me for 2 h and then immunoblotted with the indicated antibodies.
CDDO-Me forms adducts with STAT3 that are dependent on Cys259
Incubation of HeLa cell lysates with CDDO-Me-biotin showed that CDDO-Me forms complexes with STAT3 (Fig. 6A, left). Incubation of intact MDA-MB-468 cells showed binding of STAT3 to CDDO-Me-biotin and not biotin (Fig. 6A, right). In addition, pretreatment of intact MDA-MB-468 cells with different concentrations of CDDO-Me showed inhibition of the binding of CDDO-Me-biotin to STAT3 (Fig. 6A, right). We also incubated recombinant STAT3 with CDDO-Me-biotin. Analysis of the adsorbates with anti-STAT3 showed that CDDO-Me interacts directly with STAT3 (Fig. 6B). The parent compound CDDO similarly formed direct complexes with recombinant STAT3 (Fig. 6B). By contrast, the interaction was blocked when recombinant STAT3(C259A) was incubated with CDDO-Me-biotin (Fig. 6C). These findings indicate that CDDO-Me inhibits STAT3 by direct alkylation of Cys259.
Figure 6.
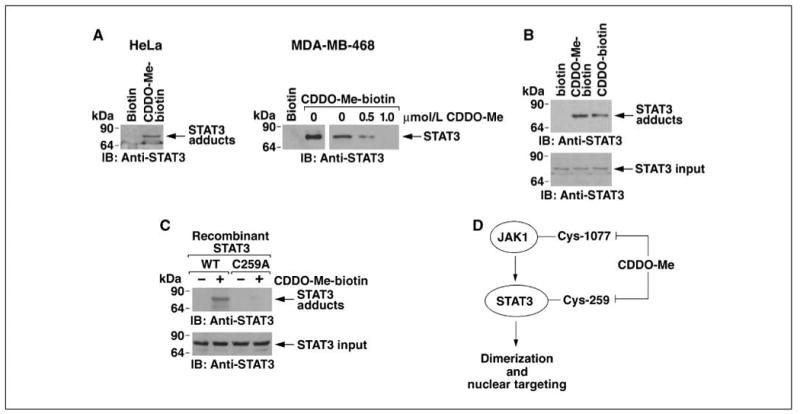
CDDO-Me binds directly to STAT3 at Cys259. A, HeLa cell lysates were incubated with 1 μmol/L of biotin or CDDO-Me-biotin for 2 h. Streptavidin-Sepharose beads were then added for 1 h. The precipitates were immunoblotted with anti-STAT3 (left). Intact MDA-MB-468 cells were pretreated with the indicated concentrations of CDDO-Me for 2 h and then incubated with 1 μmol/L CDDO-Me-biotin. Lysates were precipitated with streptavidin-Sepharose beads and the adsorbates were immunoblotted with anti-STAT3 (right). B and C, recombinant purified STAT3 was incubated with 1 μmol/L biotin, CDDO-Me-biotin, or CDDO-biotin for 30 min (B). Recombinant STAT3 or STAT3(C259A) was incubated with 1 μmol/L biotin or CDDO-Me-biotin for 30 min (C). Complexes were isolated with streptavidin beads and the precipitates were immunoblotted with anti-STAT3 (top). Input of the STAT3 proteins was assessed by immunoblotting with anti-STAT3 (bottom). D, schema depicting CDDO-Me inhibition of the JAK1→STAT3 pathway.
Discussion
The synthetic oleanane triterpenoids disrupt redox balance and thereby induce apoptosis at low micromolar concentrations (5, 7, 15, 16). The mechanism(s) responsible for such activity remain unclear. However, certain insights are being obtained from the findings that the synthetic oleanane triterpenoids can confer Michael addition with nucleophilic targets (17, 18). In this regard, a direct interaction of a synthetic oleanane triterpenoid was found spectroscopically with thiol groups of the Keap1 sensor (18). Subsequent studies have shown that CDDO and its C-28 derivatives interact directly with IκB kinase β at Cys179 in the activation loop (20, 21). Otherwise, to our knowledge, there are no reports of CDDO or its derivatives forming adducts with other protein targets. The cyclopentenone prostaglandins also contain an α,β-unsaturated carbonyl moiety and react with the IκB kinase β Cys179 residue (38). However, few targets of the cyclopentenone prostaglandins have been identified to date, even when using an unbiased proteomic approach (39). Nonetheless, the findings that the cyclopentenone prostaglandins react with H-Ras (40), heat shock protein 90 (39), and estrogen receptor α (41) provide potential leads for additional targets of the synthetic oleanane triterpenoids.
Constitutive activation of STAT3 contributes to tumorigenesis by promoting proliferation and inhibiting apoptosis (42). Tyrosine phosphorylation of STAT3 confers dimerization through binding of the SH2 domain of one monomer to a P-pY-LKTK motif on the other STAT3 monomer (31, 43–45). The present results show that CDDO-Me inhibits IL-6–induced and constitutive JAK1 activity. The results support a model in which CDDO-Me binds directly to JAK1 at Cys1077 in the kinase domain and thereby blocks JAK1 as an upstream effector of STAT3 phosphorylation (Fig. 6D). The triterpenoids have been shown to decrease pSTAT3 levels (9, 32, 33) and the present findings show that this response is associated with the direct inhibition of JAK1.
Dimerization of STAT3 is necessary for its activation and oncogenic function (25). STAT3 target genes confer diverse processes of transformation, including proliferation, apoptosis, angiogenesis, and metastasis (46). In this regard, agents that inhibit STAT3 dimerization and activation exhibit antitumor activity (30, 31). Previous studies have shown that STAT3 dimerization is also mediated by interchain disulfide bridging involving Cys259 (37). Notably, the present results show that CDDO-Me disrupts STAT3 dimers. Moreover, we show that CDDO-Me binds directly to STAT3 by forming adducts with Cys259 (Fig. 6D). Thus, CDDO-Me–mediated disruption of STAT3 dimerization is consistent with alkylation of Cys259 and thereby loss of interchain bridging. CDDO-Me treatment was also associated with down-regulation of cyclin D1 and survivin, both encoded by STAT3 target genes. Notably, the inhibition of JAK1 would be sufficient to decrease STAT3 phosphorylation and dimerization. However, the results also show that CDDO-Me directly inhibits STAT3. These findings thus collectively indicate that CDDO-Me blocks the JAK1→STAT3 pathway by directly inhibiting both JAK1 and STAT3 (Fig. 6D).
Acknowledgments
Grant support: National Cancer Institute grants CA42802, CA100707, and CA98628.
We thank Dr. Jiayuh Lin (Ohio State University) for providing the Flag-STAT3 and HA-STAT3 vectors, Dr. Robert Schreiber (Washington University) for the GFP-JAK1 vector, Dr. Nancy Reich (Stony Brook University) for the GST-STAT3 vector, and Dr. Ratna Ray (St. Louis University) for the Myc-JAK1 vector. D. Raina is a consultant to Reata.
References
- 1.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–69. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 2.Ito Y, Pandey P, Place A, et al. The novel triterpenoid CDDO induces apoptosis of human myeloid leukemia cells by a caspase-8 dependent mechanism. Cell Growth Differ. 2000;11:261–7. [PubMed] [Google Scholar]
- 3.Konopleva M, Tsao T, Ruvolo P, et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99:326–35. doi: 10.1182/blood.v99.1.326. [DOI] [PubMed] [Google Scholar]
- 4.Stadheim TA, Suh N, Ganju N, Sporn MB, Eastman A. The novel triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) potently enhances apoptosis induced by tumor necrosis factor in human leukemia cells. J Biol Chem. 2002;277:16448–55. doi: 10.1074/jbc.M108974200. [DOI] [PubMed] [Google Scholar]
- 5.Ikeda T, Kimura F, Nakata Y, et al. Triterpenoid CDDO-Im down-regulates PML/RARa expression in acute promyelocytic leukemia cells. Cell Death Differ. 2005;12:523–31. doi: 10.1038/sj.cdd.4401574. [DOI] [PubMed] [Google Scholar]
- 6.Konopleva M, Tsao T, Estrov Z, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64:7927–35. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda T, Nakata Y, Kimura F, et al. Induction of redox imbalance and apoptosis in multiple myeloma cells by the novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid. Mol Cancer Ther. 2004;3:39–45. [PubMed] [Google Scholar]
- 8.Chauhan D, Li G, Podar K, et al. The bortezomib/proteasome inhibitor PS-341 and triterpenoid CDDO-Im induce synergistic anti-multiple myeloma (MM) activity and overcome bortezomib resistance. Blood. 2004;103:3158–66. doi: 10.1182/blood-2003-08-2873. [DOI] [PubMed] [Google Scholar]
- 9.Liby K, Voong N, Williams CR, et al. The synthetic triterpenoid CDDO-imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin Cancer Res. 2006;12:4288–93. doi: 10.1158/1078-0432.CCR-06-0215. [DOI] [PubMed] [Google Scholar]
- 10.Ito Y, Pandey P, Sporn M, et al. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol. 2001;59:1094–9. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 11.Kim K, Lotan R, Yue P, et al. Identification of a novel synthetic triterpenoid, methyl-2-cyano-3, 12-dioxooleana-1,9-dien-28-oate, that potently induces caspase-mediated apoptosis in human lung cancer cells. Mol Cancer Ther. 2002;1:177–84. [PubMed] [Google Scholar]
- 12.Zou W, Liu X, Yue P, et al. c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1,9-dien-28-oate in human lung cancer cells. Cancer Res. 2004;64:7570–8. doi: 10.1158/0008-5472.CAN-04-1238. [DOI] [PubMed] [Google Scholar]
- 13.Lapillonne H, Konopleva M, Tsao T, et al. Activation of peroxisome proliferator-activated receptor γ by a novel synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res. 2003;63:5926–39. [PubMed] [Google Scholar]
- 14.Hyer ML, Croxton R, Krajewska M, et al. Synthetic triterpenoids cooperate with tumor necrosis factor-related apoptosis-inducing ligand to induce apoptosis of breast cancer cells. Cancer Res. 2005;65:4799–808. doi: 10.1158/0008-5472.CAN-04-3319. [DOI] [PubMed] [Google Scholar]
- 15.Samudio I, Konopleva M, Hail N, Jr, et al. 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J Biol Chem. 2005;280:36273–82. doi: 10.1074/jbc.M507518200. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda T, Sporn M, Honda T, Gribble G, Kufe D. The novel triterpenoid CDDO induces apoptosis by disruption of intracellular redox balance. Cancer Res. 2003;63:5551–8. [PubMed] [Google Scholar]
- 17.Couch RD, Browning RG, Honda T, et al. Studies on the reactivity of CDDO, a promising new chemopreventive and chemotherapeutic agent: implications for a molecular mechanism of action. Bioorg Med Chem Lett. 2005;15:2215–9. doi: 10.1016/j.bmcl.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 18.Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci U S A. 2005;102:4584–9. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-κB pathway by direct inhibition of IKKβ on Cys-179. J Biol Chem. 2006;281:35764–9. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 21.Yore MM, Liby KT, Honda T, Gribble GW, Sporn MB. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-κB activation through direct inhibition of IκB kinase β. Mol Cancer Ther. 2006;5:3232–9. doi: 10.1158/1535-7163.MCT-06-0444. [DOI] [PubMed] [Google Scholar]
- 22.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 23.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 24.Dechow TN, Pedranzini L, Leitch A, et al. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc Natl Acad Sci U S A. 2004;101:10602–7. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 27.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez JV, Febbo PG, Ramaswamy S, et al. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–62. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 29.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–8. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 30.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–6. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liby K, Royce DB, Williams CR, et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007;67:2414–9. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 33.Ling X, Konopleva M, Zeng Z, et al. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1,9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67:4210–8. doi: 10.1158/0008-5472.CAN-06-3629. [DOI] [PubMed] [Google Scholar]
- 34.Kharbanda S, Saleem A, Yuan ZM, et al. Nuclear signaling induced by ionizing radiation involves colocalization of the activated p56/p53lyn tyrosine kinase with p34cdc2. Cancer Res. 1996;56:3617–21. [PubMed] [Google Scholar]
- 35.Kamakura S, Oishi K, Yoshimatsu T, et al. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6:547–54. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- 36.Honda T, Janosik T, Honda Y, et al. Design, synthesis, and biological evaluation of biotin conjugates of 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid for the isolation of the protein targets. J Med Chem. 2004;47:4923–32. doi: 10.1021/jm049727e. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Shaw PE. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem Biophys Res Commun. 2004;322:1005–11. doi: 10.1016/j.bbrc.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 38.Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–8. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 39.Gharbi S, Garzon B, Gayarre J, Timms J, Perez-Sala D. Study of protein targets for covalent modification by the antitumoral and anti-inflammatory prostaglandin PGA1: focus on vimentin. J Mass Spectrom. 2007;42:1474–84. doi: 10.1002/jms.1291. [DOI] [PubMed] [Google Scholar]
- 40.Renedo M, Gayarre J, Garcia-Dominguez CA, et al. Modification and activation of Ras proteins by electrophilic prostanoids with different structure are site-selective. Biochemistry. 2007;46:6607–16. doi: 10.1021/bi602389p. [DOI] [PubMed] [Google Scholar]
- 41.Kim HJ, Kim JY, Meng Z, et al. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits transcriptional activity of estrogen receptor-α via covalent modification of DNA-binding domain. Cancer Res. 2007;67:2595–02. doi: 10.1158/0008-5472.CAN-06-3043. [DOI] [PubMed] [Google Scholar]
- 42.Yu H, Jove R. The STATs of cancer-new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 43.Turkson J, Ryan D, Kim JS, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276:45443–55. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 44.Turkson J, Kim JS, Zhang S, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3:261–9. [PubMed] [Google Scholar]
- 45.Kretzschmar AK, Dinger MC, Henze C, Brocke-Heidrich K, Horn F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J. 2004;377:289–97. doi: 10.1042/BJ20030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao SP, Bromberg JF. Touched and moved by STAT3. Sci STKE. 2006;2006:pe30. doi: 10.1126/stke.3432006pe30. [DOI] [PubMed] [Google Scholar]