

Abstract
As members of the family of heme-dependent enzymes, the heme dioxygenases are differentiated by virtue of their ability to catalyze the oxidation of l-tryptophan to N-formylkynurenine, the first and rate-limiting step in tryptophan catabolism. In the past several years, there have been a number of important developments that have meant that established proposals for the reaction mechanism in the heme dioxygenases have required reassessment. This focused review presents a summary of these recent advances, written from a structural and mechanistic perspective. It attempts to present answers to some of the long-standing questions, to highlight as yet unresolved issues, and to explore the similarities and differences of other well-known catalytic heme enzymes such as the cytochromes P450, NO synthase, and peroxidases.
The heme dioxygenase enzymes catalyze the first and rate-limiting step in the kynurenine pathway, the O2-dependent oxidation of l-tryptophan to N-formylkynurenine. This reaction is unique in heme chemistry. Somewhat confusingly, this enzyme in the early literature is termed “tryptophan pyrrolase”1−3 (and even “tryptophan peroxidase-oxidase”(4)); only later did the nomenclature converge on either tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO). Actually, both TDO and IDO catalyze the same reaction (Scheme 1), with the different denominations merely reflecting the generally wider substrate specificity of the IDOs compared to the more substrate-specific TDOs.
Scheme 1. Reaction Catalyzed by the Heme Dioxygenases.

The first report of a heme dioxygenase enzyme came more than 70 years ago,(1) but the vast majority of the spectroscopic and kinetic studies were conducted in the 1970s and 1980s. In 1996, an informative summary of this early dioxygenase literature was published(5) as part of a larger review of heme-containing oxygenases. At that time, recombinant expression systems for dioxygenases were not widely available and there was no published structural information. In the past 10 years, there have been substantial developments, including high-resolution structural information, new bacterial expression systems, important contributions from computational chemistry, and emerging mechanistic data from site-directed mutagenesis, all of which has moved the field forward and has meant that the original mechanistic proposals have required revision. This review summarizes these recent contributions. It is not an exhaustive account; instead, this article focuses specifically on structural and mechanistic aspects and attempts to present a concise, comparative summary that is relevant to those with a broader interest in heme protein structure and function.
Background
Prior to the publication of structural information (or the development of recombinant expression systems), there had been a considerable effort directed toward understanding the mechanism and reactivity of heme dioxygenases. This has been reviewed previously,(5) and we do not re-rehearse it here. It is, though, instructive to briefly summarize the main conclusions that had been drawn from these studies, because they provide context and a useful framework for the discussion that follows.
A number of different indicators had led to the convincing and well-cited idea that the dioxygenase active site contained a distal histidine residue that was catalytically important. To start with, the reaction mechanism was known to involve O2 binding to the heme: this was highly reminiscent of O2 binding to the heme-containing globins in which hydrogen bonding interactions between bound O2 and a distal histidine were well documented(6) (the peroxidase enzymes also employ similar hydrogen bonding interactions7−10). Reassuringly, there was also sequence homology with a group of IDO-like myoglobins that were also presumed to contain a distal histidine (reviewed in ref (11)). Further evidence of a distal histidine residue arose from spectroscopic data (reviewed in ref (5)) that had revealed the presence of a low-spin heme species under certain conditions. Many of these spectroscopic features (which were later observed for the recombinant proteins, too12,13) were similar to those found for bis-nitrogenous heme coordination, and it was thus concluded that the dioxygenase distal pocket contained a histidine. Perhaps most compelling of all were studies with enzyme inhibitors,(14) from which it had been concluded that the reaction mechanism was critically dependent on the presence of an active site base, presumed to be histidine, which was responsible for abstraction of the proton on the indole nitrogen in the first step of the mechanism (Scheme 2).
Scheme 2. Previous Literature Proposals5,14,15 for the Reaction Mechanism for Heme Dioxygenases.

Reaction of ferrous heme with O2 and Trp leads to a ternary complex that reacts by base-catalysed abstraction to form a peroxyindole intermediate. This species was proposed to decay by either Criegee (blue) or dioxetane (red) pathways.
On the whole, proposals for the later stages of the mechanism were less well formulated, mainly as a consequence of the difficulty of isolating reaction intermediates. The reaction had been proposed to occur through either Criegee(15) or dioxetane(16) pathways (Scheme 2).
It against is this background of information that the following discussion is based.
First Glimpse of the Dioxygenase Active Site: The Crystal Structure of Human IDO
The dioxygenase enzymes have proved to be reluctant participants in structural biology efforts, but the emergence of expression systems for different recombinant dioxygenases has meant that suitably high concentrations of protein can now be generated in some cases. Table 1 gives a summary of bacterial expression systems for various dioxygenases, and in a few cases, structures have been published.17−19 The first of these was that of recombinant human IDO,(17) which was crystallized with the inhibitor 4-phenylimidazole bound to the heme iron. The structure revealed two distinct domains (large and small) (Figure 1A) connected by a small loop of 17 residues.a
Table 1. Summary of Expression Systems for a Number of Dioxygenase Enzymes.
Figure 1.

(A) Overall structure of human IDO,(17) showing the large (red) and small (blue) domains, the loop region (yellow), the heme (in green), the proximal histidine residue (His346, red), and the bound inhibitor, 4-phenylimidazole (green). (B) Active site of human IDO(17) with the heme colored red. There is one molecule of the inhibitor 4-phenylimidazole (green) bound to the heme iron in the structure and two molecules of the crystallization buffer (CHES, not shown) in the active site. Figures 1 and 2 were prepared using Pymol.(66)
The active site (Figure 1B) confirmed some of the earlier predictions but also contained some unexpected surprises. The structure revealed the presence of a proximal histidine, which was in agreement with early predictions about the heme ligation from spectroscopy(5) and with more recent mutagenesis work in which His346 had been predicted(20) to be the ligand. Asp274 on the proximal side had been suggested from mutagenesis(20) either to bind directly to the heme, which the structure shows not to be the case, or to maintain a suitable conformation of the heme pocket. This latter hypothesis proved to be correct because there is an electrostatic interaction between this residue and Arg343 that is presumably important for maintaining the overall structure (Figure 1B).
Overall, the IDO molecule contains a large number of hydrophobic Phe and Tyr aromatic residues (not shown), including those in the distal heme pocket (Figure 1B). This was to be expected to accommodate binding of the rather hydrophobic tryptophan substrate. Most surprisingly, and in direct conflict with literature proposals about the mechanism,(5) there was no histidine residue in the distal pocket.b At least for IDO, therefore, the low-spin (presumed bis-nitrogenous(5)) species observed spectroscopically12,13,21 cannot arise from coordination of a distal histidine to the heme. Spectroscopic artifacts (often at cryogenic temperatures) relating to heme coordination are well-known in other catalytic heme enzymes, and the earlier interpretations for TDO may well also have been complicated by the fact that the heme groups of the tetramer have now been shown to be inequivalent.(22) In fact, the entire IDO active site is almost completely devoid of polar residues: Ser167 is the only candidate (Figure 1B), but this residue was later shown by mutagenesis(23) not to be essential for O2 or substrate binding or turnover. This absence of a distal histidine presented a further obvious difficulty: because the reaction mechanism was presumed to involve base-catalyzed abstraction of the indole proton (Schemes 2 and 3A), the absence of the necessary base meant that this clearly now needed reassessment. As an alternative, it was suggested(17) that abstraction of protons by the bound O2 was more likely [a suggestion first put forward by Terentis et al.(13) (Scheme 3B)]. These suggestions are developed further below.
Scheme 3. Variously Proposed Reaction Mechanisms for Heme Dioxygenases.

(A) The base-catalysed abstraction mechanism.14,15 (B) An alternative to the base-catalysed mechanism, using abstraction of protons by the bound O2.13,17 (C) Electrophilic addition.32,33,42 (D) Radical addition.32,34,38,42 The majority opinion from crystallography,(17) mass spectrometry,(23) mutagenesis,(24) and computational work(32) concludes that mechanisms A and B are unlikely. It is not yet known whether addition at C3 or C2 is most likely; both have been suggested.32−34,38,42,65.
Substrate Binding Interactions: The Crystal Structure of Bacterial TDOs
The crystal structure of the bacterial X. campestris TDO to a large extent confirmed the generally hydrophobic nature of the dioxygenase active site and revealed the high degree of structural similarity between the TDO and IDO active sites (Figure 2A). Structures for the R. metallidurans TDO(19) and S. oneidensis IDO(18) enzymes have also been published and show similar features in the active site (but substrate is not bound in these structures).
Figure 2.

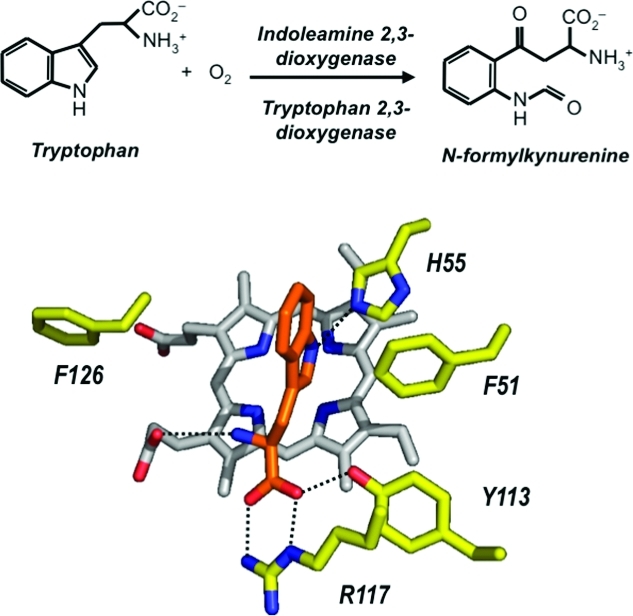
(A) Overlay of X. campestris TDO (green(18)) and hIDO (white(17)), showing the active site residues (in parentheses for hIDO). (B) Substrate binding site in X. campestris TDO,(18) showing the substrate (orange) and the associated bonding interactions.
The most significant structural information was that for the substrate-bound complex of the X. campestris enzyme (Figure 2B). This revealed, for the first time, the binding interactions holding the substrate inside the pocket. Most provocatively, in this case there is a distal histidine residue (His55) that overlays with the equivalent (Ser167) residue in human IDO and forms hydrogen bonds to the indole proton on N1! Although, in principle, this seemed to imply that the base-catalyzed abstraction mechanism may be possible (Scheme 2), site-directed mutagenesis data(24) do not support such a role because the X. campestris H55A variant is still catalytically active, albeit at a lower level (kcat that is ∼10% of the wild-type value).c In cases where His is present in the active site [for X. campestris and R. metallidurans TDO, for instance, and also assumed for hTDO (His76)], its full role is not yet established, although it might be involved in holding the substrate in a very precise orientation suitable for cleavage of the C2−C3 bond. This would fit with spectroscopic work(26) that shows that substrate binding “locks” the bound O2 into a single conformation (presumably suitably oriented for C2−C3 bond cleavage). In this context, we note that the closely related heme- and O2-dependent PrnB enzyme binds its substrate differently:(27) in this case, the C2−N1 bond of 7-chloro-l-Trp is cleaved (without insertion of O2), and crystallography shows l-typtophan binding in an orientation different from that of X. campestris TDO (Figure 3).
Figure 3.
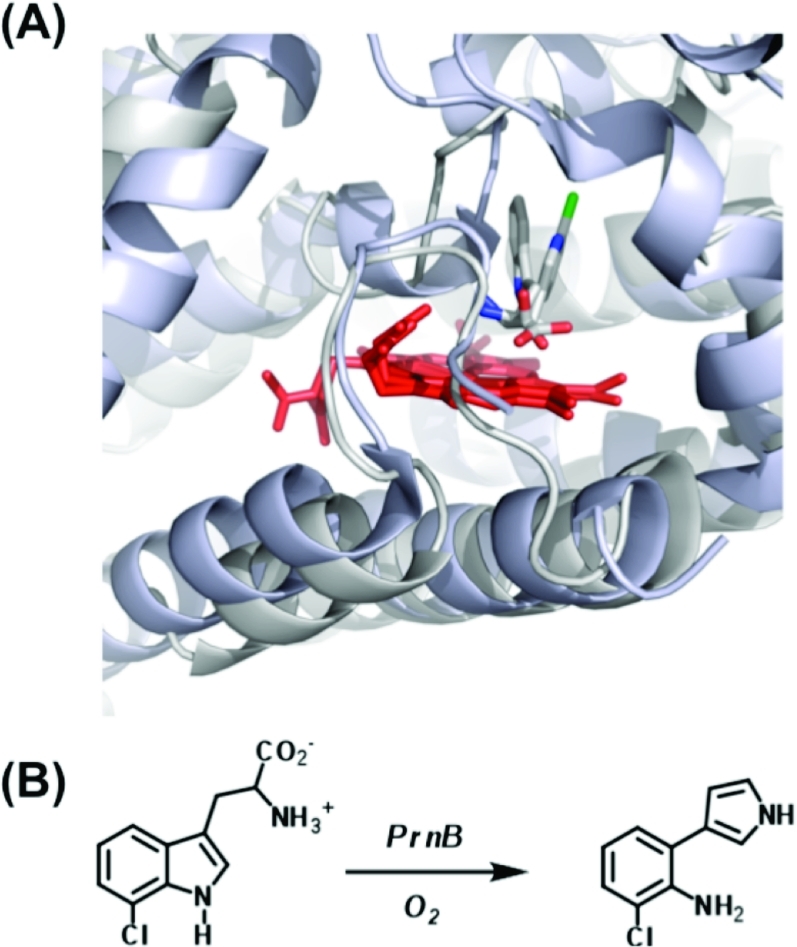
Overlay of the heme regions of X. campestris TDO (gray(18)) and PrnB (pale blue(27)), showing the different orientations of the substrate (l-Trp for TDO and 7-Cl-Trp for PrnB, both colored gray, with the Cl atom colored green). The heme groups are overlaid in red. (B) Reaction catalyzed by PrnB (compare to Scheme 1).
In X. campestris TDO, there are also ionic interactions with Arg117, also presumably present in hIDO (to Arg231) and R. metallidurans TDO (Arg134), and with the heme propionate (Figure 2B). These ionic interactions would explain why substrate binding is sensitive to substitutions at these charged positions [e.g., d-Trp, tryptamine, indole priopionic acid, and tryptophanol5,18,19,25,28−31 (Scheme 4)] and in some cases [e.g., tryptamine, indole propionic acid, and tryptophanol (Scheme 4)] essentially eliminates activity altogether.(28)
Scheme 4. Structures of the Various Tryptophan Analogues Mentioned in This Work.

Mechanism
As we highlighted in the introductory section, the briefest glance at the literature would lead one to the conclusion that there was little left to learn about the mechanism of tryptophan oxidation. The mechanism presented in Scheme 2 was first suggested more than 40 years ago(15) and has been widely reproduced. It is a curiosity of the dioxygenase literature that these proposals became deeply entrenched despite the fact that there was no experimental evidence for either of them.
One feature of the proposed mechanism (Scheme 2) that immediately stands out is that only a single initial reduction of the heme is required for turnover and that there is no further change in oxidation state of the heme iron during turnover.d If correct, this quite obviously differentiates the heme dioxygenases from other O2-dependent heme enzymes (e.g., P450s and NO synthases) in which a second reduction of the iron and protonation leads to rapid formation of an oxidized ferryl heme (Compound I), which then needs further re-reduction from a suitable reductase. This is discussed further below.
First Step: Base-Catalyzed Abstraction or Not?
The first step of the mechanism had been proposed as a base-catalyzed abstraction of the indole proton. The suggestion was first put forward by Hamilton(15) and was based on the idea that only substrates containing a proton on the indole nitrogen were active. Others(14) drew similar conclusions on the basis of the observation that 1-methyltryptophan (Scheme 4) was an inhibitor of dioxygenase activity. Because base-catalyzed abstraction is not possible for a 1-methylated substrate, the presence of an active site histidine (as the base) was assumed (Scheme 3A). As we explain above, the absence of an active site base in hIDO meant that this mechanism needed revision, and the mechanism as shown in Scheme 3B was proposed.13,17 In fact, there are chemical difficulties with both mechanisms drawn in panels A and B of Scheme 3, because the electrons in the N−H (σ) bond are perpendicular to the π-orbitals of the aromatic ring and so could not be used to directly activate the aromatic ring for electrophilic attack on oxygen.
Computational work from Chung and co-workers(32) raised the first questions about the likelihood of a base-catalyzed abstraction mechanism, and electrophilic addition was suggested as one of a number of alternative mechanisms. To date, the main experimental evidence comes from mass spectrometry work(33) that has shown that 1-Me-Trp is actually a slow substrate for several dioxygenase enzymes, thus ruling out both mechanisms in panels A and B of Scheme 3, because neither is possible with the methyl-substituted indole. This would be consistent with mutagenesis data that show that when a distal histidine is present its removal does not shut down activity completely.(24) Spectroscopic work using 1H ENDOR(26) has since shown that the indole NH group is not hydrogen bonded to the bound O2 in the ternary complex, further arguing against the mechanisms shown in panels A and B of Scheme 3. Others(34) have drawn similar conclusions from computational work, and there now seems to be a consensus that base-catalyzed abstraction does not occur.
In fact, the chemistry of indoles has a long and well-documented history, and they do not react by base-catalyzed abstraction.(35) This then raises the question of what happens instead. There are two alternative mechanisms, both of which would accommodate the reactivity of 1-Me-Trp(33) and would avoid the conceptual difficulty of needing to deprotonate an indole NH group that has a theoretical pKa that is very high (pKa ≈ 17(36)). In the first case, the lone pair on nitrogen initiates electrophilic addition to the bound O2 ligand(33) (Scheme 3C). This route was identified computationally,(32) and electrophilic addition would be consistent with the known chemistry of indoles; however, O2 is typically not a very good electrophile, and certainly in the case of the globins, the ferrous−oxy bond is best formulated as an FeIII−O2− species.(37) An alternative is radical addition (P. Ortiz de Montellano, personal communication) (Scheme 3D), again identified as a contender by computational work.33,34,38 This too would allow for reactivity of 1-Me-Trp and is appealing in the sense that it would require an FeIII−O2− formulation for the ferrous−oxy heme. It is not yet clear which mechanism, if either, is used.
Second Step: Criegee or Dioxetane?
The Criegee mechanism is well-known in the non-heme iron literature (see, for example, ref (39)) but there is no experimental evidence of either a Criegee(15) or a dioxetane(16) mechanism in the heme dioxygenases. As Scheme 2 makes clear, neither mechanism requires a formal change in oxidation state during turnover (which is unusual for heme-mediated catalysis). However, there is more than one report implicating formation of a ferryl heme species during dioxygenase turnover. The first came from Yeh and co-workers,(38) who used resonance Raman methods to observe a stretching frequency characteristic of ferryl heme (νFe=O), assigned as Compound II, during turnover in hIDO (but, curiously, not in hTDO). A similar stretching frequency has been independently observed in IDO.40,41 This has led to the suggestion(38) that simultaneous incorporation of both atoms of O2, as dictated by Scheme 2, does not occur but that instead sequential (stepwise) insertion of oxygen into the substrate and accompanying oxidation to ferryl heme is the preferred route, as depicted in Scheme 5A. Recent computational work(42) supports epoxide formation as the first oxygenation step (although pathways involving highly valent iron ferryl heme had also been suggested by previous work(32)).
Scheme 5. Alternative Mechanistic Proposal.

(A) Radical addition (instead of base-catalysed abstraction) leading to species X is followed by formation of a ferryl heme species (Compound II) and a proposed epoxide species.(38) Formation of an epoxide has also been suggested by computational work(42) (in fact, it was considered in an earlier study, but initially considered energetically unfavorable in the gas phase(32)). Electrophilic addition (Scheme 3C) could also involve epoxide formation through a similar (two-electron) mechanism. Possible ring opening of the epoxide is also indicated. (B) Proposed(42) mechanism for subsequent formation of NFK, starting from the same species X as in panel A, and also involving initial ring opening.
If correct, there are features of the mechanism shown in Scheme 5A that align with known behavior in related enzymes. Epoxide formation is, of course, well established in the P450 literature, but through direct reaction of a Compound I intermediate with the substrate. We note also that isomerization of endoperoxides by the heme enzymes prostacyclin synthase and thromboxane synthase has also been suggested to occur by homolytic cleavage of the bound endoperoxide O−O bond and formation of ferryl heme.(43) There is also an analogy with the non-heme iron enzymes (e.g., tyrosine hydroxylase(44)) in which formation of an FeIV=O species, from ferrous iron and O2, also occurs.
There is still no experimental evidence relating to the final part of the mechanism, conversion of the proposed intermediates into NFK. Computational work has, so far, provided the only information,32,42 and the most recent conclusions(42) do not support either Criegee or dioxetane mechanisms. From a chemical perspective, we note that the epoxide intermediate would be expected to undergo facile C2−O bond cleavage (because of the adjacent nitrogen lone pair), which may mean that the first step in the C2−C3 bond cleavage process involves ring opening as we show in Scheme 5A. Indeed, there is precedent for such epoxide ring opening from studies on the chemical epoxidation of tryptophan by peracids.(45) This would agree with recent computational analyses(42) that also support ring opening of the proposed epoxide intermediate, possibly assisted by the protonated amine group of the substrate (which, from spectroscopic data,(26) is thought to be within hydrogen bonding distance of the bound distal O2) (Scheme 5B). Subsequent steps in the detailed mechanism of conversion of the epoxide to NFK have been proposed(42) (Scheme 5B), but these ideas have yet to be verified experimentally. There are interesting parallels to be drawn with other systems, most notably in the PrnB enzyme mentioned above27,46 and in indole alkaloid biosynthesis,(47) because in both cases the involvement of the substrate amine group in the reaction mechanism is likewise implicated. These systems may provide important clues as the details of the dioxygenase mechanism are unravelled.
Mechanistic Similarities and Differences with Other Heme Enzymes
Oxygen activation by heme enzymes can be achieved in various ways but often occurs via formation of highly oxidized iron intermediates (Compound I and Compound II). The most well-known examples are in the cytochrome P450s, nitric oxide synthases, cytochrome c oxidases, and heme peroxidases.7,46,48−51 Typically, the reductive activation of O2 begins from the ferrous oxy intermediate and is followed by a further reduction and a protonation, to form a ferric−hydroperoxy species (which is also used in the heme oxygenase reaction mechanism52,53). Rapid heterolytic cleavage of the O−O bond leads to the “activating” Compound I species, which is also accessed via a direct reaction with hydrogen peroxide (e.g., in the peroxidase enzymes) (Scheme 6). This cycling through a high-valent Compound I species during turnover of course demands continuous reduction of the heme group, and thus the need for an appropriate reductase partner. The physiological partner reductase for the dioxygenases is not known with certainty, but for IDO, there is evidence of it being a cytochrome b5.54,55 Regardless of the source of the electrons, the evidence so far is that the dioxygenases require only a single, initiating reduction of ferric heme; further reduction and protonation of the ferrous−oxy heme would presumably not be a productive pathway for a dioxygenase, because it leads to the formation of Compound I. In fact, there is evidence from ENDOR work(26) that substrate binding shields the ferrous−oxy complex from protonation in dioxygenases, which may be one of the strategies that the dioxygenases employ to control the reactivity of the ferrous−oxy species and unproductive mechanistic pathways.
Scheme 6. Comparison of the Reaction Mechanisms in Various Heme Proteins.

Future Directions
In this review, we have focused on structural and mechanistic information. We know much more about what makes a heme dioxygenase than we did even five years ago. Literature mechanisms have slowly unravelled as new information has emerged, and new proposals are being put forward that have themselves yet to stand the test of time. Many questions and challenges remain though. As we noted at the beginning, the absence of any information about the reaction intermediates has been a long-standing difficulty that has hindered progress and the precise mechanism has yet to be established. Further work on identification of these reaction intermediates, both for the transient heme species and indeed for the substrate as it turns over to form NFK, will surely be essential in this overall quest and will require contributions from spectroscopists, enzymologists, structural biologists, and theoreticians alike. Together, this should provide the necessary fuel for further informative debate.
Acknowledgments
E.L.R. thanks Professors Larry Que, Paul Ortiz de Montellano, Masao Ikeda Saito, and Jim Naismith for helpful discussions about mechanism.
Glossary
Abbreviations
- IDO
indoleamine 2,3-dioxygenase
- TDO
tryptophan 2,3-dioxygenase
- hIDO
human indoleamine 2,3-dioxygenase
- hTDO
human tryptophan 2,3-dioxygenase
- NFK
N-formylkynurenine.
a A small anomaly of the structure was that the enzyme crystallized as a cross-linked dimer linked by a disulfide bridge (Cys308 in each monomer), even though in solution there is no evidence of such a cross-link.
b His303 in IDO had been considered as a possible candidate,12,13 but this proved to be incorrect because the crystal structure revealed that this residue was not in the active site.
c The interpretation is not completely unambiguous however, and others interpreted similar data differently.(25) In hTDO, the residue corresponding to His55 in X. campestris is thought to be His76; mutation of His76 also reduces activity and by a similar amount (kcat that is ∼10% of the wild-type value) compared to the H55 variants of X. campestris, and this has been interpreted as evidence of an essential role for His76 in TDO. There is no crystal structure yet for hTDO, to confirm the location of His76.
d There are two independent reports28,29 that the ferric form of hTDO (but not IDO) is also catalytically active, but the mechanism of this reaction has not been established.
This work was supported by The Wellcome Trust (Project Grant 083636 to E.L.R. and to S.K.C./C.G.M. and Equipment Grant WT087777MA to E.L.R.).
References
- Kotake Y.; Masayama I. (1936) The intermediary metabolism of tryptophan. XVIII. The mechanism of formation of kynurenine from tryptophan. Z. Physiol. Chem. 243, 237–244. [Google Scholar]
- Yamamoto S.; Hayaishi O. (1967) Tryptophan pyrrolase of rabbit intestine. d- and l-tryptophan-cleaving enzyme or enzymes. J. Biol. Chem. 242, 5260–5266. [PubMed] [Google Scholar]
- Tanaka T.; Knox W. E. (1959) The nature and mechanism of the tryptophan pyrrolase (peroxidase-oxidase) reaction of Pseudomonas and of rat liver. J. Biol. Chem. 234, 1162–1170. [PubMed] [Google Scholar]
- Knox W. E.; Mehler A. H. (1950) The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J. Biol. Chem. 187, 419–430. [PubMed] [Google Scholar]
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. (1996) Heme-Containing Oxygenases. Chem. Rev. 96, 2841–2887. [DOI] [PubMed] [Google Scholar]
- Springer B. A.; Sligar S. G.; Olson J. S.; Phillips G. N. Jr. (1994) Mechanisms of ligand recognition in myoglobin. Chem. Rev. 94, 699–714. [Google Scholar]
- Dunford H. B. (2010) Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology and Physiology, 2nd ed., John Wiley, Chichester, U.K. [Google Scholar]
- Dunford H. B. (1999) Heme Peroxidases, John Wiley, Chichester, U.K. [Google Scholar]
- Erman J. E. (1998) Cytochrome c peroxidase: A model heme protein. J. Biochem. Mol. Biol. 31, 307–327. [Google Scholar]
- Ortiz de Montellano P. R. (1992) Catalytic sites of hemeprotein peroxidases. Annu. Rev. Pharmacol. Toxicol. 32, 89–107. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Imai K. (1998) Evolution of myoglobin. Cell. Mol. Life Sci. 54, 979–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulou N. D.; Mewies M.; McLean K. J.; Seward H. E.; Svistunenko D. A.; Munro A. W.; Raven E. L. (2005) Redox and Spectroscopic Properties of Human Indoleamine 2,3-Dioxygenase and a His303Ala Variant: Implications for Catalysis. Biochemistry 44, 14318–14328. [DOI] [PubMed] [Google Scholar]
- Terentis A. C.; Thomas S. R.; Takikawa O.; Littlejohn T. K.; Truscott R. J. W.; Armstrong R. S.; Yeh S.-R.; Stocker R. (2002) The heme environment of recombinant human indoleamine 2,3-dioxygenase: Structural properties and substrate-ligand interactions. J. Biol. Chem. 277, 15788–15794. [DOI] [PubMed] [Google Scholar]
- Cady S. G.; Sono M. (1991) 1-Methyl-dl-tryptophan, β-(3-benzofuranyl)-dl-alanine (the oxygen analog of tryptophan), and β-[3-benzo(b)thienyl]-dl-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch. Biochem. Biophys. 291, 326–333. [DOI] [PubMed] [Google Scholar]
- Hamilton G. A. (1969) Mechanisms of two- and four-electron oxidations catalyzed by some metalloenzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 32, 55–96. [DOI] [PubMed] [Google Scholar]
- Hayaishi O.; Rothberg S.; Mehler A. H.; Saito Y. (1957) Studies on oxygenases; enzymatic formation of kynurenine from tryptophan. J. Biol. Chem. 229, 889–896. [PubMed] [Google Scholar]
- Sugimoto H.; Oda S.-i.; Otsuki T.; Hino T.; Yoshida T.; Shiro Y. (2006) Crystal structure of human indoleamine 2,3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forouhar F.; Anderson J. L.; Mowat C. G.; Vorobiev S. M.; Hussain A.; Abashidze M.; Bruckmann C.; Thackray S. J.; Seetharaman J.; Tucker T.; Xiao R.; Ma L. C.; Zhao L.; Acton T. B.; Montelione G. T.; Chapman S. K.; Tong L. (2007) Molecular insights into substrate recognition and catalysis by tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 104, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Kang S. A.; Mukherjee T.; Bale S.; Crane B. R.; Begley T. P.; Ealick S. E. (2007) Crystal Structure and Mechanism of Tryptophan 2,3-Dioxygenase, a Heme Enzyme Involved in Tryptophan Catabolism and in Quinolinate Biosynthesis. Biochemistry 46, 145–155. [DOI] [PubMed] [Google Scholar]
- Littlejohn T. K.; Takikawa O.; Truscott R. J. W.; Walker M. J. (2003) Asp274 and His346 Are Essential for Heme Binding and Catalytic Function of Human Indoleamine 2,3-Dioxygenase. J. Biol. Chem. 278, 29525–29531. [DOI] [PubMed] [Google Scholar]
- Sono M.; Dawson J. H. (1984) Extensive studies of the heme coordination structure of indoleamine 2,3-dioxygenase and of tryptophan binding with magnetic and natural circular dichroism and electron paramagnetic resonance spectroscopy. Biochim. Biophys. Acta 789, 170–187. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Fu R.; Liu A.; Hendrich M. P. (2010) EPR and Mossbauer spectroscopy show inequivalent hemes in tryptophan dioxygenase. J. Am. Chem. Soc. 132, 1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan N.; Basran J.; Efimov I.; Svistunenko D. A.; Seward H. E.; Moody P. C. E.; Raven E. L. (2008) The role of serine 167 in human indoleamine 2,3-dioxygenase: A comparison with tryptophan 2,3-dioxygenase. Biochemistry 47, 4761–4769. [DOI] [PubMed] [Google Scholar]
- Thackray S. J.; Bruckmann C.; Anderson J. L.; Campbell L. P.; Xiao R.; Zhao L.; Mowat C. G.; Forouhar F.; Tong L.; Chapman S. K. (2008) Histidine 55 of tryptophan 2,3-dioxygenase is not an active site base but regulates catalysis by controlling substrate binding. Biochemistry 47, 10677–10684. [DOI] [PubMed] [Google Scholar]
- Batabyal D.; Yeh S.-R. (2009) Substrate-Protein Interaction in Human Tryptophan Dioxygenase: The Critical Role of H76. J. Am. Chem. Soc. 131, 3260–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov R. M.; Chauhan N.; Thackray S. J.; Anderson J. L.; Papadopoulou N. D.; Mowat C. G.; Chapman S. K.; Raven E. L.; Hoffman B. M. (2010) Probing the ternary complexes of indoleamine and tryptophan 2,3-dioxygenases by cryoreduction EPR and ENDOR spectroscopy. J. Am. Chem. Soc. 132, 5494–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; van Pee K. H.; Naismith J. H. (2010) The ternary complex of PrnB (the second enzyme in the pyrrolnitrin biosynthesis pathway), tryptophan, and cyanide yields new mechanistic insights into the indolamine dioxygenase superfamily. J. Biol. Chem. 285, 21126–21133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basran J.; Rafice S. A.; Chauhan N.; Efimov I.; Cheesman M. R.; Ghamsari L.; Lloyd Raven E. (2008) A kinetic, spectroscopic, and redox study of human tryptophan 2,3-dioxygenase. Biochemistry 47, 4752–4760. [DOI] [PubMed] [Google Scholar]
- Batabyal D.; Yeh S.-R. (2007) Human tryptophan dioxygenase: A comparison to indoleamine 2,3-dioxygenase. J. Am. Chem. Soc. 129, 15690–15701. [DOI] [PubMed] [Google Scholar]
- Capece L.; Arrar M.; Roitberg A. E.; Yeh S. R.; Marti M. A.; Estrin D. A. (2010) Substrate stereo-specificity in tryptophan dioxygenase and indoleamine 2,3-dioxygenase. Proteins 78, 2961–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeds J. M.; Brown P. J.; McGeehan G. M.; Brown F. K.; Wiseman J. S. (1993) Isotope effects and alternative substrate reactivities for tryptophan 2,3-dioxygenase. J. Biol. Chem. 268, 17781–17786. [PubMed] [Google Scholar]
- Chung L. W.; Li X.; Sugimoto H.; Shiro Y.; Morokuma K. (2008) Density functional theory study on a missing piece in understanding of heme chemistry: The reaction mechanism for indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase. J. Am. Chem. Soc. 130, 12299–12309. [DOI] [PubMed] [Google Scholar]
- Chauhan N.; Thackray S. J.; Rafice S. A.; Eaton G.; Lee M.; Efimov I.; Basran J.; Jenkins P. R.; Mowat C. G.; Chapman S. K.; Raven E. L. (2009) Reassessment of the reaction mechanism in the heme dioxygenases. J. Am. Chem. Soc. 131, 4186–4187. [DOI] [PubMed] [Google Scholar]
- Capece L.; Lewis-Ballester A.; Batabyal D.; Di Russo N.; Yeh S. R.; Estrin D. A.; Marti M. A. (2010) The first step of the dioxygenation reaction carried out by tryptophan dioxygenase and indoleamine 2,3-dioxygenase as revealed by quantum mechanical/molecular mechanical studies. J. Biol. Inorg. Chem. 15, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joule J. A., and Mills K. (2000) Heterocyclic chemistry, 4th ed., Blackwell, Oxford, U.K. [Google Scholar]
- Yagil G. (1967) The proton dissociation constant of pyrrole, indole and related compounds. Tetrahedron 23, 2855–2861. [DOI] [PubMed] [Google Scholar]
- Chen H.; Ikeda-Saito M.; Shaik S. (2008) Nature of the Fe-O2 bonding in oxy-myoglobin: Effect of the protein. J. Am. Chem. Soc. 130, 14778–14790. [DOI] [PubMed] [Google Scholar]
- Lewis-Ballester A.; Batabyal D.; Egawa T.; Lu C.; Lin Y.; Marti M. A.; Capece L.; Estrin D. A.; Yeh S. R. (2009) Evidence for a ferryl intermediate in a heme-based dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 106, 17371–17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva E. G.; Neibergall M. B.; Chakrabarty S.; Lipscomb J. D. (2007) Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc. Chem. Res. 40, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa S.; Yotsuya K.; Hashiwaki Y.; Horitani M.; Sugimoto H.; Shiro Y.; Appelman E. H.; Ogura T. (2010) Identification of the Fe-O2 and the Fe=O heme species for indoleamine 2,3-dioxygenase during catalytic turnover. Chem. Lett. 39, 36–37. [Google Scholar]
- Yanagisawa S.; Horitani M.; Sugimoto H.; Shiro Y.; Okada N.; Ogura T. (2010) Resonance Raman study on the oxygenasted and the ferryl-oxo species of indoleamine 2,3-dioxygenase during catalytic turnover. Faraday Discuss. 148, 1–9. [DOI] [PubMed] [Google Scholar]
- Chung L. W.; Li X.; Sugimoto H.; Shiro Y.; Morokuma K. (2010) ONIOM study on a missing piece in our understanding of heme chemistry: Bacterial tryptophan 2,3-dioxygenase with dual oxidants. J. Am. Chem. Soc. 132, 11993–12005. [DOI] [PubMed] [Google Scholar]
- Yamane T.; Makino K.; Umezawa N.; Kato N.; Higuchi T. (2008) Extreme rate acceleration by axial thiolate coordination on the isomerization of endoperoxide catalyzed by iron porphyrin. Angew. Chem., Int. Ed. 47, 6438–6440. [DOI] [PubMed] [Google Scholar]
- Eser B. E.; Barr E. W.; Frantom P. A.; Saleh L.; Bollinger J. M. Jr.; Krebs C.; Fitzpatrick P. F. (2007) Direct spectroscopic evidence for a high-spin Fe(IV) intermediate in tyrosine hydroxylase. J. Am. Chem. Soc. 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savige W. E. (1975) New oxidation products of tryptophan. Aust. J. Chem. 28, 2275–2287. [Google Scholar]
- Zhu Y.; Silverman R. B. (2008) Revisiting heme mechanisms. A perspective on the mechanisms of nitric oxide synthase (NOS), heme oxygenase (HO), and cytochrome P450s (CYP450s). Biochemistry 47, 2231–2243. [DOI] [PubMed] [Google Scholar]
- Ames B. D.; Liu X.; Walsh C. T. (2010) Enzymatic Processing of Fumiquinazoline F: A Tandem Oxidative-Acylation Strategy for the Generation of Multicyclic Scaffolds in Fungal Indole Alkaloid Biosynthesis. Biochemistry 49, 8564–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz de Montellano P. (2005) Cytochrome P450: Structure, mechanism and biochemistry, 3rd ed., Kluwer Academic/Plenum, New York. [Google Scholar]
- Dawson J. H. (1988) Probing structure-function relations in heme-containing oxygenases and peroxidases. Science 240, 433–439. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Markris T. M.; Sligar S. G.; Schlichting I. (2005) Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. (2010) Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T.; Iwasaki M.; Sugiyama R.; Unno M.; Ikeda-Saito M. (2010) Dioxygen activation for the self-degradation of heme: Reaction mechanism and regulation of heme oxygenase. Inorg. Chem. 49, 3602–3609. [DOI] [PubMed] [Google Scholar]
- Matsui T.; Unno M.; Ikeda-Saito M. (2010) Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions. Acc. Chem. Res. 43, 240–247. [DOI] [PubMed] [Google Scholar]
- Vottero E.; Mitchell D. A.; Page M. J.; MacGillivray R. T. A.; Sadowski I. J.; Roberge M.; Mauk A. G. (2006) Cytochrome b5 is a major reductant in vivo of human indoleamine 2,3-dioxygenase expressed in yeast. FEBS Lett. 580, 2265–2268. [DOI] [PubMed] [Google Scholar]
- Maghzal G. J.; Thomas S. R.; Hunt N. H.; Stocker R. (2008) Cytochrome b5, Not Superoxide Anion Radical, Is a Major Reductant of Indoleamine 2,3-Dioxygenase in Human Cells. J. Biol. Chem. 283, 12014–12025. [DOI] [PubMed] [Google Scholar]
- Littlejohn T. K.; Takikawa O.; Skylas D.; Jamie J. F.; Walker M. J.; Truscott R. J. W. (2000) Expression and purification of recombinant human indoleamine 2,3-dioxygenase. Protein Expression Purif. 19, 22–29. [DOI] [PubMed] [Google Scholar]
- Austin C. J. D.; Astelbauer F.; Kosim-Satyaputra P.; Ball H. J.; Willows R. D.; Jamie J. F.; Hunt N. H. (2009) Mouse and human indoleamine 2,3-dioxygenase display some distinct biochemical and structural properties. Amino Acids 36, 99–106. [DOI] [PubMed] [Google Scholar]
- Fukumura E.; Sugimoto H.; Misumi Y.; Ogura T.; Shiro Y. (2009) Cooperative Binding of l-Trp to Human Tryptophan 2,3-Dioxygenase: Resonance Raman Spectroscopic Analysis. J. Biochem. 145, 505–515. [DOI] [PubMed] [Google Scholar]
- Ren S.; Liu H.; Licad E.; Correia M. A. (1996) Expression of rat liver tryptophan 2,3-dioxygenase in Escherichia coli: Structural and functional characterization of the purified enzyme. Arch. Biochem. Biophys. 333, 96–102. [DOI] [PubMed] [Google Scholar]
- Dick R.; Murray B. P.; Reid M. J.; Correia M. A. (2001) Structure-function relationships of rat hepatic tryptophan 2,3-dioxygenase: Identification of the putative heme-ligating histidine residues. Arch. Biochem. Biophys. 392, 71–78. [DOI] [PubMed] [Google Scholar]
- Manandhar S. P.; Shimada H.; Nagano S.; Egawa T.; Ishimura Y. (2002) Subunit structure of recombinant rat liver l-tryptophan 2,3-dioxygenase. Int. Congr. Ser. 1233, 161–169. [Google Scholar]
- Li J. S.; Han Q.; Fang J.; Rizzi M.; James A. A.; Li J. (2007) Biochemical mechanisms leading to tryptophan 2,3-dioxygenase activation. Arch. Insect Biochem. Physiol. 64, 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglino A.; Lombardo F.; Arca B.; Rizzi M.; Rossi F. (2008) Purification and biochemical characterization of a recombinant Anopheles gambiae tryptophan 2,3-dioxygenase expressed in Escherichia coli. Insect Biochem. Mol. Biol. 38, 871–876. [DOI] [PubMed] [Google Scholar]
- Hu X.; Bao Z.; Hu J.; Shao M.; Zhang L.; Bi K.; Zhan A.; Huang X. (2006) Cloning and characterization of tryptophan 2,3-dioxygenase gene of Zhikong scallop Chlamys farreri (Jones and Preston 1904). Aquacult. Res. 37, 1187–1194. [Google Scholar]
- Guallar V.; Wallrapp F. H. (2010) QM/MM methods: Looking inside heme proteins biochemistry. Biophys. Chem. 149, 1–11. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA. [Google Scholar]