

Abstract
Volume overload (VO) caused by aortocaval fistula (ACF) is associated with oxidative/inflammatory stress. The resulting inflammation, matrix metalloproteinase (MMP) activation, and collagen degradation is thought to play a pivotal role in left ventricular (LV) dilatation and failure. Since mitochondria are also targets for inflammation and oxidative stress, we hypothesized that there would be bioenergetic dysfunction with acute VO. In Sprague-Dawley rats subjected to 24 hrs of ACF, there was a two-fold increase in LV pressure-volume area in vivo, consistent with increased LV myocardial oxygen usage and increased bioenergetic demand in cardiomyocytes. Isolated cardiomyocytes from ACF LVs demonstrated increased hydrogen peroxide and superoxide formation and increased MMP activity. Subsarcolemmal mitochondria (SSM) showed a 40% decrease in state 3 respiration and proteomic analysis of SSM demonstrated decreased levels of complexes I-V in ACF. Immunohistochemical analysis revealed disruption of the subsarcolemmal location of the SSM network in ACF. To test for a potential link between SSM dysfunction and loss of interstitial collagen, rats were treated with the MMP-inhibitor PD166793 prior to ACF. MMP-inhibitor preserved interstitial collagen, integrin-α5 and the SSM structural arrangement. In addition, the decrease in state 3 mitochondrial respiration with ACF was prevented by PD166793. These studies established an important interaction between degradation of interstitial collagen in acute VO and the disruption of SSM structure and function which could contribute to progression to heart failure.
Keywords: volume overload, oxidative stress, matrix metalloproteinase, subsarcolemmal mitochondria
1. Introduction
The volume overload (VO) caused by aortocaval fistula (ACF) in the rat is marked by acute inflammation and oxidative stress that results in matrix metalloproteinase (MMP) activation and interstitial collagen degradation [1-3]. These extracellular myocardial events have been proposed to play a pivotal role in an adverse eccentric LV remodeling and progression to heart failure through a process of myocyte and myofiber slippage along with cardiomyocyte elongation. A number of lines of evidence support a role for MMPs in the progressive left ventricular (LV) dilatation and dysfunction leading to heart failure [4]. Treatment with a mast cell stabilizer or MMP inhibitor in the ACF rat has been shown to inhibit MMP activity and attenuate collagen loss and LV dilatation [5,6]. In the mouse with 4 weeks of ACF, MMP inhibition improves LV diastolic and systolic dysfunction in vivo and decreases oxidative stress [7]. In contrast, inhibition of the profibrotic cytokine transforming growth factor (TGF)-β in mice with coronary ligation, which induces a VO on the area opposite the myocardial infarction, results in greater LV dilatation, less collagen production, and increased TNF-α and MMP expression [8]. Taken together, these studies suggest that increased inflammation and the oxidative stress with a VO leads to MMP activation, interstitial collagen loss, LV dilatation and failure.
Inflammation and oxidative stress can also cause alterations in mitochondria, sensitive targets and sources of oxidative stress, which in turn can adversely affect cellular bioenergetics. Volume overload produces an increase in pressure-volume area (PVA), which requires higher myocardial oxygen consumption (MVO2) and ATP consumption [9]. This is consistent with the finding that MVO2 is increased as high as 1.5-fold in patients with VO over normal hearts [10,11]. However, this increase in energy demand is associated with decreased mitochondrial state 3 respiration in papillary muscle strips taken from patients with VO due to isolated mitral valve regurgitation [12]. This combination of increased energy demand and mitochondrial dysfunction may place the heart in a particularly vulnerable state during the evolution of VO-induced cardiac failure. In support of this concept, studies of mitochondria from rats with 12 weeks of VO are more vulnerable to calcium overload and hypoxia/reoxygenation injury, and are increasingly sensitive to generation of reactive oxygen species [13,14]. We have recently shown that patients with isolated MR and normal LV systolic have significant myofibrillar loss and increased formation of reactive oxygen and nitrogen species (ROS/RNS), along with aggregates of small mitochondria in cardiomyocytes. These findings suggest a potential novel interaction between bioenergetics and activation of MMPs in the cardiomyocyte of the VO heart [15]. Thus, in current investigation we sought to establish a link between MMP activation and bioenergetic function in the acute stress of VO using the well established model of ACF in the rat.
2. Materials and Methods
2.1. Reagents and antibodies
All inorganic salts were purchased from Sigma-Aldrich (St.-Louis, MO); rabbit anti-cytochrome c antibody was obtained from Cell Signaling Technology (Danvers, MA); anti-voltage-dependant anion channel (VDAC) antibody was from MitoSciences (Eugene, OR); anti-cytochrome c oxidase subunit 4 (CcOX4) antibody was purchased from Cell Signaling Technology (Danvers, MA); rabbit anti-collagen III antibody were obtained from Acris Antibodies GmbH (Herford, Germany); rabbit anti-collagen I antibody and anti-mitochondrial transcription factor A (mTFA) antibody were purchased from Abcam (Cambridge, MA); rabbit anti-integrin α5 antibody were purchased from Millipore Corporation (Billerica, MA); complete Mini protease inhibitor cocktail was obtained from Roche Diagnostics GmbH (Mannheim, Germany); protease inhibitor cocktail (catalog number P8340), phosphatase inhibitor cocktails 1 and 2 (catalog numbers P2850 and P5726 correspondingly) were purchased from Sigma-Aldrich (St.- Louis, MO); 2′,7′-dichlorodihydrofluorescein-diacetate (H2DCFDA) and dihydroethidium (DHE) were obtained from Invitrogen Corporation (Carlsbad, CA).
2.2. Animal experiments
Sprague-Dawley rats (200-250g) at 12 weeks of age were subjected to sham and ACF as previously described in our laboratory [2,3]. Separate sets of sham and ACF rats were sacrificed 24 hrs after surgery for studies of isolated cardiomyocytes (N=3 per group) and heart mitochondria (N=5 per group). Another set of sham and ACF rats were studied for in vivo hemodynamic and echo measurements prior to sacrifice and this tissue was used for immunohistochemistry and protein analysis (N=5 per group). To examine the effects of MMP inhibition, another set of sham (N=10) and ACF (N=9) rats were fed a standard chow, with or without an MMP-inhibitor PD166793 (Tocris Cookson Inc., MO, 1mg/kg body weight/day for 1 week) and sacrificed 24 hrs after ACF [6].
Echochardiography
Echocardiography and hemodynamics were performed prior to sacrifice using the Visualsonics imaging system (Vivo 770, Toronto, Canada) combined with simultaneous high-fidelity LV pressure catheter recordings (Millar Inst. Houston, TX). With the rat under isoflurane anesthesia high fidelity LV pressure catheter was advanced into the LV cavity via a right carotid cut-down. LV pressure and echocardiaographic dimensions were obtained simultaneously using software included in the Visualsonics system. LV volume was calculated from traced m-mode LV dimensions using the Teicholz formula.
Where V = volume, LVID = LV internal dimension. These LV pressure-volume data were analyzed for LV PVA and stroke work using the PVAN (Millar Inst. Houston, TX) software package.
2.3. Confocal microscopy
Cardiomyocytes were isolated from Sham and ACF rats, as described previously in our laboratory [2]. Cells were plated on four-chambered slides (Nalge, Naperville, IL), two chambers per group, and washed twice with culture media (1xMEM, Invitrogen Corporation), which was treated with either 50μmol/L H2DCFDA or 20μmol/L DHE. Image acquisition was performed within 20 minutes using the Leica DMIRBE SP1 confocal microscope (Leica Microsystems Inc., Bannockburn, IL). For quantitative analysis of the fluorescence signal, mean fluorescence intensity was assessed in three images (5x objective) acquired from each chamber. The intensity values were averaged for each group, and resulting data was averaged between experiments performed on 3 different days.
2.4. Immunohistochemistry
Samples of LV tissue were embedded in OCT compound and frozen in liquid nitrogen-cooled methlybutane. Sections were cut at 5 microns, blocked with 5% normal serum, and incubated with anti-CcOX4 (MitoSciences, 1:100), a combination of anti-collagen I (Abcam, 1:100) and anti-collagen III (Acris, 1:100), or anti-integrin α5 (Millipore, 1:500) overnight at 4°C. Fluorescent secondary antibodies with the appropriate host combinations (Invitrogen Corporation, Carlsbad CA) were used to stain for visualization. To identify nuclei, sections were mounted using Vectashield with DAPI (1.5 μg/ml; Vector Laboratories, Burlingame, CA). Image acquisition was performed on the Leica DM6000 epifluorescence microscope with SimplePCI software (Compix, Inc., Cranberry Township, PA). Immunohistochemistry for collagen type I and III was obtained from cross-sectional LV orientation which included the large contribution of the perivascular and perimysial areas. Images were adjusted appropriately to remove background fluorescence. Immunohistochemistry for integrin-α5 was performed with the same conditions and images were acquired using an identical protocol. The apparent high intensities do not reflect the relative abundance of integrin to other proteins. The presentation of the images is not internally calibrated, especially with respect to the collagen I and III, but presented to demonstrate relative changes of the specific proteins under the different treatment strategies.
2.6 Interstitial collagen analysis
Interstitial CVF was determined on picrosirius red-stained sections. Interstitial collagen was quantified as previously described in our laboratory [2].
2.5. Transmission electron microscopy
Heart LV tissue was fixed overnight in Karnovsky’s solution (2% paraformaldehyde/2.5% glutaraldehyde in 0.1M phosphate buffer). After post-fixation with 1% Osmium Tetroxide in PBS for one hour, the tissue was dehydrated with a graded series of ethanol to 100% and infiltrated with a step-procedure of 100% propylene oxide to 100% embedding media (Embed 812, Electron Microscopy Sciences, Fort Washington, PA). After blocks were polymerized, 90nm-sections were cut, mounted on nickel mesh grids, and post-stained with uranyl acetate and lead citrate. Sections were viewed in a Tecnai Twin 120kv TE microscope (FEI, Hillsboro, OR). Digital images were analyzed for qualitative changes in mitochondria.
2.7. Gel zymography
Gel zymography experiments were performed according to the manufacturer’s protocol (Invitrogen Corporation). Briefly, cardiomyocytes isolated from sham (N=6) and 24hr ACF rats (N=5) were gently homogenized and lysed using RIPA buffer without reducing agents. Samples (20μg) were mixed with Novex®Tris-Glycine SDS Sample Buffer and loaded on precast 10% gelatin gels (Invitrogen Corporation). After electrophoresis, gels were renatured and developed. Images of resulting gels were taken and bands were quantified using Scion Image Software (Scion Corporation, Frederick, MD).
2.8. In situ zymography
Gelatinolytic activity of MMPs was demonstrated in unfixed cryostat sections (7-μm thick) using DQ-gelatin as a substrate (EnzChek; Molecular Probes, Eugene, OR). Cryostat sections of heart were dried for 10 min. DQ-gelatin was dissolved in a concentration of 1 mg/mL in water and then diluted 10 times in 1% (w/v) agarose (Sigma) in the DAPI-containing PBS solution. The resulting mixture (40μL) was placed on top of the sections and covered with a coverslip. After gelling to the agar at 4°C, the incubation of the sections was performed for 24 hrs at 37°C. Fluorescence was detected with the excitation at 460-500nm and emission at 512-542 nm.
2.9. Isolation of heart mitochondria
Heart subsarcolemmal mitochondria (SSM) were isolated from LV tissue (70mg) as previously described in our laboratory [16]. The pellet resulting from the centrifugation of LV homogenate at 1000xg for 5 minutes (4°C) was considered to contain the intermyofibrillar mitochondria (IFM) and was divided into two aliquots: one was mixed with 25mg/ml Nagarse (Sigma) solution in the isolation buffer for 5 minutes followed by the incubation with 2.5% BSA for 4 minutes and centrifugation at 1000xg for 4min. The supernatant was centrifuged at 6000xg for 10min and the resulting pellet was washed twice in isolation buffer and used for respiration measurements. The other aliquot of the IFM-containing pellet was lyzed in the buffer containing 0.75mol/L aminocaproic acid, 0.05mol/L BisTris (Blue-Native (BN) extraction buffer), 3% n-dodecyl-L-maltoside (DM) and collected for the BN proteomics.
2.10. Assessment of mitochondrial function
Respiratory activity of isolated heart mitochondria was analyzed using the protocol described in our laboratory [16]. The oxygen concentration in the Perplex chamber (1ml) was measured using a Clark-type electrode (Hansatech Instruments, Norfolk, UK). State 2 mitochondrial respiration was initiated upon an addition of the glutamate/malate (5mmol/L) as substrates. Respiratory state 3 rate of oxygen consumption (nmol O2 min−1·mg protein−1) was measured in the presence of 1.5mmol/L ADP. State 4 was evaluated as a stabilized rate of oxygen consumption after utilization of 15μmol/L adenosine diphosphate (ADP). Measurement of the cytochrome c oxidase (complex IV, CcOX) activity in isolated mitochondria was performed as described by Wharton et al [17]. Analysis of complex IV activity was performed on LV tissue (50-70mg), which was homogenized in BN extraction buffer in the presence of 3% DM and incubated on ice for 3 hours. The resulting suspension was centrifuged at 10000xg for 10min (4°C). Protein concentration in the supernatant was measured by the Lowry method [18] and further analysis of complex IV activity was performed using the supernatant (5μg protein). Citrate synthase (CS) activity was measured spectrophotometrically as described by Shepherd et al [19]. The BN proteomics on SSM mitochondria was performed according to Brookes et al [20]. For the BN proteomics on myofibrillar mitochondria, IFM-containing lysates were used from LV tissue homogenates. LV tissue (30-50μg) was homogenized in extraction buffer containing 3% DM and Complete Mini protease inhibitor cocktail, incubated on ice for 4 hours, and centrifuged at 10000xg for 10 min. Aliquots of supernatants (60μg protein) were mixed with 5% Coomassie brilliant blue G-250 in 0.5mol/L aminocaproic acid and loaded on the 5-12% gradient gel. Protein separation in 1D BN/PAGE was performed at 30V overnight as described previously by Brookes et al [20].
2.11. Western blotting
LV tissue was homogenized in RIPA buffer containing 1% SDS, 2% protease inhibitor cocktail (Sigma) and 1% (v/v) phosphatase inhibitor cocktail 1 and 2 (Sigma). Solubilized LV protein (10μg) was separated using 1D SDS/PAGE and transferred on to nitrocellulose membranes at 25V at 4°C overnight. The membrane was blocked with 5% fat-free milk followed by incubation with 1:1000 dilution of anti-cytochrome c, anti-mTFA, or anti-VDAC lgG. After washing, the membrane was blocked with corresponding secondary HRP-conjugated lgG and the chemiluminescnence detection of proteins was performed in the presence of SuperSignal HRP-substrates (Thermo Scientific, Rockford, IL). Quantitation of the chemiluminescence signal on the membrane was performed using Alpha Innotech software (Cell Biosciences, Santa Clara, CA). Signal amount on the western blot membrane was normalized by the total protein amount on the 1D SDS/PAGE after staining with Coomassie brilliant blue G-250.
2.12. Analysis of thiol oxidation
The relative level of reactive thiols in LV homogenates (15μg protein) was analyzed using the labeling with N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl)-conjugated iodoacetamide (BODIPY-IAM, Invitrogen Corporation) as previously described [21].
3.0. Statistics
All data are expressed as mean ± SEM. A Student’s t-test was used to determine statistical difference between sham and ACF groups for LV PVA and stroke work, oxidant production in cardiomyocytes, and mitochondrial respiration and respiratory complexes. Statistical significance was set at p<0.05.
3. Results
3.1. Left ventricular function in acute VO
We previously reported that 24 hours of ACF produced a three-fold increase in LV end-diastolic (LVED) pressure as well as significant increases in LVED dimension, LV fractional shortening, and rate-corrected LV circumferential shortening [2]. In the current investigation, acute VO produced an increase in LV pressure volume area (PVA) and LV stroke work in ACF vs. sham rats (Figure 1). Thus, a combined increase in contractility and PVA was consistent with increased MVO2 and ATP consumption in acute VO.
Figure 1. LV Pressure-Volume loops and LV stroke work in sham and 24hr ACF rats.
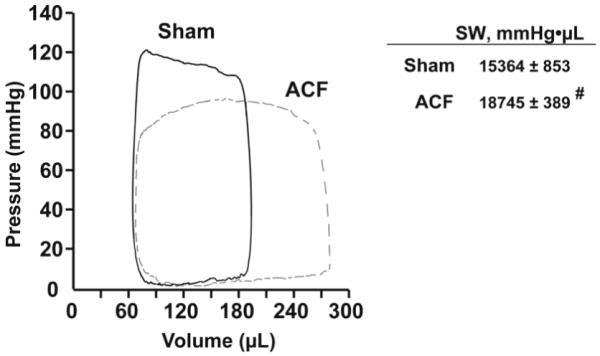
LV pressure-volume loops were obtained from simultaneous high-fidelity LV pressure catheter and echo-derived LV volumes. Representative sham (solid line) and ACF (dashed line) LV pressure volume loops demonstrate the characteristic increase in pressure volume area with volume overload. On the right, LV stroke work (SW) is increased in the ACF rats. Data is presented as mean ± SE, # p<0.05 vs. sham (n=5 per group).
3.2. Increased oxidative stress in cardiomyocytes in acute VO
Cardiomyocytes were probed with H2DCFDA and DHE as indirect indices of H2O2 and superoxide formation, respectively. ACF LV cardiomyocytes demonstrated significant 30% and 25% increases DCF and DHE fluorescence consistent with increased levels of H2O2 and superoxide in ACF vs sham cardiomyocytes (Figures 2A and B). Oxidation of protein cysteine thiols was analyzed as a further index of oxidative stress in LV homogenates using fluorescent labeling of protein thiols with BODIPY-IAM. Protein cysteine residues modified by oxidative modifications cannot react with BODIPY-IAM resulting in a decrease in fluorescent signal. As shown in Figure 2C, the intensity of the fluorescent signal in 1D SDS gel was lower in ACF vs. shams. Densitometry analysis of the fluorescent signal (Figure 2D) showed an approximate 20% decrease in signal density in ACF group vs. shams indicating a global decrease in reactive protein thiols in the ACF LV.
Figure 2. Oxidative stress and protein modifications of LV proteins in acute VO in 24hr ACF rats.

Panel A: Confocal fluorescence microscopy mages of isolated cardiomyocytes from sham and 24hr ACF rats probed with H2DCFDA and DHE dyes for assessment of the H2O2 and O2·− formation, respectively. Panel B: Quantitation of the DCF and DHE fluorescence intensity in cardiomyocytes from sham (white bars) and ACF (grey bars) rats. *p<0.05 vs. sham N=3 rats per group. Panel C: Analysis of protein cysteine thiol oxidation in LV homogenates with BODIPY-IAM labeling. Labeled proteins were separated using 1D SDS-PAGE, and BODIPY fluorescence was detected using Typhoon imager. Panel D: Quantitative analysis of the BODIPY fluorescence in 1D gel demonstrating decreased BODIPY-IAM labeling of LV proteins in ACF group vs. sham, consistent with the increased oxidation of LV protein cysteine thiols in acute VO. *p<0.05, n=6 per group.
3.3. Cardiomyocyte MMP activity and LV in situ zymography
We previously reported increase in MMP-2 activity in ACF LV tissue homegenate2 [2]. In the current investigation, we demonstrated an increase in cardiomyocyte MMP activity by both in situ zymography and gelatinolytic activity of cardiomyocytes isolated from ACF vs. sham LVs (Figure 3). Gel zymography performed on isolated cardiomyocytes revealed increased gelatinolytic activities of ~125 and ~115 kD in ACF, which is consistent with neutrophil gelatinase-associated lipocalin (NGAL)/MMP-9 complex, with MMP-9 complexed with NGAL monomer and dimer.
Figure 3. Activation of MMPs in LV tissue and cardiomyocytes in acute VO.
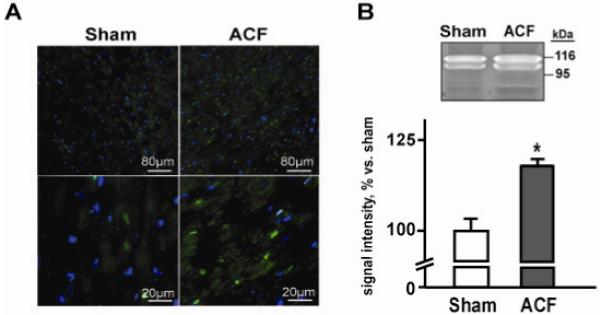
Panel A. Representative images of gelatinolytic activity assessed by in situ zymography in LV tissue of sham and ACF rats at low (upper panels) and higher magnification (lower panels). Activated MMPs are shown in green, nuclei are blue. Panel B. Representative image of gel zymography of gelatinolytic activity of cardiac myocytes isolated from a sham and ACF rat. Quantification of the bands is shown below. * p < 0.05 vs. sham, n = 6; ACF, n = 5.
3.4. Mitochondrial changes in VO
Analysis of oxygen consumption was performed in both isolated SSM and IFM. In SSM, State 3 respiration and RCR were decreased in ACF vs. sham rats (Table 1), which was also consistent with a decrease in SSM CcOX activity in ACF vs. shams (Figure 4B). However in IFM, O2 consumption (Table 1) as well as citrate synthase CcOX activities (Figure 4C and D) did not differ in ACF vs. sham LVs.
Table 1.
Parameters of mitochondrial respiration in isolated SSM and IFM
| State 2 (nmoles O2/mg/min) |
State 3 (nmoles O2/mg/min) |
State 4 (nmoles O2/mg/min) |
RCR | ||
|---|---|---|---|---|---|
| SSM | sham | 22.9±3.0 | 113.0±10.8 | 35.1±2.7 | 5.1±0.3 |
| ACF | 19.6±2.3 | 55.4±16.5# | 23.1±7.9 | 2.8±0.7# | |
| IFM | sham | 36.8±13.3 | 106.8±10.5 | 39.0±11.4 | 3.8±0.9 |
| ACF | 21.4±2.6 | 119.3±3.9 | 25.6±5.9 | 4.9±0.1 |
p<0.05 vs. sham (paired t-test)
Figure 4. Enzymatic activities of mitochondrial proteins in isolated LV mitochondria and total LV tissue in acute VO.
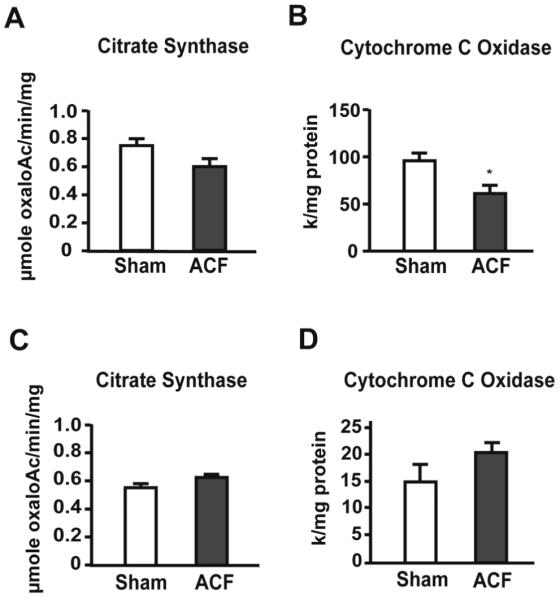
Panels A and C display citrate synthase activity in isolated SSM (A) and LV tissue homogenates (C). Panels B and D demonstrate activity of cytochrome c oxidase in SSM (B) and LV tissue homogenates (D). * = p<0.05, n = 6 per group.
Respiratory chain complexes (RCC) were analyzed in isolated SSM and IFM-containing fractions, as well as in LV homogenates by 1D BN/PAGE. In the SSM, there was no major change in the overall RCC subunit protein composition (Figure 5A). However, the RCC levels in ACF SSM were significantly lower in ACF vs. sham rats (Figure 5B and C). In contrast, protein patterns as well as levels of RCCs in both IFM fractions and LV homogenates did not differ in ACF and sham rats (data not shown). This is consistent with the lack of change in citrate synthase and CcOX activities measured in LV homogenates, which largely reflects IFM (Figure 4 C and D). Taken together, these results suggested that SSM and IFM are differentially affected in acute VO.
Figure 5. Analysis of respiratory chain complexes in isolated subsarcolemmal mitochondria (SSM, A-C) using Blue-Native proteomics in acute VO.

Protein extracts from SSM pellet were separated in 1D BN/PAGE. Panel A shows representative images of gel containing SSM respiratory complexes. Panel B demonstrates densitometry profiles of the protein bands on corresponding 1D BN gels. Panel C displays quantification of protein densities in corresponding 1D BN gels. White bars represent sham and grey bars to ACF rats. * = p<0.05 vs. sham.
3.5. Effect of VO on distribution of SSM, interstitial collagen, and integrin α5
There was a 50% decrease in interstitial collagen in ACF rats, which was significantly improved by MMP inhibitor (Figure 6). LV tissue from sham and ACF rats was stained with anti-CcOX4 (green), anti-collagen I, anti-collagen III or integrin α5 antibodies (Figure 7). There was a generalized decrease in collagen I and III, CcOX4, and integrin α5 in ACF vs. sham rats (Figure 7A), as quantitated by PASR analysis (Figure 6). When viewed at higher magnification (Figure 7B), SSM (green) were aligned with collagen fibers (red) and integrin α5 (red) in sham rats; while in ACF rats, there was a marked decrease in collagen, integrin-α5 and CcOX4 in cardiomyocyte subsarcolemmal areas, suggesting a disruption of the SSM connections with the matrix-integrin network with acute VO.
Figure 6. Volume per cent interstitial collagen in sham and ACF rats.

There is a significant loss of interstitial collagen by picrosirius red-staining in ACF (n = 5) vs. sham rats (n = 5) that is improved in PD 166793 ACF rats (n = 9). PD 166793 treatment has no effect on volume per cent collagen in sham rats (n = 8). * = p<0.05 vs shams, # = p < 0.05 vs ACF
Figure 7. Acute VO alters the distribution of subsarcolemmal mitochondria (SSM) interstitial collagen, and integrin α5 in LV.

Sections of LV tissue are stained with collagen I and III, CcOX4, and integrin α5 (Intg α5) antibodies using fluorescent microscopy. There is loss of collagen I and III, CcOX4, and Intg α5 that is restored by PD 166793 and infliximab (Panel A). In Panel B, overlay of collagen I and III and Intg α5 with CcOX4 and DAPI at higher power demonstrates a prominent loss of CcOX4 and Intg α5 in the subsarcolemmal area of cardiomyocytes in the ACF LV, which is restored by PD 166793.
To determine whether the loss of collagen in acute VO contributed to the structural and functional changes in SSM, pretreatment with PD166793 prevented loss of collagen I and III and integrin α5 and preserved the collagen/integrin-α5/SSM network in ACF rats (Figure 7B). In the ACF group, MMP-inhibitor treatment significantly improved state 3 mitochondrial respiration vs. untreated ACF rats (82 ± 5 vs. 48 ± 12 nmole O2/mg/min p<0.05), despite decreasing state 3 mitochondrial respiration in MMP inhibitor-treated vs. untreated sham rats (62 ± 4 vs. 113 ± 11 nmole O2/mg/min, p<0.05). In addition, a 50% decrease in the RCR of ACF vs. shams (2.8 ± 0.7 in ACF vs. 5.1 ± 0.3 p < 0.05) was normalized by treatment with MMP-inhibitor in ACF rats (5.3 ± 0.6 p < 0.05). Importantly, RCR was unaffected by MMP-inhibitor treatment in sham rats (5.4 ± 0.8) consistent with the requirement for a hyperactive state of MMP activation to cause mitochondrial dysfunction.
3.6. Transmission electron microscopy analysis of SSM and IFM mitochondria
Using transmission electron microscopy, sections of LV free wall from sham and ACF rats were analyzed in transverse (Figure 8) and longitudinal sections (Figure 9). Transverse TEM images of LV free wall showed a tight alignment of SSM below the sarcolemma in sham samples, whereas in ACF LVs, there were fewer SSM located underneath the sarcolemmal membrane (Figure 8). In addition, there SSM lost their compact structure and alignment and there was an increase in empty space directly below the sarcolemmal membrane.
Figure 8. Transmission electron microscopic transverse images of LV cardiomyocytes from sham and ACF rats.

TEM images of LV cardiomyocyte in sham rats demonstrates the tight arrangement of mitochondria below the sarcolemmal membrane (pointed with arrows). In the ACF rats, there is a decrease in density of SSM with more space between them, consistent with decreased subsarcolemmal CcOX4 immunostaining in Figure 5.
Figure 9. Disorganization of intermyofibrillar mitochondria in ACF rat LV.

Longitudinal TEM images of sham LV demonstrate the linear array of mitochondria between myofibrils, while the ACF LV demonstrates disorganization and clustering as well as swelling of interfibrillar mitochondria.
Longitudinal TEM images in sham LVs displayed the normal organized arrays of myofibrils with IFM (Figure 9). In contrast, ACF hearts displayed profound myofibrillar and mitochondrial disarray. IFM in ACF LVs appeared swollen and were disorganized and aggregated in clusters rather than elongated and linear along the myofibrils. Analysis of IFM swelling in the presence of 150μmole/L Ca2+ showed no significant difference in the LV IFM sensitivity to Ca2+ between sham and 1d ACF animal groups (data not shown). However, LV VDAC protein level was increased 25%, while cytochrome c and mTFA did not differ in ACF vs. shams (Figure 10).
Figure 10. Western blot analysis of mitochondrial proteins in LV homogenate.
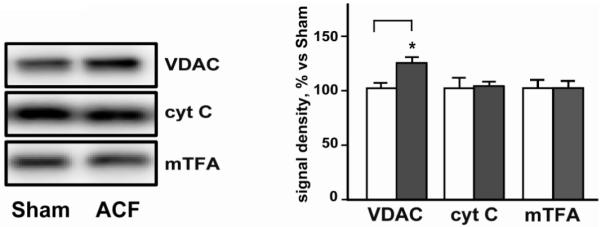
Representative western blot of mitochondrial proteins VDAC, cytochrome c (cyt C), and anti-mitochondrial transcription factor A (mTFA) demonstrates a selective increase in VDAC levels in ACF LV vs. sham. Panel to the right displays quantification of the mitochondrial proteins from Western blot in sham (white bars) and ACF (grey bars) group. * = p<0.05, N=6 per group.
Discussion
The current study shows that disruption of interstitial collagen and integrin-α5 is associated with a disruption SSM structure and function in acute VO. LV hyper-function combined with an increase in the LV PVA and LV stroke work is consistent with an increase in LV MVO2 and ATP usage. A loss of extracellular matrix has been the hallmark of the pure VO of ACF [1-6], which if linked to bioenergetic dysfunction in the context of increased ATP demand, may be an important mechanism in the progression to heart failure in chronic VO.
There is emerging evidence for an important interaction of the extracellular matrix and muscle cell mitochondria [22]. A recent study has demonstrated a highly ordered co-localization of SSM with myonuclei and surrounding collagen fibers in perimysial junctional plates of skeletal muscle fiber bundles [23]. In transgenic mice with collagen VI deficiency, there are marked structural mitochondrial alterations including abnormal cristae and modified matrix density, in addition to mitochondrial dysfunction and decreased skeletal muscle force [24]. It is of interest that a collagen VI deficiency has been identified in the pathogenesis of congenital muscular dystrophy and mitochondrial dysfunction in humans [22]. Interstitial collagen and other ECM cell surface and cell membrane proteins are structurally and functionally connected to the cardiomyocyte cytoskeleton through the transmembrane integrins. A knockdown of fibronectin mRNA expression induces mitochondria-derived signaling that results in apoptosis in rat mesangial cells [25]. In rabbit fibroblasts, knockdown of integrin α5 results in oxidant production, MMP activation, and mitochondrial dysfunction [26]. Furthermore, signal transduction through integrin α5/β3 has been implicated in the elevated expression of MMPs and the enhanced invasion of melanoma cells through basement membrane matrices [27].
The current study shows that loss of collagen and integrin-α5 is associated with disruption SSM organization along with decreased State 3 respiration, respiratory chain complexes expression, and CcOX activity. SSM comprise numerous tightly joined mitochondria with intermitochondrial junctions directly beneath the sarcolemma, as demonstrated in TEM images in Figure 6. In the SSM region, actomyosin filaments as well as other organelles are absent, so almost all the space is occupied by mitochondria, which also gives the intense sarcolemma-associated CcOX-4 staining on immunochistochemistry images in Figure 5B. Prevention of LV interstitial collagen loss with the global MMP inhibitor preserves SSM and integrin-α5 organization and improves SSM state 3 respiration and RCR.
In the present study, we also demonstrate increased H2O2 and superoxide within cardiomyocytes along with increased MMP activity within ACF cardiomyocytes and an increase in LV thiol oxidation (>20%). We have previously shown that patients with isolated MR and normal LV systolic have significant myofibrillar loss and increased formation of reactive oxygen and nitrogen species (ROS/RNS), along with aggregates of small mitochondria in cardiomyocytes. These findings suggest to us a potential novel interaction between bioenergetics and activation of MMPs in the cardiomyocyte of the VO heart. There is now emerging evidence that cardiomyocyte MMPs may degrade cellular proteins including troponin in ischemia reperfusion injury of the heart [28] and that their activation may occur in mitochondria [29]. Connexin 43, a transmembrane protein important in the formation of gap junctions and cell-cell coupling, has recently been localized in SSM [30] but not IFM, and is susceptible to MMP degradation [31]. In addition, disruption of integrin α5-mediated myocyte-ECM communication may make the cardiomyocyte SSM population particularly vulnerable to cardiomyocyte ROS-mediated MMP activation. Further, there is diffuse patchy gelatinase activation within the cardiomyocyte by in situ zymography, which can degrade the network of cardiac cytoskeletal proteins (actin, desmin etc.) that support the IFM [32] resulting in progressive respiratory defects in VO.
Subsarcolemmal mitochondrial function has been related to ATP production for electrolyte and protein transport across the sarcolemma; however, the more plentiful IFM largely provide energy for cardiomyocyte contraction and thus have a higher respiratory capacity than SSM [33,34]. Within 24 hours of ACF, IFM respiratory complexes, respiratory function, and Ca2+-sensitivity are unaffected. However, TEM images demonstrate swelling and clustering of LV IFM with a loss of their orientation along myofibrils, which may result from MMP digestion of intermediate filaments that support IFM. The increase in LV VDAC levels cannot be attributed to an increase in IFM number in the absence of cytochrome c and mTFA changes. In a model of hepatic inflammation, increased VDAC levels are concomitant with mitochondrial swelling and ~50% loss of mitochondrial membrane potential [35]. VDAC along with the adenine nucleotide transporter and cyclophilin D are components of the mitochondrial permeability transition pore (MPTP), which when open has been associated with mitochondrial matrix swelling and eventual rupture of the outer mitochondrial matrix membrane [36]. Future studies will determine whether ROS-mediated MMP activation targets MPTP and whether there is a difference in susceptibility for MPTP opening in subsarcolemmal versus interfibrillar mitochondria in the stretch of VO.
The important finding of the current study is that from the outset of VO, disruption of ECM-cell attachment is associated with increased cardiomyocyte oxidative stress and MMP activation, decreased function of SSM, and structural abnormalities in IFM. The oxidative stress with VO is consistent with graded increases in TNF-α and ROS production with increased amplitudes of mechanical stretch of cardiomyocytes in vitro [37,38] and MMP-2 activation [39]. Treatment of rats with MMP-inhibitor prevented dissolution of interstitial collagen and disarrangement of SSM and integrin α5 in LV, indicating a connection between interstitial and intracellular MMPs and the changes in SSM. It is important to note that this study only addresses the acute stage of VO. Whether intracellular MMP activation is also important in the chronic phases of VO must be addressed in future studies. However, the loss of collagen throughout the course of VO, along with a disruption of integrin and focal adhesion complex [40], raises the intriguing possibility that intracellular MMP activation may play an important role in adverse cardiomyocyte remodeling and bioenergetic function in the course of VO.
Acknowledgements
This study is supported by NHLBI Grants RO1 HL54816 (LJD) and Specialized Center of Clinically Orientated Research in Cardiac Dysfunction P50HL077100 (LJD).
Footnotes
Disclosures: None declared
References
- [1].Cox MJ, Hawkins UA, Hoit BD, Tyagi SC. Attenuation of oxidative stress and remodeling by cardiac inhibitor of metalloproteinase protein transfer. Circulation. 2004;109:2123–8. doi: 10.1161/01.CIR.0000127429.53391.78. [DOI] [PubMed] [Google Scholar]
- [2].Chen Y, Pat B, Zheng J, Cain L, Powell P, Shi K, et al. Tumor necrosis factor-alpha produced in cardiomyocytes mediates a predominant myocardial inflammatory response to stretch in early volume overload. J Mol Cell Cardiol. 2010;49:70–8. doi: 10.1016/j.yjmcc.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA, Dell’Italia LJ. Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol. 2007;49:811–21. doi: 10.1016/j.jacc.2006.06.083. 20. [DOI] [PubMed] [Google Scholar]
- [4].Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiolog Rev. 2007;87:1285–342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- [5].Brower GL, Janicki JS. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Card Fail. 2005;11:548–56. doi: 10.1016/j.cardfail.2005.05.005. [DOI] [PubMed] [Google Scholar]
- [6].Chancey AL, Brower GL, Peterson JT, Janicki JS. Effects of matrix metalloproteinase inhibition on ventricular remodeling due to volume overload. Circulation. 2002;105:1983–88. doi: 10.1161/01.cir.0000014686.73212.da. [DOI] [PubMed] [Google Scholar]
- [7].Cox MJ, Hawkins UA, Hoit BD, Tyagi SC. Attenuation of oxidative stress and remodeling by cardiac inhibitor of metalloproteinase transfer. Circulation. 2004;109:2123–28. doi: 10.1161/01.CIR.0000127429.53391.78. [DOI] [PubMed] [Google Scholar]
- [8].Frantz S, Kai H, Adamek A, Wolf J, Sallam A, Maier SKG, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008;103:485–92. doi: 10.1007/s00395-008-0739-7. [DOI] [PubMed] [Google Scholar]
- [9].Vendelin M, Bovendeerd PH, Saks V, Engelbrecht J. Cardiac mechanoenergetics in silico. Neuro Endocrinol Lett. 2002;23:13–20. [PubMed] [Google Scholar]
- [10].Strauer BE. Cardiac energetics in clinical heart disease. Basic Res Cardiol. 1987;82:389–402. doi: 10.1007/978-3-662-11289-2_38. [DOI] [PubMed] [Google Scholar]
- [11].Strauer BE, Beer K, Heitlinger K, Hofling B. Left ventricular systolic wall stress as a primary determinant of myocardial oxygen consumption: comparative studies in patients with normal left ventricular function, with pressure and volume overload and with coronary heart disease. Basic Res Cardiol. 1977;72:306–13. doi: 10.1007/BF01906378. [DOI] [PubMed] [Google Scholar]
- [12].Santosh S, Pawan K, Karpagam P, Kaushala M, Neela P. Defect in oxidative phosphorylation in LV papillary muscle mitochondria of patients undergoing mitral valve replacement. Mitochondrion. 2006;6:89–93. doi: 10.1016/j.mito.2006.02.001. [DOI] [PubMed] [Google Scholar]
- [13].Marcil M, Ascah A, Matas J, Belanger S, Deschepper CF, Burelle Y. Compensated volume overload increases the vulnerability of heart mitochondria without affecting their functions in the absence of stress. J Mol Cell Cardiol. 2006;41:998–1009. doi: 10.1016/j.yjmcc.2006.08.117. [DOI] [PubMed] [Google Scholar]
- [14].Matas J, Young NT, Bourcier-Lucas C, Ascah A, Marcil M, Deschepper CF, et al. Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy. J Mol Cell Cardiol. 2009;46:420–30. doi: 10.1016/j.yjmcc.2008.10.020. [DOI] [PubMed] [Google Scholar]
- [15].Ahmed MI, Gladden JD, Litovsky SH, Lloyd SG, Gupta H, Inusah S, et al. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60% J Am Coll Cardiol. 2010;55:671–79. doi: 10.1016/j.jacc.2009.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brookes PS, Zhang J, Dai L, Zhou F, Parks DA, Darley-Usmar VM, et al. Increased sensitivity of mitochondrial respiration to inhibition by nitric oxide in cardiac hypertrophy. J Mol Cell Cardiol. 2001;33:69–82. doi: 10.1006/jmcc.2000.1276. [DOI] [PubMed] [Google Scholar]
- [17].Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–50. [Google Scholar]
- [18].Lowry O, Rosebrough N, Farr A, Randall R. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- [19].Shepherd JA, Garland GP. Citrate synthase from rat liver. Methods Enzymol. 1969;13:11–19. [Google Scholar]
- [20].Brookes PS, Pinner A, Ramachandran A, Coward L, Barnes S, Kim H, et al. High throughput two-dimensional blue-native electrophoresis: A tool for functional proteomics of mitochondria and signaling complexes. Proteomics. 2002;2:969–77. doi: 10.1002/1615-9861(200208)2:8<969::AID-PROT969>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- [21].Hill BG, Reily C, Oh JY, Johnson MS, Landar A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic Biol Med. 2009;47:675–83. doi: 10.1016/j.freeradbiomed.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rizzuto R. The collagen mitochondrial connection. Nat Genet. 2003;35:300–01. doi: 10.1038/ng1203-300. [DOI] [PubMed] [Google Scholar]
- [23].Passerieux E, Rossignol R, Chopard A, Carnino A, Marini JF, Letellier T, et al. Structural organization of the perimysium in bovine skeletal muscle: Junctional plates and associated intracellular subdomains. J Struct Biol. 2006;154:206–16. doi: 10.1016/j.jsb.2006.01.002. [DOI] [PubMed] [Google Scholar]
- [24].Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:367–71. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- [25].Wu D, Chen X, Guo D, Hong Q, Fu B, Ding R, et al. Knockdown of fibronectin induces mitochondriadependent apoptosis in rat mesangial cells. J Am Soc Nephrol. 2005;16:646–57. doi: 10.1681/ASN.2004060445. [DOI] [PubMed] [Google Scholar]
- [26].Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;158:357–68. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Seftor RE, Seftor EA, Gehlsen KR, Stetler-Stevenson WG, Brown PD, Ruoslahti E, et al. Role of the alpha v beta 3 integrin in human melanoma cell invasion. Proc Natl Acad Sci U S A. 1992;89(5):1557–61. doi: 10.1073/pnas.89.5.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kandasamy AD, Chow AK, Ali MAM, Schulz R. Matrix metalloproteinase-2 and myocardial oxidative stress injury: beyond the matrix. Cardiovasc Res. 2010;85:413–23. doi: 10.1093/cvr/cvp268. [DOI] [PubMed] [Google Scholar]
- [29].Moshal KS, Tipparaju SM, Vacek TP, Kumar M, Singh M, Frank IE, et al. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am J Physiol. 2008;295:H 890–97. doi: 10.1152/ajpheart.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado, et al. Presence of Connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol. 2009;104:141–7. doi: 10.1007/s00395-009-0007-5. [DOI] [PubMed] [Google Scholar]
- [31].Tyagi N, Vacek JC, Givvimani S, Sen U, Tyagi SC. Cardiac specific deletion of N-methyl-d-aspartate receptor 1 ameliorates mtMMP-9 mediated autophagy/mitophagy in hyperhomocysteinemia. J Recept Signal Transduct Res. 2010;30(2):78–87. doi: 10.3109/10799891003614808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150(6):1283–98. doi: 10.1083/jcb.150.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Riva A, Tandler B, Loffredo F, Vazquez E, Hoppel C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am J Physiol. 2004;289:H868–72. doi: 10.1152/ajpheart.00866.2004. [DOI] [PubMed] [Google Scholar]
- [34].Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731. [PubMed] [Google Scholar]
- [35].Tang XH, Gao J, Chen J, Xu LZ, Tang YH, Zhao XN, et al. Mitochondrial modulation is involved in the hepatoprotection of Limonium sinense extract against liver damage in mice. J Ethnopharmacol. 2008;120:427–31. doi: 10.1016/j.jep.2008.09.022. [DOI] [PubMed] [Google Scholar]
- [36].Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the holy grail of cardioprotection. Basic Res Cardiol. 2010;105:151–4. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
- [37].Pimentel DR, Amin JK, Miller T, Viereck J, Oliver-Krasinki J, Baliga R, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res. 2001;89:453–60. doi: 10.1161/hh1701.096615. [DOI] [PubMed] [Google Scholar]
- [38].Frank D, Kuhn C, Brors B, Hanselmann C, Lüdde M, Katus HA, et al. Gene expression pattern in biomechanically stretched cardiomyocytes evidence for a stretch-specific gene program. Hypertension. 2008;51:309–18. doi: 10.1161/HYPERTENSIONAHA.107.098046. [DOI] [PubMed] [Google Scholar]
- [39].Wang TL, Yang YH, Chang H, Hung CR. Angiotensin II signals mechanical stretch-induced cardiac matrix metalloproteinase expression via JAK-STAT pathway. J Mol Cell Cardiol. 2004;37:588–95. doi: 10.1016/j.yjmcc.2004.06.016. [DOI] [PubMed] [Google Scholar]
- [40].Sabri A, Rafiq K, Seqqat R, Kolpakov MA, Dillon R, Dell’Italia LJ. Sympathetic activation causes focal adhesion signaling alteration in early compensated volume overload attributable to isolated mitral regurgitation in the dog. Circ Res. 2008;102:1127–36. doi: 10.1161/CIRCRESAHA.107.163642. [DOI] [PMC free article] [PubMed] [Google Scholar]