

Abstract
Background
The LV dilatation of isolated mitral regurgitation (MR) is associated with an increase in chymase and a decrease in interstitial collagen and extracellular matrix (ECM). In addition to pro-fibrotic effects, chymase has significant antifibrotic actions because it activates matrix metalloproteinases (MMP) and kallikrein and degrades fibronectin. Thus, we hypothesize that chymase inhibitor (CI) will attenuate ECM loss and LV remodeling in MR.
Methods and Results
We studied dogs with four months of untreated MR (MR, n=9) or treated with CI (MR+CI, n=8). Cine magnetic resonance imaging (MRI) demonstrated a >40% increase in LV end-diastolic volume in both groups, consistent with a failure of CI to improve a 25% decrease in interstitial collagen in MR. However, LV cardiomyocyte fractional shortening was decreased in MR vs. normals (3.71 ± 0.24% vs. 4.81 ± 0.31%, P<0.05) and normalized in MR+CI dogs (4.85 ± 0.44%). MRI with tissue tagging demonstrated an increase in LV torsion angle in MR+CI vs. MR dogs. CI normalized the significant decrease in fibronectin and FAK phosphorylation, and prevented cardiomyocyte myofibrillar degeneration in MR dogs. In addition, total titin and its stiffer isoform were increased in the LV epicardium and paralleled the changes in fibronectin and FAK phosphorylation in MR+CI dogs.
Conclusions
These results suggest that chymase disrupts cell surface-fibronectin connections and FAK phosphorylation that can adversely affect cardiomyocyte myofibrillar structure and function. The greater effect of CI on epicardial vs. endocardial titin and non collagen cell surface proteins may be responsible for the increase in torsion angle in chronic MR.
Keywords: mitral valve, cardiac volume, contractility, collagen, cell adhesion molecules
INTRODUCTION
Chronic isolated mitral regurgitation (MR) in the dog is characterized by left ventricular (LV) dilatation, loss of interstitial collagen and non-collagen extracellular matrix (ECM) proteins, and progressive cardiomyocyte elongation and dysfunction.1–4 In both the acute (2–4 weeks) and chronic (4 and 6 months) MR dog, there is a significant decrease in LV interstitial collagen.1–3 In addition, there is downregulation of multiple non-collagen ECM scaffolding genes and transforming growth factor β (TGFβ) signaling,4 suggesting that LV dilatation is due to collagen loss and an overall decrease in ECM supporting structure.
Isolated MR is associated with increased LV mast cell infiltration and degranulation,1–2 resulting in release of chymase, a proteolytic enzyme which is increased at both early and late stages of MR.2 Chymase mediates the majority of angiotensin II (Ang II) production in the human5 and the MR dog6. However, Ang II type 1 receptor blockade does not attenuate collagen loss nor prevent eccentric LV or cardiomyocyte remodeling in MR dogs.7 In addition to its Ang II-forming capacity, chymase directly activates matrix metalloproteinases (MMPs) 8,9 and degrades fibronectin,10 resulting in cell death through the loss of cell-ECM connections. 11 In addition, chymase activates kallikrein,12 thereby increasing bradykinin, which reduces fibronectin and collagen I and III mRNA expression in adult cardiac fibroblasts.13
Chymase has been linked to fibrosis through its Ang II forming capacity. However, in the pure stretch of MR, the persistent elevation of mast cell chymase may contribute to interstitial collagen loss and the decrease in ECM, resulting in adverse LV and cardiomyocyte remodeling in MR. We hypothesize that blockade of chymase will prevent disruption of the ECM, resulting in improved LV remodeling and cardiomyocyte function in chronic MR.
MATERIALS AND METHODS
Experimental Procedures
Mitral valve regurgitation was induced at Auburn University (AU) College of Veterinary Medicine in mongrel dogs of either sex (19–26 kg) by chordal rupture as previously described.1–4,6,7 Dogs were randomly assigned to two groups: (1) four months of MR untreated (n=9; male = 5; female = 4) and (2) four months of MR treated with chymase inhibitor (CI) (4-{1-[(4-methylbenzo[6] thiophen-3-yl)methyl]benzimidazole-2-ylthio}butanoic acid: TEI-F00806; Teijin Pharma Ltd, Tokyo, Japan, at a dose of 100 mg/kg PO, twice daily; n=8; male = 2; female = 6) started 24 hours after MR induction. The effective oral dose of chymase inhibitor was determined by demonstrating that pretreatment with CI (oral dose 100 mg/kg, twice daily) significantly decreased an increase in interstitial fluid (ISF) chymase activity during ischemia reperfusion of the left anterior descending artery in the open chest dog using in vivo cardiac microdialysis to measure ISF chymase activity.14 Hemodynamic data was collected at baseline and after 4 months of MR. Dogs were transferred to the University of Alabama at Birmingham (UAB) for sacrifice. Drug was withheld on the day of the terminal experiments. A third cohort of normal dogs (n=13; male = 7; female = 6) were analyzed by magnetic resonance imaging (MRI) before sacrifice and were also used for cardiomyocyte isolation and tissue morphometry. All dogs in this study were between two and four years of age. This study was approved by the Animal Services Committees at UAB and AU.
Terminal Study: Instrumentation
Animals were maintained at a deep plane of general anesthesia using 1–2% isofluorane in 100% oxygen. The heart was arrested with KCl, quickly extirpated, placed in ice-cold Krebs solution, and the coronaries flushed with the same solution. LV sections were flash frozen in liquid nitrogen or perfusion fixed with 3% paraformaldehyde.
Isolated Cardiomyocyte Studies
Cardiomyocytes were isolated from the tissue by recirculating perfusion buffer supplemented with 2mg/mL collagenase type II (Invitrogen, Carlsbad, California) as previously described.3 The [Ca2+]I was measured with the fluorescent indicator fluo 3-acetoxymethyl ester (fluo- 3-AM; Molecular Probes, Eugene, Oregon) and the contractile activity (cell fractional shortening) of field-stimulated (5 ms square pulses with constant voltage at ~20% above threshold and 1000 ms cycle length) cardiomyocytes was measured with a video-edge detector (Crescent Electronics, Sandy, Utah) as previously described.3
Cine-MRI and Tissue Tagging
MRI was performed on a 1.5 Tesla GE Signa Horizon (Milwaukee, WI) instrument optimized for cardiac application.
Cine MRI
Steady state free precession (SSFP) technique was used to measure absolute LV volumes and ejection fraction. Shortest repetition time (TR) and echo-time (TE) were used (typical values: 4.5 ms and 1.9 ms, respectively). Slice thickness was 10 mm, with a zero interslice gap. True temporal resolution was 25–30 ms in all cases. LV volumes were calculated from summated serial endocardial and epicardial contours as previously described.4 Left atrial volumes were calculated using a standard area length method from 2 and 4 chamber views.
LV time volume curves
The LV volume at each time frame was computed and an LV volume time curve was constructed and differentiated with respect to time. End-diastole was defined as the maximum-volume time frame, and end-systole was defined as the minimum-volume time frame. Early diastole was defined as the first half of the diastolic interval. The early diastolic filling rate was defined as the maximum derivative (ml/sec) and corrected for EDV (EDV/sec).15
Early Mitral annular Velocity
The mitral annular velocity was computed by tracking the mitral annulus in a four-chamber view from user-defined landmark points at end-diastole and end-systole using a non-rigid registration algorithm. A mitral annular displacement curve was constructed and differentiated with respect to time to obtain a mitral annular velocity curve.15
Tagged Cine MRI
A grid tagged, fast gradient echo, cine sequence was used with TR of 8.4 ms, TE of 4.8 ms on identical slice prescription as above. True temporal resolution was 50 ms or less. Tag line spacing in the grid was 7×7 mm. Two-dimensional torsion was measured by tracking a circular mesh of points in a basal and apical slice. The mesh was identified the first time based on user-defined contours and tracked through the remaining imaged phases using improved HARP tracking.16 Normalized torsion, T, was computed from the rotation angle, ϕ, in the basal and apical slices by the following formula:17
where ρ is the epicardial radius and L is the distance between the basal and apical slices.
Collagen Analysis
Interstitial collagen was quantified with Picric Acid Sirius Red F3BA (PASR) in epicardial and endocardial sections as previously reported.3 Total LV endocardial collagen was identified by the colorimetric hydroxyproline method as previously described.4
Assessment of Cardiomyocyte Myofibrillar Degeneration
Hematoxylin & eosin (H&E) stained sections were assessed for myofibrillar degeneration using light microscopy. Longitudinal orientated tissue was graded on a scale of 1–5, 1 being <20%, 2 = 20–40%, 3 = 40–60%, 4 = 60–80% and 5 = >80% of cardiomyocyte degeneration per high powered field (40× objective, 1600× total magnification) in 20–30 fields. All analyses were performed in a blinded manner.
Western Blot
LV mid wall tissue was dounced in RIPA buffer containing EDTA, protease and phosphatase cocktail inhibitors (all from PIERCE). Lysates (10–40μg) were separated on a 4–20% gradient Bis/Tris gel (Invitrogen) by sodium dodecyl sulfate (SDS)- polyacrylamide gel electrophoresis, transferred to a PVDF membrane and probed with antibodies to: laminin (Abcam, 1:500), fibronectin (Calbiochem, 1:1000), phospho-FAK (Tyr397) (Millipore, 1:200), FAK (BD Transduction Laboratories, 1:200) or β-actin (Abcam, 1:4000) diluted in Blotto B (Santa Cruz) for 1hr (room temperature = RT) or overnight (4°C) then with appropriate horse-radish-peroxidase-conjugated secondary antibodies (1:2000–1:5000). Bands were visualized by enhanced chemiluminescence (PIERCE), scanned and quantitated in Scion Image v4.0.3.2 (Scion Corporation), and normalized to β-actin.
Titin gel electrophoresis
LV tissue lysates were separated on 1–2% vertical SDS-agarose gels and stained with Coomassie Blue.18 Wet gels were scanned and analyzed with one-dimensional scan software (Scanalytics). The integrated optical density of N2BA titin, N2B titin, total titin (N2BA + N2B), and myosin heavy chain (MHC) were determined as a function of the volume of the solubilized protein sample that was loaded (a range of volumes was loaded on each gel). The slope of the linear range of the relation between integrated optical density (IOD) and loaded volume was obtained for each protein. This slope was converted to IOD/mg of tissue (samples were solubilized at a fixed ratio of tissue weight and volume of solubilization solution).
MMP-2 activity assay and gel zymography
LV endocardial midwall tissue (50–100mg) was homogenized in 50mM Tris-HCl pH7.6, 0.01% Triton X-100, 15mM NaCl, 0.5mM CaCl2, 20μM ZnCl2 and centrifuged at 16,000 × g (30mins at 4°C). Lysates were assayed using the Biotrack MMP-2 activity assay System (RPN 2631, Amersham Biosciences). MMP-2 activity was normalized to total protein. For gel zymography, lysates were mixed with an equal volume of Tris–Glycine SDS Sample buffer (2X) (Novex, Invitrogen) and separated on a 10% (v/v) Tris-glycine polyacrylamide gel containing 0.1% (w/v) gelatin. Gels were developed and stained with Colloidal Blue Staining Kit according to the protocol for Novex Zymogram Gels (Invitrogen). Dog serum served as a positive control for both MMP-2 and MMP-9.
TGFβ1 activity assay
LV endocardial tissue (50–100mg) was homogenized [50mM HEPES, 50mM Na4O7P2, 100mM NaF, 10mM EDTA pH7.4 + cOmplete protease cocktail inhibitor (Roche)] and centrifuged at 16,000 × g (20mins at 4°C). Lysates were assayed with the TGFβ1 ELISA kit (R&D Systems) according to the manufacturer’s protocol. TGFβ1 activity was normalized to total protein.
Bradykinin assay
LV endocardium (100–200mg) was homogenized in 10mM Tris-HCl pH7.5 (1:2) containing cOmplete protease inhibitor cocktail (Roche) and centrifuged at 10,000 × g (15mins at 4°C). The supernatant was acidified with an equal volume of 1% trifluoroacetic acid (TFA) in water and loaded onto a purification C18 Sep – Pak cartridge (Waters Chromatography Division). Sample was loaded and the cartridge was washed with 6ml of 1% TFA in water. Peptides were eluted with 3ml TFA- Acetonitrile- water (0.4:60:39.6) and lyophilized overnight, then reconstituted in sample buffer from the Bradykinin EIA kit (S-1135, Peninsula Laboratories, USA), and measured according to the manufacturer’s protocol. Bradykinin levels were normalized to total tissue weight.
Statistics
Data are presented as mean ± standard error (SEM). Statistical analysis was performed on all groups by one-way analysis of variance and the Holm-Sidak post-hoc test for significance using Sigma Stat version 3.5. P<0.05 was considered significant. Analysis of covariance (adjusting for gender) was performed on torsion angle, fractional shortening, and titin expression and there was no effect of gender on outcome.
RESULTS
LV morphometry and hemodynamics (Table 1)
Table 1.
LV morphometry and hemodynamics in Normal and MR dogs.
| Normal | MR | MR+CI | |
|---|---|---|---|
| Body weight, kg | 21±1 | 23±1 | 22±1 |
| LV:Body weight, g/kg | 3.9±0.2 | 4.6±0.2 * | 4.7±0.2 * |
| RV:Body weight, g/kg | 1.6±0.1 | 1.7±0.1 | 1.6±0.1 |
| Sample size, n | 13 | 9 | 8 |
| Baseline | MR | MR+CI | |
| Heart rate, bpm | 100±3 | 109±4 | 109±8 |
| Cardiac output, L/min | 3.6±0.2 | 3.3±0.2 | 3.5±0.3 |
| LV end-systolic pressure, mmHg | 105±3 | 109±3 | 105±2 |
| Systemic vascular resistance, dyne cm sec−5 | 2011±122 | 2207±225 | 1942±206 |
| Pulmonary artery mean pressure, mmHg | 10±1 | 11±1 | 12±1 |
| Pulmonary artery wedge pressure, mmHg | 3±0.4 | 5±1 | 5±1 |
| Pulmonary vascular resistance, dyne cm sec−5 | 170±14 | 176±24 | 167±15 |
| Sample Size, n | 17 | 9 | 8 |
Values are mean ± SEM
P < 0.05 indicates significant difference from Normal group
Baseline values represent hemodynamics in all MR dogs (n = 17) prior to induction of MR. Values are mean ± SEM
P < 0.05 indicates significant difference from Baseline
MR and MR+CI dogs had a similar increase in LV:body weight without an increase in right ventricular mass (Table 1). Mean heart rate, cardiac output, mean arterial pressure, pulmonary arterial wedge pressure, and pulmonary and systemic vascular resistance did not differ from normal in MR and MR+CI dogs. The failure of CI to decrease LV mass in MR dogs, as previously reported with renin angiotensin system blockade,1,7 may be due to the failure of CI to decrease mean arterial pressure or systemic vascular resistance.
LV remodeling and function (Tables 2)
Table 2.
MRI-derived indices of LV remodeling in Normal and 4 month MR dogs.
| Normal | MR | MR+CI | |
|---|---|---|---|
| LV end-diastolic volume, mL | 58±3 | 83±10 * | 87±10* |
| LV end-systolic volume, mL | 38±2 | 48±6 | 51±7 |
| LV stroke volume, mL | 20±2 | 35±5 * | 36±4 * |
| LV ejection fraction, % | 34±2 | 44±2 * | 42±3 * |
| LV end-diastolic dimension, cm | 3.6±0.2 | 4.3±0.1 * | 4.3±0.2 * |
| LV end-systolic dimension, cm | 3.3±0.1 | 3.6±0.1 * | 3.6±0.2 * |
| Left atrial volume, mL | 144±1 | 340±75 * | 344±64 * |
| LV early diastolic filling rate (ml/sec) | 148±24 | 240±48 | 269±48 |
| LV early diastolic filling rate (EDV/sec) | 2.5±0.4 | 2.9±0.5 | 3.2±0.4 |
| Peak early mitral annular velocity (mm/sec) | 30±3.5 | 30±12 | 52±14 |
| Peak early mitral annular velocity (% LA length/sec) | 53±6 | 48±20 | 81±22 |
| Normalized Peak LV Systolic Torsion Angle (°) | 2.54±0.84 | 3.15±0.36 | 4.16±0.31 *# |
| Sample Size, n | 7 | 9 | 8 |
Values are mean ± SEM
P < 0.05 indicates significant difference from Normal group
P < 0.05 indicates significant difference from MR group
LA = Long axis
LV end-diastolic volume and dimension were increased >40% in MR and MR+CI vs. normal dogs (Table 2). In both MR groups there was at least a two-fold increase in LV stroke volume and left atrial volume in the absence of any other valvular disease. These findings coupled with significant MR by analysis of cine-MR imaging in each dog documented a significant amount of regurgitation in all MR dogs. MRI-derived indices of diastolic function including early diastolic filling from time volume curves and early diastolic mitral annular velocity tended to be increased compared to normal in both MR and MR+CI dogs (Table 2). MRI tissue tagging-derived peak LV systolic torsion angle was increased in MR+CI vs. MR and normal dogs (Table 2).
Effect of chymase inhibition on ECM, FAK and bradykinin
CI had no effect on the decrease in LV endocardial and epicardial collagen (Fig 1A) and LV endocardial collagen by hydroxyproline (Fig. 1B) in MR dogs. However, MMP-2 and MMP-9 activity did not differ from normal in MR and MR+CI dogs (Fig 1C and 1E). TGFβ1 activity was significantly decreased in MR dogs but did not differ from normal in MR+CI dogs (Fig 1D). Laminin was significantly decreased in the LV endo- but not epicardium of MR and MR+CI dogs (Fig 2); while fibronectin was decreased in the MR endocardium and preserved with CI. There was a significant decrease in LV endocardial FAK phosphorylation in MR dogs, which was normalized with CI (Fig 3). In contrast, LV epicardial fibronectin was increased in MR dogs and MR+CI dogs compared to normal. LV epicardial phospho-FAK did not differ between normal and MR but was significantly increased in MR+CI dogs (Fig 3). There was a 12-fold elevation in LV bradykinin levels in MR dogs vs. normal (P<0.05) which was normalized in MR+CI dogs (Fig 1F).
FIGURE 1. Effect of chymase inhibition on collagen homeostasis.

Quantitation of interstitial collagen volume % in PASR-stained sections (A) and of total collagen by hydroxyproline in LV endocardium (B). Quantitative assay of MMP-2 activity (C), TGFβ1 activity (D), representative gel zymography demonstrating MMP-2 and MMP-9 (E), and bradykinin levels (F) in LV endocardial tissue from normal, MR and MR+CI hearts. Values mean ± SEM. * = P < 0.05 vs. Normal; # P < 0.05 vs. MR
FIGURE 2. Effect of chymase inhibition on non-collagen ECM proteins.

Representative western blots of laminin and fibronectin protein in normal (n=7), MR (n=7) and MR+CI (n=8) LV endocardium and epicardium. Densitometric analysis of laminin and fibronectin protein expression normalized to β-actin. Values mean ± SEM. * =P < 0.05 vs. Normal; # P < 0.05 vs. MR
FIGURE 3. Effect of chymase inhibition on focal adhesion kinase phosphorylation.

Representative western blot of phospho FAK (tyrosine 397) and total FAK (band size 125kD) in normal (n=7), MR (n=7) and MR+CI (n=8) LV endocardium and epicardium. Densitometric analysis of phospho FAK and total FAK protein expression is normalized to β-actin. Values mean ± SEM. *=P<0.05 vs. Normal; #=P < 0.05 vs. MR
Isolated cardiomyocyte function (Table 3)
Table 3.
Isolated Cardiomyocyte Function in Normal and MR dogs.
| Normal | MR | MR+CI | |
|---|---|---|---|
| Fractional Shortening, % | 4.81±0.31 | 3.71±0.24 * | 4.85±0.44 |
| Calcium transients, F0/Fmax | 1.57±0.09 | 1.31±0.05 * | 1.26±0.01 * |
| Number of dogs (total number of cells sampled) | 8 (91) | 8 (134) | 8 (125) |
Values are mean ± SEM
P < 0.05 indicates significant difference from Normal
MR hearts had myofibrillar loss characterized by the appearance of peri-nuclear vacuoles and cellular debris (Fig 4B and 4D) vs. normal hearts (Fig 4A). This was significantly improved in MR+CI hearts (Fig 4C and 4E). Furthermore, cardiomyocyte fractional shortening was significantly decreased in MR vs. normal and normalized in MR+CI cardiomyocytes (Table 3). However, calcium transients were equally depressed in both MR and MR+CI cardiomyocytes vs. normal cells.
FIGURE 4. Myofibrillar degeneration in MR.

Representative H&E-stained sections of LV myocardium in normal (A), MR (B) and MR+CI (C) at 1600X (total magnification). (D) Higher magnification (4000X total) of an MR heart demonstrating increased peri-nuclei space (arrowheads) and lipid accumulation (blue arrows). (E) Myofibrillar degeneration is graded in all hearts. Values mean ± SEM. * = P < 0.05 vs. Normal; #=P<0.05 vs. MR
Effect of chymase inhibition on cardiomyocyte sarcomeric titin
There was a significant increase in total titin levels in the LV epicardium of MR+CI vs. normal and MR dogs (Fig 5A and 5B). In addition, there was a significant increase in LV epicardial N2B titin isoform in MR+CI dogs vs. normal dogs (Fig 5B). In contrast, there were no significant changes in LV endocardial titin (Fig 5C).
FIGURE 5. Cardiomyocyte sarcomeric titin in MR.
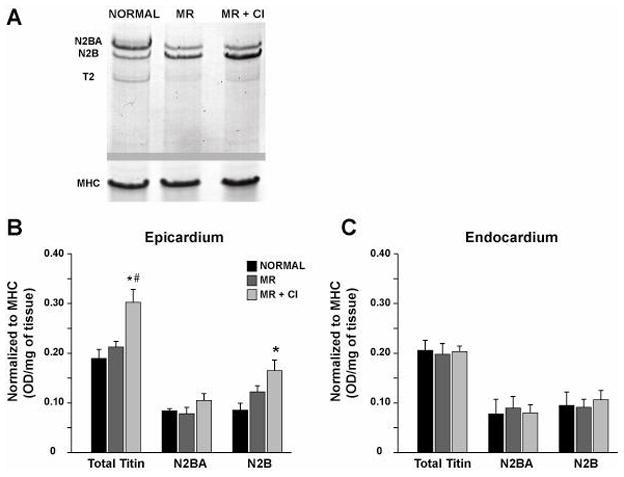
LV tissue lysates separated by vertical SDS-agarose gel electrophoresis in normal (n=7), MR (n=8) and MR+CI (n=8) dogs. (A) Representative blot of total titin (N2BA and N2B isoforms) and the titin degradation product T2 in the epicardium. Bands are normalized to myosin heavy chain (MHC) for quantitative analysis of LV epicardium (B) and endocardium (C). Values mean ± SEM. * = P < 0.05 vs. Normal; # P < 0.05 vs. MR
DISCUSSION
In the present study, we demonstrate that chronic chymase inhibition does not prevent collagen loss or eccentric LV remodeling in the MR dog. However, chymase inhibition preserves LV fibronectin and FAK phosphorylation, which significantly attenuates myofibrillar degeneration and improves cardiomyocyte function and LV torsion in isolated MR.
In MR and MR+CI dogs, there are equivalent increases in LVED volume, left atrial volume and stroke volume, which is consistent with similar 25% LV interstitial collagen loss in both groups. This is not associated with an increase in MMP-2 or MMP-9 activity in either group. Previous studies in canine MR have demonstrated a 6-fold increase in MMP-9 activity 6 hours after MR induction, which returns to normal after 14 days,19 suggesting an acute rather than prolonged activation of MMPs. In our MR dogs, a decrease in TGFβ1 activity is not improved by chymase inhibition, which can explain the persistent decrease in LV collagen or laminin in MR+CI dogs. Taken together, the early loss of interstitial collagen in MR may be related to elevated MMP activity, while long term collagen loss may be due to decreased ECM synthesis. This may explain the failure of CI to improve LV dilatation. Further, other mast cell contents such as TNF-α, which can activate MMPs and has been shown to be upregulated in MR,20 may be unaffected by CI.
Chymase inhibition does prevent the loss of LV endocardial fibronectin in MR, in keeping with its ability to directly degrade fibronectin.10 It is of interest that LV bradykinin in MR hearts is increased 12-fold and normalized by CI. The increase in LV bradykinin may be due to chymase activation of kallikrein.12 Bradykinin has been shown to reduce fibronectin mRNA and protein expression by adult cardiac fibroblasts;13 while knockdown of kallikrein results in increased fibronectin expression in kidney mesangial cells.21 Thus, it is tempting to speculate that chymase mediates fibronectin loss by direct degradation as well as indirectly decreasing its synthesis through increased bradykinin formation in the MR heart.
Chymase degradation of fibronectin has also been shown to cause a decrease in FAK phosphorylation, resulting in smooth muscle cell death.11 We have previously demonstrated a decrease in FAK tyrosine phosphorylation and disruption of the focal adhesion complex in two and four week MR dogs.22 In the current study, there is a concordant decrease in fibronectin and FAK phosphorylation in the LV endocardium of four month MR dogs. However, there is no evidence of increased apoptosis by TUNEL staining in acute22 or four month MR dogs. Interestingly, LV epicardial fibronectin is increased in both MR and MR+CI dogs, and this is associated with preserved or increased FAK phosphorylation, respectively. An intact focal adhesion complex is essential for cell growth, survival and myofibril assembly. As opposed to the volume overloaded MR heart, increased FAK phosphorylation has been documented in animal models of pressure overload.23 Interestingly, mice with selective inactivation of cardiomyocyte FAK demonstrate increased myofibrillar degeneration, elongation and thinning of cardiomyocytes, and eccentric LV remodeling and heart failure after six months.24 Indeed, there is increased myofibrillar degeneration in MR hearts in the presence of decreased fibronectin and FAK phosphorylation, all of which are improved by CI. Thus, the decrease in FAK phosphorylation in MR and myofibrillar loss may be initiated by chymase-mediated breakdown of fibronectin in the cardiac interstitium.
Previous studies have implicated myofibrillar loss in the pathophysiology of contractile dysfunction in isolated MR.25 In the current investigation, isolated cardiomyocyte function is significantly depressed in MR dogs and improved with CI without improving intracellular calcium transients. These data suggest that improved cardiomyocyte contractile performance is associated with preservation of myofibrillar structure. In addition, β-receptor blockade has also been shown to improve cardiomyocyte function and myofibrillar density in the MR dog.25 In MR+CI dogs, cardiomyocyte functional improvement is not accompanied by an increase in LV ejection fraction but rather demonstrate an increase in LV torsion angle above both normal and MR dogs.
One of the major myofibrillar proteins in the heart is the large sarcomeric protein titin, which exists as two isoforms – N2BA and N2B.26 Previous studies demonstrate a relationship between increased LV diastolic stiffness and total titin levels and a shift from the more compliant N2BA isoform to the stiffer N2B in animal models of heart failure27 and pressure overload,28 and in patients with aortic stenosis.29 However, MRI-derived indices of diastolic function are not compromised in MR+CI dogs despite increases in LV epicardial N2B titin, most likely counterbalanced by the persistent decrease in interstitial collagen. There is emerging evidence that titin also affects systolic function by determining lattice spacing of myofilaments and calcium sensitivity.26 In the current study, LV endocardial total titin does not differ among normal, MR and MR+CI dogs; while LV epicardial titin is increased with a shift to the stiffer N2B isoform in MR+CI dogs. We speculate that this gradient of titin expression from endocardium to epicardium can explain the significant increase in LV torsion in MR+CI dogs.
Torsion is the wringing motion of the LV along its long axis during systole, induced by contracting myofibers, which are aligned 180° from endocardium to epicardium. Studies in the intact animal demonstrate that acute increases in preload and contractility are associated with increased LV torsion.15,30 Studies in the mouse heart demonstrate a gradient of myosin phosphorylation from endocardium to epicardium that facilitate torsion,31 because the direction of torsion during systole follows the direction of the epicardial fibers.17,32 However, changes in myofiber orientation and function, as well as collagen and/or other matrix proteins from endo- to epicardium may affect torsion. Taken together, myofibrillar preservation combined with increased fibronectin and titin N2B expression from endocardium to epicardium in MR + CI dogs may indeed drive the higher torsion angle. Further, FAK is located at the Z disk at the insertion of titin in the cardiomyocyte and there is evidence that the N2B region of titin interacts with integrins and FAK. 33 Future studies will elucidate the mechanism that causes a titin isoform shift in the myocardium and whether this is dependent on an intact fibronectin:FAK interaction.
The LV endocardium is subjected to higher wall stress than the epicardium during a hemodynamic load. Thus, it is not unexpected that there is a greater loss of laminin in LV endocardium compared to LV epicardium of MR hearts, which is also paralleled by greater decreases in LV endocardial fibronectin. Taken together, better structural integrity and function of the LV epicardium vs. endocardium may account for the greater torsion angle in the MR+CI dogs. Indeed, it has been postulated that an increased LV torsion angle in patients with aortic stenosis may represent an important functional compensation for a decrease in LV endocardial blood flow and function in concentric LV hypertrophy.17,32 Whether this represents a major compensatory mechanism for LV functional preservation over time in the MR heart remains an open question.
The current study suggests that increased chymase activity in the cardiac interstitium sets in motion a cascade of events that degrade fibronectin, disrupts FAK, and leads to increased myofibrillar degeneration. It is of interest that we recently report a similar marked cardiomyocyte myofibrillar loss in patients with isolated MR despite a well preserved LV ejection fraction of greater than 60%. 34 Blockade of chymase preserves the fibronectin:FAK interaction and prevents myofibrillar loss. The improvement in cardiomyocyte shortening and LV torsion with CI may be related to the maintenance of a functional ECM-cell interface that promotes an increase in titin-dependent cardiomyocyte shortening in MR.
Acknowledgments
FUNDING SOURCES
This study is supported by the Office of Research and Development, Medical Service, Department of Veteran Affairs (LJD), HL62881 (HG) and Specialized Centers of Clinically Orientated Research grant in Cardiac Dysfunction P50HL077100 and R01HL79040 (AH) and in part by Teijin Pharmaceuticals Ltd, Tokyo, Japan (LJD).
Footnotes
DISCLOSURES
None.
References
- 1.Dell’Italia LJ, Balcells E, Meng QC, Su X, Schultz D, Bishop SP, Machida N, Straeter-Knowlen I, Hankes GH, Dillon R, Cartee RE, Oparil S. Volume overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in the dog. Am J Physiol. 1997;273:H961–70. doi: 10.1152/ajpheart.1997.273.2.H961. [DOI] [PubMed] [Google Scholar]
- 2.Stewart JA, Wei C-C, Brower GL, Rynders PE, Hankes GH, Dillon AR, Lucchesi P, Janicki JS, Dell’Italia LJ. Cardiac mast cell and chymase mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35:311–319. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 3.Pat B, Killingsworth C, Denney T, Zheng J, Powell P, Tillson M, Dillon AR, Dell’Italia LJ. Dissociation between cardiomyocyte function and remodeling with β-adrenergic receptor blockade in isolated canine mitral regurgitation. Am J Physiol Heart Circ Physiol. 2008;295:H2321–7. doi: 10.1152/ajpheart.00746.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng J, Chen Y, Pat B, Dell’Italia LA, Tillson M, Dillon AR, Powell PC, Shi K, Shah N, Denney T, Husain A, Dell’Italia LJ. Microarray identifies extensive downregulation of noncollagen extracellular matrix and profibrotic growth factor genes in chronic isolated mitral regurgitation in the dog. Circulation. 2009;119:2086–95. doi: 10.1161/CIRCULATIONAHA.108.826230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. 1990;265:22348–57. [PubMed] [Google Scholar]
- 6.Dell’Italia LJ, Meng QC, Balcells E, Straeter-Knowlen IM, Hankes GH, Dillon AR, Cartee RE, Orr R, Bishop SP, Oparil S, Elton TS. Increased ACE and chymase-like activity in cardiac tissue of dogs with chronic mitral regurgitation. Am J Physiol. 1995;269:H2065–731. doi: 10.1152/ajpheart.1995.269.6.H2065. [DOI] [PubMed] [Google Scholar]
- 7.Perry GJ, Wei CC, Hankes GH, Dillon SR, Rynders P, Mukherjee R, Spinale FG, Dell’Italia LJ. Angiotensin II receptor blockade does not improve left ventricular function and remodeling in subacute mitral regurgitation in the dog. JACC. 2002;39:1374–79. doi: 10.1016/s0735-1097(02)01763-1. [DOI] [PubMed] [Google Scholar]
- 8.Fang KC, Raymond WW, Blount JL, Caughey GH. Dog mast cell alpha-chymase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92 bonds of the catalytic domain. J Biol Chem. 1997;272:25628–35. doi: 10.1074/jbc.272.41.25628. [DOI] [PubMed] [Google Scholar]
- 9.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–6. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 10.Okumura K, Takai S, Muramatsu M, Katayama S, Sakaguchi M, Kishi K, Jin D, Miyazaki M. Human chymase degrades human fibronectin. Clin Chim Acta. 2004;347:223–5. doi: 10.1016/j.cccn.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Leskinen MJ, Lindstedt KA, Wang Y, Kovanen PT. Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol. 2003;23:238–43. doi: 10.1161/01.atv.0000051405.68811.4d. [DOI] [PubMed] [Google Scholar]
- 12.Forteza R, Lauredo I, Abraham WM, Conner GE. Bronchial tissue kallikrein activity is regulated by hyaluronic acid binding. Am J Respir Cell Mol Biol. 1999;21:666–74. doi: 10.1165/ajrcmb.21.6.3651. [DOI] [PubMed] [Google Scholar]
- 13.Kim NN, Villegas S, Summerour SR, Villarreal FJ. Regulation of cardiac fibroblast extracellular matrix production by bradykinin and nitric oxide. J Mol Cell Cardiol. 1999;31:457–66. doi: 10.1006/jmcc.1998.0887. [DOI] [PubMed] [Google Scholar]
- 14.Wei CC, Hase N, Bradley WE, Shi K, Kobayashi T, Killingsworth CR, Walcott GP, Powell P, Husain A, Dell’Italia LJ. Upregulation of cardiac interstitial chymase after canine myocardial ischemia and reperfusion. FASEB J. 2008;22:730.28. (Meeting abstract) [Google Scholar]
- 15.Feng W, Nagaraj H, Gupta H, Lloyd SG, Aban I, Perry GJ, Calhoun DA, Dell’Italia LJ, Denney TS., Jr A dual propagation contours technique for semi-automated assessment of systolic and diastolic cardiac function by CMR. J Cardiovasc Mag Res. 2009;11:30. doi: 10.1186/1532-429X-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osman NF, Kerwin WS, McVeigh ER, Prince JL. Cardiac motion tracking using CINE harmonic phase (HARP) magnetic resonance imaging. Magn Reson Med. 1999;42:1048–60. doi: 10.1002/(sici)1522-2594(199912)42:6<1048::aid-mrm9>3.0.co;2-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russel IK, Gotte MJW, Bronzwaer JG, Knaapen P, Paulus WJ, van Rossum AC. Left ventricular torsion. JACC: Cardiovas Imag. 2009;2:648–55. doi: 10.1016/j.jcmg.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 2003;24:1695–702. doi: 10.1002/elps.200305392. [DOI] [PubMed] [Google Scholar]
- 19.Nagatomo Y, Carabello BA, Coker ML, McDermott PJ, Nemoto S, Hamwaki M, Spinale FG. Differential effects of pressure or volume overload on myocardial MMP levels and inhibitory control. Am J Physiol. 2000;278:H151–61. doi: 10.1152/ajpheart.2000.278.1.H151. [DOI] [PubMed] [Google Scholar]
- 20.Oral H, Sivasubramanian N, Dyke DB, Mehta RH, Grossman PM, Briesmiester K, Fay WP, Pagani FD, Bolling SF, Mann DL, Starling MR. Myocardial proinflammatory cytokine expression and left ventricular remodeling in patients with chronic mitral regurgitation. Circulation. 2003;107:831–7. doi: 10.1161/01.cir.0000049745.38594.6d. [DOI] [PubMed] [Google Scholar]
- 21.Pawluczyk IZ, Tan EK, Lodwick D, Harris KP. Kallikrein gene ‘knock-down’ by small interfering RNA transfection induces a profibrotic phenotype in rat mesangial cells. J Hypertens. 2008;26:93–101. doi: 10.1097/HJH.0b013e3282f0ca68. [DOI] [PubMed] [Google Scholar]
- 22.Sabri A, Rafiq K, Seqqat R, Kolpakov MA, Dillon R, Dell’italia LJ. Sympathetic activation causes focal adhesion signaling alteration in early compensated volume overload attributable to isolated mitral regurgitation in the dog. Circ Res. 2008;102:1127–36. doi: 10.1161/CIRCRESAHA.107.163642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samuel JL, Barrieux A, Dufour S, Dubus I, Contard F, Koteliansky V, Farhadian F, Marotte F, Thiery JP. Accumulation of fetal fibronectin mRNAs during the development of rat cardiac hypertrophy induced by pressure overload. J Clin Invest. 1991;88:1737–46. doi: 10.1172/JCI115492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng X, Kraus MS, Wei H, Shen TL, Pariaut R, Alcaraz A, Ji G, Cheng L, Yang Q, Kotlikoff MI, Chen J, Chien K, Gu H, Guan JL. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–27. doi: 10.1172/JCI24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsutsui H, Spinale FG, Nagatsu M, Schmid PG, Ishihara K, DeFreyte G, Cooper G, Carabello BA. Effects of chronic beta-adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J Clin Invest. 1994;93:2639–48. doi: 10.1172/JCI117277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuda N, Granzier HL, Ishiwata S, Kurihara S. Physiological functions of the giant elastic protein titin in mammalian striated muscle. J Physiol Sci. 2008;58:151–9. doi: 10.2170/physiolsci.RV005408. [DOI] [PubMed] [Google Scholar]
- 27.Bell SP, Nyland L, Tischler MD, McNabb M, Granzier H, LeWinter M. Alterations in the determinants of diastolic suction during pacing tachycardia. Circ Res. 2000;87:235–40. doi: 10.1161/01.res.87.3.235. [DOI] [PubMed] [Google Scholar]
- 28.Warren CM, Joradan MC, Roos KP, Krzesinski PR, Greaser ML. Titin isoform expression in normal and hypertensive myocardium. Cardiovasc Res. 2003;59:86–94. doi: 10.1016/s0008-6363(03)00328-6. [DOI] [PubMed] [Google Scholar]
- 29.Williams L, Howell N, Pagano D, Andreka P, Vertesaljai M, Pecor T, Frenneaux M, Granzier H. Titin isoform expression in aortic stenosis. Clin Sci (Lond) 2009;117:237–42. doi: 10.1042/CS20080248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong S-J, Hees PS, Huang W-M, Buffer SA, Weiss JL, Shapiro EP. Independent effects of preload, afterload and contractility on left ventricular torsion. Am J Physiol. 1999;277:H1053–60. doi: 10.1152/ajpheart.1999.277.3.H1053. [DOI] [PubMed] [Google Scholar]
- 31.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras AH, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–41. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 32.Van Der Toorn A, Barenbrug P, Snoep G, Van Der Veen FH, Delhaas T, Prinzen FW, Maessen J, Arts T. Transmural gradients of cardiac myofiber shortening in aortic valve stenosis patients using MRI tagging. Am J Physiol. 2002;283:H1609–15. doi: 10.1152/ajpheart.00239.2002. [DOI] [PubMed] [Google Scholar]
- 33.Kruger M, Linke WA. Titin-based mechanical signaling in normal and failing myocardium. J Mol Cell Card. 2009;46:490–8. doi: 10.1016/j.yjmcc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed M, Gladden JD, Litovsky S, Lloyd SG, Gupta H, Inusah S, Denney T, Powell P, McGiffin D, Dell’Italia LJ. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction > 60% JACC. 2010;55:671–79. doi: 10.1016/j.jacc.2009.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]