

Abstract
Objectives
We assessed myocardial damage in patients with chronic isolated mitral regurgitation (MR) and LV ejection fraction (EF) > 60%.
Background
MR patients typically have decreased LVEF after mitral valve (MV) repair despite normal pre-operative LVEF.
Methods
27 patients with isolated MR had LV biopsies taken at time of MV repair. Magnetic resonance imaging with tissue tagging was performed in 40 normal subjects and in MR patients pre- and 6 months post-MV repair.
Results
LVEF (66 ± 1 to 54 ± 2% p<0.0001) and LV end-diastolic volume index (108 ± 5 to 78 ± 5 ml/m2 p<0.0001) decreased, while LV end-systolic volume index (ESVI) was 60% above normal pre- and post-MV repair (p<0.05). LV circumferential and longitudinal strain rates decreased below normal post-MV repair (6.38 ± 0.30 vs. 5.11 ± 0.25 p=0.0009, and 7.51 ± 0.50 vs. 5.31 ± 0.30, %RR, p<0.0001), as LVES stress (σ)/LVESVI ratio was depressed at baseline and post-MV repair vs. normals (0.25 ± 0.02 and 0.28 ± 0.01 vs. 0.33 ± 0.02, p < 0.01). LV biopsies demonstrated cardiomyocyte myofibrillar degeneration vs. normals (p=0.0016). Immunostaining and immunoblotting demonstrated increased xanthine oxidase (XO) in MR vs. normals (p<0.05). Lipofuscin deposition was increased in cardiomyocytes of MR vs. normals (0.62 ± 0.04 vs. 0.33 ± 0.04, % area, p <0.01).
Conclusions
Decreased LV strain rates and LVES σ/ESVI post-MV repair indicate contractile dysfunction, despite pre-surgical LVEF > 60%. Increased oxidative stress could cause myofibrillar degeneration and lipofuscin accumulation resulting in LV contractile dysfunction in MR.
Keywords: Mitral regurgitation, oxidative stress, magnetic resonance imaging, tissue tagging
Background
Chronic left ventricular (LV) volume overload from isolated mitral regurgitation (MR) results in increased LV diastolic wall stress and eccentric hypertrophy, fostering an initial adaptive LV chamber enlargement (1). A combination of increased preload and ejection into the low pressure left atrium fosters favorable loading conditions which falsely elevate LV ejection fraction (EF) despite underlying cardiomyocyte contractile impairment (2). Therefore, in order to preserve LV systolic function and improve survival, corrective mitral valve (MV) surgery is recommended if LVEF falls below 60% (3).
There is currently no effective medical therapy to attenuate the progressive LV remodeling resulting from the chronic volume overload stress in isolated MR, and mechanisms of LV myocardial remodeling are poorly understood (3). It is of interest that echocardiographic studies of patients with isolated MR have reported that LV end-diastolic (ED) dimension decreases as LV end-systolic (ES) dimension remains unchanged resulting in a decrease in LVEF in patients with isolated MR after mitral valve repair, even when LVEF is > 55% (4–7). Thus, it is well accepted that LVEF may belie the degree of dysfunction in the setting of MR. Indeed, some studies have reported myocardial dysfunction from LV muscle strips, derangement of calcium handling proteins, and increased cytokines in patients with isolated MR despite LVEF > 55% (8–10)—all of which can be associated with and/or attributed to increased oxidative stress. Xanthine oxidase (XO) is well characterized as a major contributor to free radical generation and oxidative stress in the cardiovascular system (11). Furthermore increased XO has been implicated in cardiovascular disease states including heart failure (12). Recent studies have demonstrated that excessive mechanical stretch in the lung increases XO activity which plays a prominent role in acute lung injury (13). Whether XO plays a role in adverse LV remodeling and functional impairment resulting from myocardial stretch in isolated MR has not previously been investigated. Accordingly, we obtained LV biopsies at the time of surgery to evaluate the level of myocardial oxidative stress and cardiomyocyte damage in patients who were within conventional guidelines for valve repair (LVEF>60%). In addition, we utilized magnetic resonance imaging (MRI) with tissue tagging and 3-dimensional analysis to define LV function at the myocardial level pre-and post-MV repair.
Methods
Study Subjects
The study protocol was approved by the University of Alabama at Birmingham Institutional Review Board and informed consent was obtained from all participants. The study group consisted of 27 patients (age mean 53 ± 3, median 54, range 34–71) with severe isolated MR secondary to degenerative mitral valve disease who were referred for corrective MV surgery and all had estimated LVEF > 60%. Severe MR was documented on echocardiogram/Doppler studies and cine-MRI in all cases. All patients had cardiac catheterization prior to surgery and patients with obstructive coronary artery disease (> 50% stenosis), aortic valve disease, or concomitant mitral stenosis were excluded from the study. Patients underwent MRI with tissue tagging prior to surgery and six months after MV repair. At the time of surgery, LV myocardial tissue was taken from the lateral endocardial wall of the LV at the level of the tips of the papillary muscles in all patients. MRI with tissue tagging was also performed in control volunteers (age 40 ± 3, median 38, range 21–62 years) who had no prior history of cardiovascular disease and were not taking any cardiovascular medications. It is important to note that six control subjects were between the ages of 45 and 50 and nine were over 50 years of age.
Magnetic resonance Imaging
Magnetic resonance imaging was performed on a 1.5-T MRI scanner (Signa GE, Milwaukee, Wisconsin) optimized for cardiac application. Electrocardiographically gated breath-hold steady-state free precision technique was used to obtain standard (2-, 3-, and 4-chamber short-axis) views using the following parameters: slice thickness of the imaging planes 8 mm, field of view 44 × 44, scan matrix 256 × 128, flip angle 45°, repetition/echo times 3.8/1.6 ms).
Three-dimensional (3D) LV geometric parameters were measured from endocardial and epicardial contours manually traced on cine-MR images acquired near end-diastole and end-systole. The contours were traced to exclude the papillary muscles. Cubic B-spline surfaces were fit to the endocardial and epicardial contours for each time frame. Three-dimensional wall thickness was computed by measuring the distance from a point on the endocardial surface to the closest point on the epicardial surface along a line perpendicular to the epicardial surface.
Tagged magnetic resonance images were acquired on the same scanner with repetition/echo times 8/44 ms, and tag spacing 7mm. Three-dimensional LV strain was measured from tagged images at end-systole, which was defined by visual inspection of the image data as the time frame with maximum contraction. Strain computations were conducted using an in-house software package (14,15). Two-dimensional strain rates were measured using harmonic phase (HARP) analysis (16–18). HARP analysis measures the local, two-dimensional strain of the myocardium based on the local spatial frequency of the tag lines. During myocardial contraction, the tag lines become closer to each other and the tag frequency increases in proportion to that contraction. Strain rates were computed at midwall segments as defined by Cerqueira et al(19).
Calculations
Three-dimensional wall thickness was computed at the same segments by measuring the distance from a point on the epicardial surface to the closest point on the epicardial surface along a line perpendicular to the epicardial surface. The radius of curvature to wall thickness ratio (R/T) was computed as the reciprocal of the product of the endocardial circumferential curvature (κ) and wall thickness (T). End-systolic wall stress was computed according to the formula: where P is mean arterial LV blood pressure measured by a cuff measurement at the time of the MR scan. Mean arterial pressure (MAP) was calculated as MAP = (systolic blood pressure + 2[diastolic pressure])/3. (20)
Surgical Methods
All patients underwent mitral valve repair. The operation was performed through a median sternotomy and employed standard hypothermic cardiopulmonary bypass and cold blood cardioplegia. A variety of methods were used to repair the mitral valve including leaflet resection, chordal replacement or a combination of each and all patients had implantation of a flexible annuloplasty ring. The adequacy of repair was assessed by intraoperative transesophageal echocardiography.
Histopathological Analysis
Control LV myocardial specimens for immunohistochemistry of XO and lipofuscin were obtained at time of autopsy in patients of comparable age with no evidence of cardiac disease on autopsy (ages 24, 28, 31, 34, 45, 49, 49 years). Biopsies from MR patients were immersion-fixed in 10% neutral-buffered formalin and embedded in paraffin. 5μm sections were stained with Hematoxylin and Eosin for examination by light microscopy, using high power (40 x objectives, 1500x total magnification). A semi-quantitative method, with a grading scale of 0 to 4, was used to evaluate ten randomly selected fields for presence of myofibrillar degeneration. A score of 0 represented no degeneration; 1. 1-<25% degeneration; 2. 25-<50% degeneration; 3. 50-<75% degeneration; 4 – 75–100% degeneration. A mean grade for each biopsy was used to express myofibrillar degeneration observed in the LV tissue with all measurements performed in a blinded manner.
Immunohistochemistry
Xanthine Oxidase
5μm sections were mounted on slides, deparaffinized in xylene and rehydrated in graded solutions of ethanol. After blocking with 5% normal serum, sections were incubated with XO antibody (NeoMarkers, Fremont, CA; 1:50) for 1 hour at room temperature, followed with Alexa Fluor 488-conjugated secondary antibody incubation (Molecular Probes, Eugene, OR; 1:150) for 1 hour at room temperature. Slides were mounted with Vectashield Mounting Medium with DAPI for nuclear staining (Vector Laboratories, Burlingame, CA). Image acquisition and intensity measurements were performed on a Leica DM6000 epifluorescence microscope with SimplePCI software (Compix, Inc., Cranberry Township, PA). Primary antibody absorbed with purified enzyme served as a negative control for each biopsy section to measure background fluorescence.
Nitrotyrosine
Slides were prepared and mounted as for XO (see above). After blocking with 5% normal serum, sections were incubated with nitrotyrosine antibody (Upstate, Lake Placid, NY; 1:100) overnight at 4°C, followed with Alexa Fluor 594-conjugated secondary antibody incubation (Molecular Probes, Eugene, OR; 1:200) for 1 hour RT. Primary antibody absorbed with 10mM nitrotyrosine served as a negative control for each biopsy section to measure background fluorescence.
Lipofuscin
5μm sections were stained with lipofuscin stain (AFIP method, Laboratory Methods in Histotechnology, Armed Forces Institute of Pathology, 1994) for imaging. Quantitative analysis was accomplished by light microscopy at medium power (20x objective, 700x total magnification), using a 540-nm (green) filter to provide grayscale contrast for lipofuscin granules. Using images collected by the digital camera, we determined the percent lipofuscin of 30 to 40 randomly selected fields in each section, and the mean value was calculated. All measurements were performed in a blinded manner. Results are presented as the mean standard error with values computed from the average of individual measurements obtained from each biopsy.
Western Blots
LV biopsies were obtained from 5 patients undergoing valve repair (4 men, 1 woman, mean age 52 ± 7 years). Normal LV tissue (4 men, 1 woman, mean age 46 ± 4) was obtained from Imgenex Corporation (San Diego, CA). Samples were homogenized in RIPA buffer containing protease inhibitors. Cell debris and fragments were then removed by centrifugation and the Bio-Rad Bradford assay was performed to determine protein concentration. Fifteen μg total protein was separated on a Tris-Hcl gel and then transferred to a nitrocellulose membrane. Membranes were then probed with a goat polyclonal antibody directed against human xanthine oxidoreductase (Santa Cruz Biotechnology, Santa Cruz, CA). Bound primary antibodies were detected with horseradish peroxidase conjugated secondary antibodies rabbit-anti goat IgG, followed by a chemiluminescent system. Densitometry analysis was conducted using ImageJ software. Membranes were stripped and reprobed with a rabbit polyclonal antibody directed towards calsequestrin (Abcam, Cambridge, MA). All reported densitometry values are normalized to the loading control calsequestrin.
Statistical Analysis
Values are presented as mean ± standard deviation. Fisher’s exact test was used to compare the gender while a repeated measures logistic regression was used to compare the other binary characteristics. Continuous patient characteristics and MRI functional data were analyzed with repeated measures ANOVA models. Using the MIXED procedure in SAS, these models allowed for comparison of the three groups (Normals, Baseline MR, and six month post-MV repair) while adjusting for the within patient correlation. To avoid inflating the probability of a Type I error for the MRI variables, the Bonferroni-Holm step down test procedure was utilized to adjust the significance level accordingly. This method is used to adjust the level of significance 0.05, by the number of tests. When comparing biopsy myofibrillar degeneration grades between MR patients and controls, Wilcoxon Rank Sum test, a nonparametric analog of the t test, was used to compare the differences in the mean ranks of the measurements between the MR and control groups. Simple linear regression was used to test associations between biopsy findings (i.e. XO, lipofuscin) and patient characteristics including age and MRI parameters. P values less than 0.05 were considered significant. All statistical analysis was performed using SAS version 9.1.3.
Results
Patient Characteristics (Table 1)
Table 1.
Patient Characteristics
| Characteristics | Normal | Pre-Surgery | Post-Surgery |
|---|---|---|---|
| Sample Size | 39 | 27 | 27 |
| Age, years | 40 ± 11 | 53 ± 12* | - |
| Male, % | 48 | 85 | 85* |
| BSA, m2 | 1.92 ± 0.23 | 2.02 ± 0.21 | 1.99 ± 0.22 |
| Heart Rate, beats/min | 76 ± 11 | 71 ± 11 | 80 ± 16 |
| Mean Systolic BP, mmHg | 116 ± 12 | 123 ± 15 | 120 ± 17 |
| Mean Diastolic BP, mmHg | 74 ± 11 | 74 ± 10 | 77 ± 9 |
| ACE-Inhibitor/AR Blocker (%) | 0 | 22 | 17 |
| Beta-blocker (%) | 0 | 26 | 35 |
| Calcium Channel blocker (%) | 0 | 7 | 0 |
BSA, body surface area; BP, blood pressure; ACE, Angiotensin converting enzyme; AR, Angiotensin receptor; Values are ± standard error of the mean
p <0.05 vs. normal control group.
There were no significant differences in body surface areas, heart rates, or blood pressures among control and pre- and post-MV repair groups. Mean age of control subjects was 40 ± 2 and MR patients was 53 ± 2. All MR patients were New York Heart Association class I (45%), or had only very mild symptoms (Class II, 55%) prior to surgery. No MR patients had atrial fibrillation. All patients had pre-operative LVEF > 60%. Nine patients (33%) had pre-surgical LV end systolic dimension (LVESD) ≥ 40mm at time of surgery. Seven patients (26%) had LVESD < 40mm and were asymptomatic at time of surgery.
LV Geometry and Function by MRI with Tissue Tagging (Table 2)
Table 2.
Magnetic Resonance Imaging Variables
| Variables | Normal | Pre-Surgery | Post-Surgery |
|---|---|---|---|
| Sample size | 39 | 27 | 27 |
| LV EF, % | 65 ± 6 | 66 ± 7 | 54 ± 9 *† |
| LV ESD, mm | 35 ± 4 | 42 ± 6 * | 43 ± 8 |
| LVEDD, mm | 51 ± 4 | 62 ± 5* | 54 ± 7† |
| LV ESV index, ml/m2 | 23 ± 6 | 37 ± 14 * | 37 ± 18 * |
| LV EDV index, ml/m2 | 66 ± 11 | 108 ± 28 * | 78 ± 24 † |
| LV SV index, ml/m2 | 43 ± 7 | 71 ± 18 * | 41 ± 9 † |
LVEF, LV ejection fraction; ESD, end-systolic dimension; ESV, end-systolic volume;
EDV, end-diastolic volume, SV, stroke volume. Values are ± standard error of the mean.
p <0.05 vs. normal control group.
p <0.05 vs. pre-MV repair.
LVED volume index was increased by > 60% in pre-MV repair patients vs. controls (108 ± 5 vs. 66 ± 2 ml/m2 p<0.05), decreased post-MV repair (78 ± 5 ml/m2 p<0.0001) and did not differ from controls. Pre-MV repair LVES volume index was increased above controls (37 ± 3 vs. 23 ± 1 ml/m2, p=0.0018) and did not change six months post-MV repair. LVED and LVES dimensions changed in similar directions as LV volumes.
LVEF decreased from 66 ± 1 to 54 ± 2% (p <0.001) post-MV repair. LV systolic circumferential and longitudinal strain rates did not differ in control vs. MR patients (Figure 1A). LV strain rates not only decreased six months post-MV repair but were also reduced below controls (6.38 ± 0.30 vs. 5.11 ± 0.25 p=0.0009, and 7.51 ± 0.50 vs. 5.31 ± 0.30 %/%RR, p<0.0001 respectively, Figure 1A). LVES radius/wall thickness (R/T) ratios (Figure 1B) did not differ significantly between groups (p=0.192). In contrast, LV mass/volume ratio was decreased (0.76 ± 0.03 vs. 0.67 ± 0.03, p<0.01) and LV 3D radius/wall thickness was increased (3.72 ± 0.15 vs. 4.32 ± 0.21, p<0.01) pre-MV repair and returned to normal post-MV repair, consistent with a reversal of eccentric LV remodeling. Furthermore, when compared to normal subjects, the LV contracted to a higher ESV at a relatively small increase in mean arterial pressure that did not achieve statistical significance (91 ± 2 vs. 97 ± 2 mmHg, p = 0.15) but did increase post-MV repair (100 ± 2 mmHg, p < 0.01, Figure 2). LVES wall stress was increased pre- (p = 0.053) but did increase post-MV repair (p < 0.01) vs. normals. However, LVES stress/ESVI ratio was decreased pre- and post-MV repair vs. normals (0.25 ± 0.02 and 0.28 ± 0.01 vs. 0.33 ± 0.02, p < 0.01).
Figure 1. LV systolic strain rates and remodeling pre- and six months post-MV repair.

LV circumferential and longitudinal systolic strain rates (A) are significantly decreased post-MV repair vs. pre-surgery and vs. normal controls. 3-D LV end-systolic (B) radius/wall thickness ratios did not differ between normal and post-surgery groups. RR denotes R to R interval. Graphs displaying LV mass/volume (C) and LV end-diastolic radius/wall thickness demonstrate reversal of eccentric remodeling following MV repair. * = p <0.05 vs. control group. † = p <0.05 vs. pre-MV repair.
Figure 2. Indices of LV afterload and function pre- and six months post-MV repair.
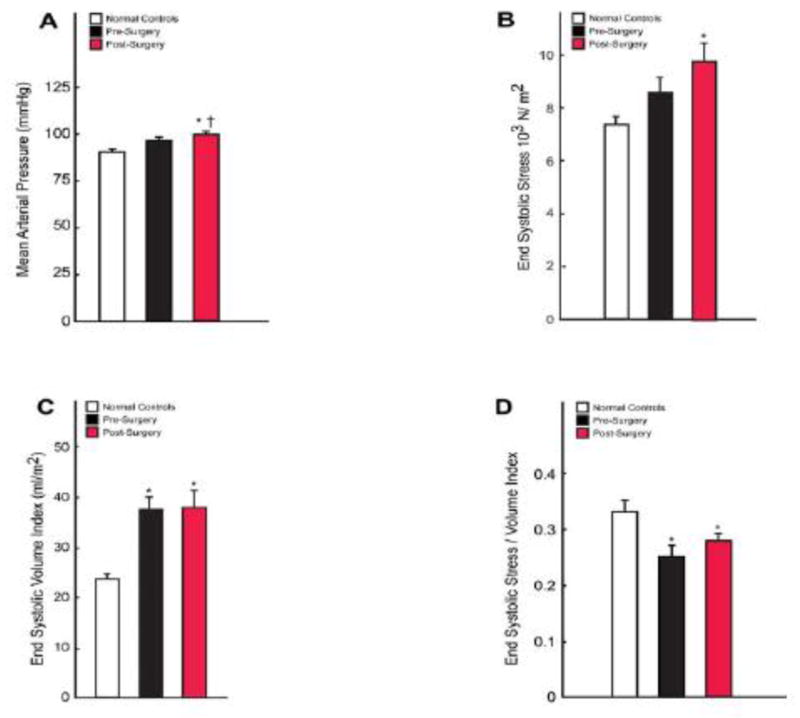
Graphs displaying mean arterial pressures (A), LV end-systolic wall stress (B), LV end systolic volume index (C), and LV end systolic stress/end-systolic volume index (D) in normals and pre- and post-surgical MR patients.
Myocardial Biopsy Histology Findings
MR hearts demonstrated marked myofibrillar degeneration (2.32 ± 0.23 vs. 1.25 ± 0.13, p=0.0016 [mean degeneration grade 1–4]), as demonstrated in Figure 3a–c. Immunostaining of biopsies for XO/XDH (Figure 3d–f) demonstrated mean intensity increase in MR hearts vs. controls (88 ± 7 vs. 33 ± 4%, p <0.01). XO was diffusely distributed and clearly associated with myofibrillar Z bands. In MR patients however, XO showed a punctate, perinuclear pattern (arrows), with distribution in close proximity to areas of myofibrillar degeneration. Immunostaining for lipofuscin (Figure 3g–i) demonstrated marked deposition in MR hearts compared to controls (0.59 ± 0.04 vs. 0.33 ± 0.05 volume%, p <0.005). Of note, no significant association was noted between age and lipofuscin deposition in MR patients. Nitrotyrosine staining was prominent in areas of lipofuscin accumulation and myofibrillar degeneration (Figure 3). Scanning electron microscopy in a representative patient with MR demonstrated the electron dense bodies that are consistent with lipofuscin located in a perinuclear distribution (Figure 4). There were no significant associations noted on linear regression analyses between biopsy measures of oxidative stress (XO, lipofuscin) and various MRI measures of LV function in MR patients.
Figure 3. Myofibrillar loss and oxidative stress in patients with isolated MR.

Myocardial biopsy findings in controls (n=10) (a,d,g) and MR patients (n=27) demonstrating myofibrillar degeneration (b,c), increased xanthine oxidase (e,f), and increased lipofuscin (h,i). Nitrotyrosine staining in the LV of an MR patient demonstrating increased staining in areas of lipofuscin accumulation (j) and a corresponding image (k) with immunoabsorbed antibody with no uptake of antibody and only autofluorescence of lipofuscin. Scale bar = 20 μm. * = p <0.05 vs. normal control group. Scale bar = 20 μm.
Figure 4. Lipofuscin in electron micrographs of patients with isolated MR.
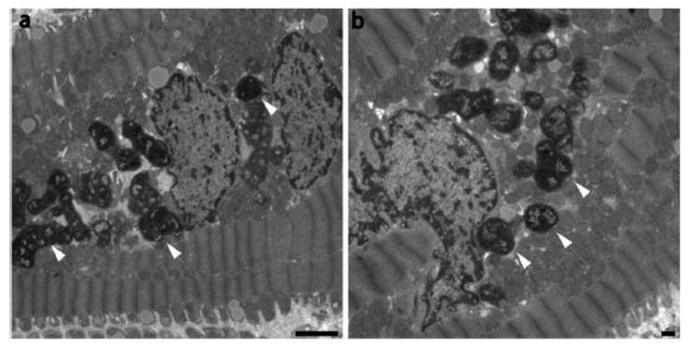
Transmission electron microscopy of endomyocardial biopsy samples demonstrating marked lipofuscin deposition (arrows) in the hearts of MR patients.
Western Blot Analysis for Xanthine Oxidase
XO is produced by either a transient or permanent modification of xanthine dehydrogenase (XDH). On western blot analysis (Figure 5), XDH and transiently modified XO (145kD band) were increased >5-fold in density in MR patients vs. controls (p=0.0125). Permanently modified XO is represented by a 125kD and an 85kD band. The 125kD band demonstrated a >5 fold increase in MR vs. normal controls (p=0.00098) while the 85kD band did not differ significantly between groups (p=0.128). Overall, total XDH/XO expression (145kD + 125kD +85kD) was increased 2.6 fold (p=0.032) in MR patients vs. control subjects.
Figure 5. Protein quantification of xanthine oxidase in LV of patients with isolated MR.

Western blot analysis for xanthine oxidoreductase (A) depicts a band at 145kD which represents both XDH and transiently modified XO and bands at 125kD and 85kD which represent permanently modified XO. Densitometry depicting average band intensity normalized to respective calsequestrin loading control (B). *=p<0.05 vs. normal control group.
Discussion
In this study we demonstrate LV systolic dysfunction six-month post-MV repair in patients with isolated degenerative MR despite pre-surgical LVEF > 60%. Decreased LV strain rates in the absence of increases in indices of LVES wall stress, along with a decreased LV end-systolic stress/end-systolic volume ratio, strongly suggests underlying myocyte dysfunction post-MV repair. This contention is further supported by the presence of marked myocyte myofibrillar degeneration and lipofuscin accumulation in hearts of isolated MR patients prior to surgery.
The decrease in LVEF post-MV repair has been attributed to a decrease in preload and a relative increase in afterload due to the correction of regurgitation into the low pressure left atrium (2,21). However, other studies report a stable LVEF with chordal preservation (22). In the current study, in which all patients had MV repair, MRI-derived LVEDVI and 3D radius/wall thickness ratio decreased as LV mass/volume ratio returned to normal—all indicative of a reversal of eccentric cardiac hypertrophy. However, the LVES stress/ESVI ratio, which has been shown to predict adverse outcomes in patients with isolated MR (23), was decreased both pre- and post-MV repair suggesting a decrease LV contractility. LVES wall stress was increased at baseline and at 6 months post repair. Further, when compared to normals, the LV contracted to a higher ESVI in the presence of a relatively small increase in mean arterial pressure, which also suggests decreased LV contractility at baseline and at 6 months post MV repair.
Previous studies in patients with isolated MR and LVEF > 55% have demonstrated decreases in the force-frequency effect in isolated LV muscle strips (9) and in LV calcium handling proteins (8), indicative of mechanical and biochemical markers of heart failure. In another study, LV TNF-α was increased in patients with isolated MR and well preserved LVEF (13). In our patients, there was marked deposition of lipofuscin, a non degradable material primarily composed of oxidatively modified protein and lipid degradation residues (24). Lipofuscin accumulation is usually seen in the senile heart and is considered to be an irreversible end product of excessive oxidative stress that overwhelms protective mechanisms (25). Lipofuscin accumulation has been shown to have deleterious effects on cellular function including triggering of mitochondrial pro-apoptotic pathways in cardiomyocytes and fibroblasts (26,27) and its accumulation in the heart is irreversible (24). Thus, it is tempting to speculate that the cumulative effects of prolonged oxidative stress and lipofuscin accumulation in the volume overloaded heart could account for LV contractile dysfunction at baseline and at 6 months post MV repair. It must be noted however that LV function in patients with aortic regurgitation and aortic stenosis may continue to improve for years following surgery, thus the definitive impact of our pre-op findings may be unknown at this time (28,29).
Another marker of heart failure was cardiomyocyte vacuolization with extensive myofibrillar degeneration. This has also been reported in the clinically relevant model of MR in the dog (30). Myofibrillar degeneration can occur as a result of increased oxidative stress and indeed degraded myofibrils may also constitute a part of lipofuscin. We also found increased levels of xanthine oxidoreductase (XOR), which when transformed from its parent enzyme XDH into its oxidase form XO, generates superoxide and hydrogen peroxide upon conversion of xanthine to hypoxanthine and hypoxanthine to uric acid, respectively (11). XO activity was upregulated in the LV of humans with dilated cardiomyopathy and intracoronary infusion of allopurinol improved LV contractile performance without increasing myocardial oxygen consumption(12). In vitro studies demonstrated that XO depresses myofilament sensitivity to calcium and that it colocalized with nitric oxide synthase-1 in the sarcoplasmic reticulum in the mouse cardiomyoctye, which could regulate excitation-contraction coupling as well as myofilament oxidative damage (31,32). Interestingly, staining of nitrotyrosine—a marker of oxidative and nitrosative damage—was increased in areas of myofibrillar degeneration and lipofuscin accumulation in the MR hearts. XO is a major source of superoxide and its combination with NO produces peroxynitrite. Thus, the proximity of XO to these markers of excessive nitration in the myocyte suggested a pathophysiological role for XO in the cardiomyocyte myofibrillar degeneration.
The current study cannot directly determine cause and effect of XO-mediated oxidative damage and the decrease in LV function post MV-repair. Nevertheless, the marked lipofuscin accumulation and myofibrillar degeneration in LV endomyocardial biopsies are indeed markers of heart failure that were present in patients with otherwise normal LV strain rates and LVEF prior to surgery. This underscores the unreliability of ejection phase indices in reflecting underlying LV myocardial damage on ours and other studies (8–10). These findings may be of use in defining the optimal timing of mitral valve surgery. Future studies are required to determine whether XO and persistent oxidative stress are causative in maladaptive LV remodeling and offer potential therapeutic targets in ameliorating LV damage in patients with isolated MR.
Acknowledgments
Funding Sources
This study was supported by: SCCOR in Cardiac Dysfunction P50HL077100.
Abbreviations
- ED
End diastolic
- EF
Ejection fraction
- ES
End systolic
- LV
Left ventricular
- MRI
Magnetic resonance imaging
- MV
Mitral valve
- XO
Xanthine oxidase
- XDH
Xanthine dehydrogenase
Footnotes
Disclosures
None
References
- 1.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borer JS, Bonow RO. Contemporary approach to aortic and mitral regurgitation. Circulation. 2003;108:2432–8. doi: 10.1161/01.CIR.0000096400.00562.A3. [DOI] [PubMed] [Google Scholar]
- 3.Bonow RO, Carabello BA, Chatterjee K, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing Committee to Revise the 1998 guidelines for the management of patients with valvular heart disease) developed in collaboration with the Society of Cardiovascular Anesthesiologists endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol. 2006;48:e1–148. doi: 10.1016/j.jacc.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 4.Le Tourneau T, de Groote P, Millaire A, et al. Effect of mitral valve surgery on exercise capacity, ventricular ejection fraction and neurohormonal activation in patients with severe mitral regurgitation. J Am Coll Cardiol. 2000;36:2263–9. doi: 10.1016/s0735-1097(00)01015-9. [DOI] [PubMed] [Google Scholar]
- 5.Madaric J, Watripont P, Bartunek J, et al. Effect of mitral valve repair on exercise tolerance in asymptomatic patients with organic mitral regurgitation. Am Heart J. 2007;154:180–5. doi: 10.1016/j.ahj.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura T, Ohtaki E, Tanaka K, et al. Echocardiographic prediction of left ventricular dysfunction after mitral valve repair for mitral regurgitation as an indicator to decide the optimal timing of repair. J Am Coll Cardiol. 2003;42:458–63. doi: 10.1016/s0735-1097(03)00649-1. [DOI] [PubMed] [Google Scholar]
- 7.Shyu KG, Chen JJ, Lin FY, et al. Regression of left ventricular mass after mitral valve repair of pure mitral regurgitation. Ann Thorac Surg. 1994;58:1670–3. doi: 10.1016/0003-4975(94)91656-x. [DOI] [PubMed] [Google Scholar]
- 8.Leszek P, Korewicki J, Klisiewicz A, et al. Reduced myocardial expression of calcium handling protein in patients with severe chronic mitral regurgitation. Eur J Cardiothorac Surg. 2006;30:737–43. doi: 10.1016/j.ejcts.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Mulieri LA, Leavitt BJ, Martin BJ, Haeberle JR, Alpert NR. Myocardial force-frequency defect in mitral regurgitation heart failure is reversed by forskolin. Circulation. 1993;88:2700–4. doi: 10.1161/01.cir.88.6.2700. [DOI] [PubMed] [Google Scholar]
- 10.Oral H, Sivasubramanian N, Dyke DB, et al. Myocardial proinflammatory cytokine expression and left ventricular remodeling in patients with chronic mitral regurgitation. Circulation. 2003;107:831–7. doi: 10.1161/01.cir.0000049745.38594.6d. [DOI] [PubMed] [Google Scholar]
- 11.Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cappola TP, Kass DA, Nelson GS, et al. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104:2407–11. doi: 10.1161/hc4501.098928. [DOI] [PubMed] [Google Scholar]
- 13.Abdulnour RE, Peng X, Finigan JH, et al. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am J Physiol Lung Cell Mol Physiol. 2006;291:L345–53. doi: 10.1152/ajplung.00453.2005. [DOI] [PubMed] [Google Scholar]
- 14.Denney TS, Jr, Gerber BL, Yan L. Unsupervised reconstruction of a three-dimensional left ventricular strain from parallel tagged cardiac images. Magn Reson Med. 2003;49:743–54. doi: 10.1002/mrm.10434. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Denney TS., Jr Left ventricular motion reconstruction with a prolate spheroidal B-spline model. Phys Med Biol. 2006;51:517–37. doi: 10.1088/0031-9155/51/3/004. [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Chen J, Ji S, et al. Harmonic phase MR tagging for direct quantification of Lagrangian strain in rat hearts after myocardial infarction. Magn Reson Med. 2004;52:1282–90. doi: 10.1002/mrm.20276. [DOI] [PubMed] [Google Scholar]
- 17.Osman NF, Kerwin WS, McVeigh ER, Prince JL. Cardiac motion tracking using CINE harmonic phase (HARP) magnetic resonance imaging. Magn Reson Med. 1999;42:1048–60. doi: 10.1002/(sici)1522-2594(199912)42:6<1048::aid-mrm9>3.0.co;2-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osman NF, Prince JL. Visualizing myocardial function using HARP MRI. Phys Med Biol. 2000;45:1665–82. doi: 10.1088/0031-9155/45/6/318. [DOI] [PubMed] [Google Scholar]
- 19.Cerqueira MD, Weissman NJ, Dilsizian V, et al. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart: a statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation. 2002;105:539–42. doi: 10.1161/hc0402.102975. [DOI] [PubMed] [Google Scholar]
- 20.Corin WJ, Sutsch G, Murakami T, Krogmann ON, Turina M, Hess OM. Left ventricular function in chronic mitral regurgitation: preoperative and postoperative comparison. J Am Coll Cardiol. 1995;25:113–21. doi: 10.1016/0735-1097(94)00354-s. [DOI] [PubMed] [Google Scholar]
- 21.Goldfine H, Aurigemma GP, Zile MR, Gaasch WH. Left ventricular length-force-shortening relations before and after surgical correction of chronic mitral regurgitation. J Am Coll Cardiol. 1998;31:180–5. doi: 10.1016/s0735-1097(97)00453-1. [DOI] [PubMed] [Google Scholar]
- 22.Solomon NA, Pranav SK, Naik D, Sukumaran S. Importance of preservation of chordal apparatus in mitral valve replacement. Expert Rev Cardiovasc Ther. 2006;4:253–61. doi: 10.1586/14779072.4.2.253. [DOI] [PubMed] [Google Scholar]
- 23.Carabello BA, Nolan SP, McGuire LB. Assessment of preoperative left ventricular function in patients with mitral regurgitation: value of the end-systolic wall stress-end-systolic volume ratio. Circulation. 1981;64:1212–7. doi: 10.1161/01.cir.64.6.1212. [DOI] [PubMed] [Google Scholar]
- 24.Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33:611–9. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 25.Terman A, Brunk UT. Ceroid/lipofuscin formation in cultured human fibroblasts: the role of oxidative stress and lysosomal proteolysis. Mech Ageing Dev. 1998;104:277–91. doi: 10.1016/s0047-6374(98)00073-6. [DOI] [PubMed] [Google Scholar]
- 26.Powell SR, Wang P, Divald A, et al. Aggregates of oxidized proteins (lipofuscin) induce apoptosis through proteasome inhibition and dysregulation of proapoptotic proteins. Free Radic Biol Med. 2005;38:1093–101. doi: 10.1016/j.freeradbiomed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Terman A, Dalen H, Brunk UT. Ceroid/lipofuscin-loaded human fibroblasts show decreased survival time and diminished autophagocytosis during amino acid starvation. Exp Gerontol. 1999;34:943–57. doi: 10.1016/s0531-5565(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 28.Borer JS, Herrold EM, Hochreiter C, et al. Natural history of left ventricular performance at rest and during exercise after aortic valve replacement for aortic regurgitation. Circulation. 1991;84:III133–9. [PubMed] [Google Scholar]
- 29.Villari B, Vassalli G, Monrad ES, Chiariello M, Turina M, Hess OM. Normalization of diastolic dysfunction in aortic stenosis late after valve replacement. Circulation. 1995;91:2353–8. doi: 10.1161/01.cir.91.9.2353. [DOI] [PubMed] [Google Scholar]
- 30.Spinale FG, Ishihra K, Zile M, DeFryte G, Crawford FA, Carabello BA. Structural basis for changes in left ventricular function and geometry because of chronic mitral regurgitation and after correction of volume overload. J Thorac Cardiovasc Surg. 1993;106:1147–57. [PubMed] [Google Scholar]
- 31.Khan SA, Lee K, Minhas KM, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez NG, Gao WD, Marban E. Novel myofilament Ca2+-sensitizing property of xanthine oxidase inhibitors. Circ Res. 1998;83:423–30. doi: 10.1161/01.res.83.4.423. [DOI] [PubMed] [Google Scholar]