

Abstract
Nerve growth factor (NGF) and the ubiquitous second messenger cyclic AMP(cAMP) are both implicated in neuronal differentiation. Multiple studies indicate that NGF signals to at least a subset of its targets via cAMP, but the link between NGF and cAMP has remained elusive. Here, we have described the use of small molecule inhibitors to differentiate between the two known sources of cAMP in mammalian cells, bicarbonate- and calcium-responsive soluble adenylyl cyclase (sAC) and G protein-regulated transmembrane adenylyl cyclases. These inhibitors, along with sAC-specific small interfering RNA, reveal that sAC is uniquely responsible for the NGF-elicited rise in cAMP and is essential for the NGF-induced activation of the small G protein Rap1 in PC12 cells. In contrast and as expected, transmembrane adenylyl cyclase-generated cAMP is responsible for Rap1 activation by the G protein-coupled receptor ligand PACAP (pituitary adenylyl cyclase-activating peptide). These results identify sAC as a mediator of NGF signaling and reveal the existence of distinct pathways leading to cAMP-dependent signal transduction.
The signaling mechanisms underlying neuronal differentiation have been extensively studied using the rat pheochromocytoma PC12 cell line as a model system. Upon treatment with nerve growth factor (NGF),2 PC12 cells differentiate into sympathetic-like neurons (1). Among the early steps in this signaling pathway is sustained activation of the mitogen-activated protein kinase cascade via two small GTP-binding proteins, Ras and Rap1, with Ras contributing to an early phase of mitogen-activated protein kinase activation and Rap1 mediating a later, sustained phase (2, 3). Interestingly, both Rap1 activation and the subsequent neuritogenesis can be elicited by cell-permeable analogues of cyclic adenosine monophosphate (cAMP) (4, 5). Consistent with involvement of this ubiquitous second messenger, several groups have reported transient NGF-induced cAMP elevations in both PC12 cells and primary neuronal cultures (6–10), and it was reported that NGF-induced activation of Rap1 is mediated by protein kinase A (cAMP-dependent protein kinase), a direct target of cAMP (11). Yet, the existence of a link between NGF and cAMP in neuronal differentiation has remained a subject of controversy, and the mechanism leading from NGF to the second messenger has never been adequately described.
Until recently, the only known sources of cAMP in mammalian cells were the transmembrane adenylyl cyclases (tmACs), which are integral plasma membrane proteins regulated by heterotrimeric G proteins. In PC12 cells, tmACs can be activated by pituitary adenylyl cyclase-activating peptide (PACAP) via its specific G protein-coupled receptor PAC1 (12). Similar to NGF, PACAP activates Rap1 and elicits neurite outgrowth (13–15), but all attempts to link NGF or its receptors to tmAC activation have been unsuccessful. We have identified an additional, ubiquitously expressed (17, 18) source of cAMP in mammalian cells, “soluble” adenylyl cyclase (sAC) (16). In contrast to tmACs, sAC is not regulated by heterotrimeric G proteins but is, instead, regulated by bicarbonate (19) and calcium (20, 21) ions. Also, in contrast to tmACs, sAC is not bound to the plasma membrane but is localized to various intracellular compartments (22), where it is thought to be the source of local pools of cAMP (23, 24). In sperm, bicarbonate regulation of sAC mediates a number of maturation events essential for fertilization (25, 26). In somatic tissues, bicarbonate regulation of sAC in epididymis permits clear cells to respond to luminal pH (27), and calcium regulation of sAC in neutrophils is essential for the tumor necrosis factor-induced respiratory burst (28). Tumor necrosis factor-induced respiratory burst has been proposed to occur via Rap1, and tumor necrosis factor activation of Rap1 in neutrophils appears to be mediated by sAC (28). We explored whether sAC might represent the source of cAMP leading to the activation of Rap1 in other contexts unexplained by heterotrimeric G protein activation of tmACs. In this report, we have demonstrated that sAC is present in PC12 cells and is, in fact, the link between NGF and cAMP leading to Rap1 activation. Furthermore, NGF activation of sAC is dependent upon elevations of intracellular calcium. Thus, we postulate the existence of a new paradigm in signal transduction, where the activation of a receptor tyrosine kinase stimulates cAMP production via sAC. Our findings reveal that cAMP can be generated by distinct sources, yet still converge onto a single effector cascade.
Materials and Methods
Cell Culture
PC12 cells were maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum, 5% donor horse serum, and antibiotics at 37 °C in 5% CO2. The cells were incubated in low serum (2% fetal bovine serum, 1% donor horse serum) for 16 h prior to treatment with 100 ng/ml NGF (Harlan Bioproducts), 200 μm PACAP (Sigma), or 60 mm KCl. For the bicarbonate experiments, the cells were incubated in HEPES-buffered bicarbonate-free medium for 1 h and then stimulated in medium containing 44 mm NaHCO3 at a constant pH. The inhibitor KH7 (corresponding to compound 1185–0408 from ChemDiv Inc.) (50 μm) and the P-site inhibitor 2′,5′-dideoxyadenosine (Sigma) (50 μm) were added 15 min prior to treatment with NGF, PACAP, or 1 mm 8-bromo-cAMP (Calbiochem). The intracellular calcium chelator BAPTA-AM (Sigma) (10 μm) was added 30 min prior to treatment with NGF.
Small Interfering RNA (siRNA) Reagents and Transfection
siRNA against sAC corresponded to nucleotides 692–710 relative to the first nucleotide of the start codon of rat sAC (GenBank™ accession number AF081941). As the control, we utilized the commercially available siRNA control nonsilencing sequence (Qiagen). Early passage PC12 cells were transfected with siRNA (control or sAC-specific) using the Gene Silencer transfection reagent (Gene Therapy Systems, San Diego, CA) following the manufacturer's protocol. Briefly, siRNA and the transfection reagent were both diluted in OptiMEM (Invitrogen) and then mixed for 15 min. The cells were incubated in fresh low serum medium and overlaid with the transfection mix. After 12–16 h, the medium was changed to normal serum-containing Dulbecco's modified Eagle's medium (see “Cell Culture”). The cells were used for experiments ∼48 h after siRNA transfection.
Immunocytochemistry
Cells were plated on coverslips coated with 0.01% poly-l-lysine (Sigma). For sAC immunolocalization, cells were washed in phosphate-buffered saline, fixed in 4% paraformaldehyde for 30 min, permeabilized in 0.1% Triton X-100 for 15 min, and then blocked for 3 h in 2% bovine serum albumin. Immunostaining for sAC was performed using R41 monoclonal antibody generated against the human 48-kDa isoform of sAC antigen and mapped to amino acids 450–466 of human sAC (22). Primary antibody staining was done overnight at 4 °C in 2% bovine serum albumin and 0.01% Triton X-100. The staining was visualized by incubation with goat anti-mouse Alexa Fluor 488 (Molecular Probes) for 1 h at room temperature. The coverslips were washed with phosphate-buffered saline and mounted with gelvatol/1,4-diazobicyclo-(2,2,2)octane (Sigma). Confocal images were acquired with a confocal system (model LSM 510; Carl Zeiss MicroImaging, Inc.) at the Rockefeller University Bioimaging Center.
cAMP Accumulation Assay
PC12 cells were seeded at a density of 2.0 × 105 in 24-well plates, serum-starved for 16 h (see “Cell Culture”), treated with 100 ng/ml NGF or 60 mm KCl and 500 μm 3-isobutyl-l-methylxanthine (Sigma) for 2 min or 200 μm PACAP and 3-isobutyl-l-methylxanthine for 5 min, and then lysed in 0.1 M HCl. Each condition contained the same concentration of vehicle (Me2SO). cAMP was quantitated in cell lysates using the Correlate-EIA cAMP assay (Assay Design, Inc.), and the results were analyzed using Prism software.
Rap1 and Ras Activation Assays
Activated Rap1 was isolated from cell lysates using an adapted protocol (29). Briefly, treated cells were lysed in 1 ml of ice-cold lysis buffer, and lysates were cleared by low speed centrifugation. After a small sample of each supernatant was set aside for total protein control, the remaining portions of supernatants were incubated with 40 μl of agarose-coupled GST-RalGDS-Rap1 binding domain (Upstate Biotechnology) for 1 h. The beads were rinsed three times with lysis buffer, and protein was eluted with Laemmli sample loading buffer. Activated Ras was isolated from cell lysates in a similar fashion using agarose-coupled GST-Raf1-Ras binding domain (Upstate Biotechnology). The amount of activated Rap1 or Ras bound to the beads was detected by Western blotting using Rap1 or Ras antisera (see next paragraph).
Western Blotting
Samples were boiled for 5 min, separated under reducing conditions using SDS-PAGE, transferred to polyvinylidene difluoride membrane, and blocked in 5% nonfat dry milk. The blots were probed with polyclonal antiserum against Rap1 (Santa Cruz Biotechnol-ogy), monoclonal antibody against Ras (Upstate Biotechnology), or monoclonal antibody against 48-kDa human sAC antigen. Anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibodies were used and proteins were detected by enhanced chemiluminescence (Bio-Rad). Spot densitometry was performed on multiple Western blots using a Fluorchem 8800 Imager (Alpha Innotech, San Leandro, CA), and the results were analyzed using Prism software.
Results
NGF Signals via Calcium to Raise Intracellular cAMP and Activate Rap1
Although cAMP was first suggested to mediate the actions of NGF in PC12 cells over 25 years ago (8), the link between NGF and this second messenger and its importance to NGF signaling have remained subjects of controversy. Recent studies revealing that sustained activation of Rap1 by NGF is dependent upon protein kinase A (cAMP-dependent protein kinase) (11) confirm the importance of cAMP, yet the question of its source remains.
We measured increased cAMP levels in PC12 cells within 2 min of NGF addition (Fig. 1A). It was recently postulated that neurotrophins may elevate this second messenger through extracellular signal-regulated kinase (ERK)-dependent inhibition of cAMP-catabolizing phosphodiesterases (9). However, because we performed our experiment in the presence of the broad specificity phosphodiesterase inhibitor 3-isobutyl-l-methylxanthine, modulation of phosphodiesterase activity was not likely to be involved. These data implicate modulation of adeny-lyl cyclase activity for the actions of NGF.
FIGURE 1. NGF elevates cAMP and activates Rap1 in a calcium-dependent manner.
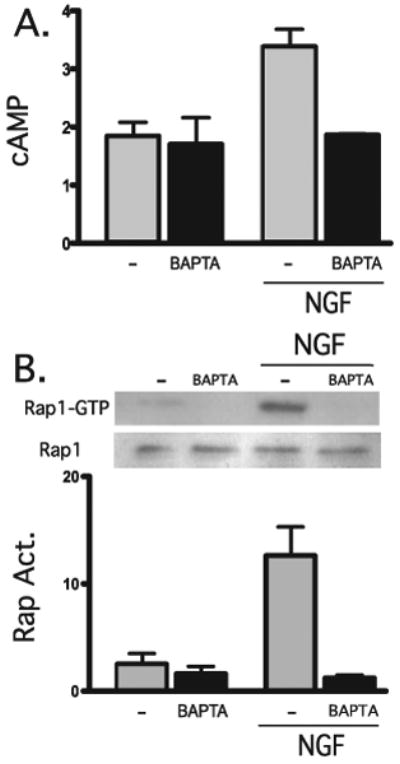
A, accumulated cAMP was measured in cells in the presence or absence of NGF (100 ng/ml) for 2 min, in the presence or absence of BAPTA-AM (1 μm, 15-min pretreatment). Shown are averages of triplicate determinations of a representative experiment (repeated three times). B, PC12 cells were incubated for 15 min in the presence or absence of BAPTA-AM (1 μm) and then incubated for an additional 15 min in the presence or absence of NGF (100 ng/ml). Activated Rap1 (Rap Act.), pulled down by GST-RalGDS (top) or total Rap1 (bottom) were detected by Western blot using Rap1-specific antisera. Western blot data from three separate experiments were quantified by spot densitometry (bar graph); activated Rap1 was calculated as a percentage of total Rap1 for each condition. The results were analyzed using Prism software.
Importantly, we found the cAMP elevation in response to NGF to be abolished by preincubation with the intracellular calcium chelator BAPTA-AM (Fig. 1A). Furthermore, calcium chelation with BAPTA-AM also inhibited the NGF-induced sustained activation of Rap1 (Fig. 1B). These results indicate that both the NGF-induced stimulation of adenylyl cyclase activity and the subsequent Rap1 activation occur in a calcium-dependent manner.
NGF-induced Rap1 Activation Is Not Affected by Inhibition of tmACs
There are two known sources of cAMP in mammalian cells, heterotrimeric G protein-regulated tmACs and the bicarbonate- and calcium-responsive sAC. TmACs are ubiquitously expressed, and their presence in PC12 cells was demonstrated biochemically and molecularly (30, 31). In fact, they were shown to mediate the actions of PACAP (which is known to activate Rap1) in PC12 cells (14). To test whether NGF-induced activation of Rap1 is also mediated by tmACs, we assayed Rap1 activation by NGF and PACAP in the presence of a selective tmAC inhibitor.
Several adenosine analogues, known as P site ligands, have been characterized as inhibitors of tmAC activity (32, 33). These compounds inhibit the activity of each tmAC isoform with low micromolar affinities, but they display diminished potency toward sAC (34). We found that the P site ligand 2′5′dideoxyadenosine (2′5′ddAdo) inhibits sAC activity with >100-fold reduced affinity relative to tmACs (data not shown). In PC12 cells, the tmAC-selective P site inhibitor 2′5′ ddAdo completely blocked PACAP-induced Rap1 activation, but it had no effect on NGF-induced activation (Fig. 2). These data suggested that NGF signals through another cAMP source, prompting us to explore the involvement of sAC.
FIGURE 2. PACAP (but not NGF) activates Rap1 via tmACs.
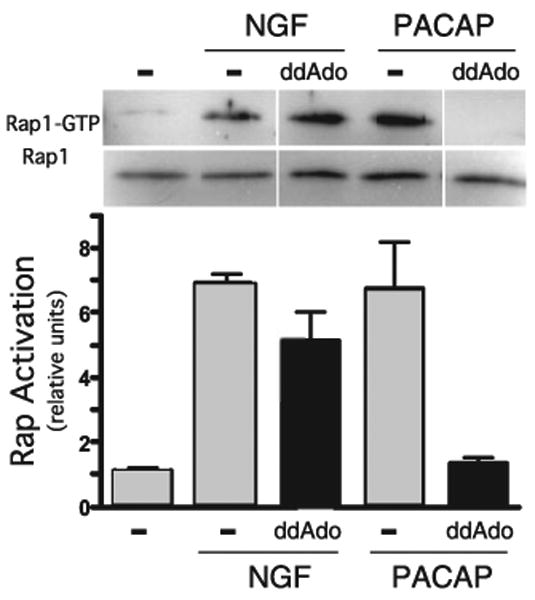
PC12 cells pretreated for 15 min with 2′,5′-ddAdo (50 μm) or Me2SO (vehicle control) were stimulated for 15 min with NGF (100 ng/ml) or PACAP (200 nm). Rap1 activation was quantified as in Fig. 1 by GST pull-down followed by Western blot. Western blot data from three separate experiments were quantified by spot densitometry (bar graph); activated Rap1 was calculated as a percentage of total Rap1 for each condition. The results were analyzed using Prism software.
Presence and Localization of sAC in PC12 Cells
Similar to tmACs, sAC is widely expressed (17, 18, 35). We confirmed sAC mRNA expression in PC12 cells by reverse transcription-PCR (data not shown) and protein expression by immunoblotting, using a sAC-specific monoclonal antibody. Recent studies in our (data not shown) and others' laboratories (17, 35) reveal complex patterns of alternative splicing leading to multiple isoforms of sAC protein in somatic tissues. Western blotting identified a single protein band of ∼70 kDa (Fig. 3A) corresponding to a sAC isoform detected in other cell lines (35). We also used our antibody to examine sAC localization in PC12 cells. We and others previously demonstrated sAC to be localized to the cytoplasm, as well as the mitochondria, centrosomes, and nuclei in multiple cell lines and tissues (22, 24, 36). In PC12 cells, we observed predominantly cytoplasmic staining, with the strongest intensity around the periphery (Fig. 3B), consistent with the proximity to plasma membrane-bound NGF receptors.
FIGURE 3. Presence and functionality of sAC in PC12 cells.
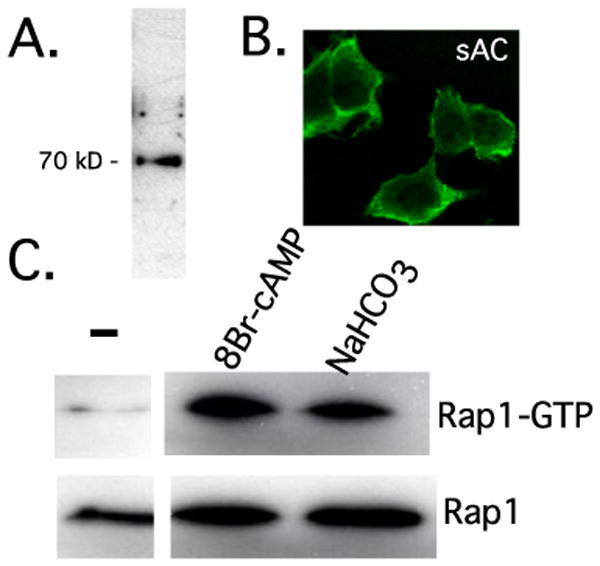
A, Western blot using a monoclonal antibody specific for sAC (R21). B, localization of sAC was examined by immunostaining using sAC monoclonal antibody R41 and visualized using confocal microscopy. C, activation of Rap1 by sAC-generated cAMP. Cells were starved for 1 h in bicarbonate-free medium and then stimulated for 5 min in medium containing 44 mm NaHCO3−. Rap1 GTP loading was quantified by GST pull-down/Western blot. PC12 cells were treated with the nonhydrolyzable analogue 8-bromo-cAMP (1 mm) as the positive control for Rap1 activation.
We next explored whether sAC could generate functionally relevant cAMP in PC12 cells. Among mammalian adenylyl cyclases, sAC is uniquely bicarbonate-stimulated (19, 21, 36, 37), and cAMP production via sAC represents the only known signaling effect initiated by bicarbonate (24). Generation of cAMP can lead to activation of Rap1 (5, 11, 38, 39), and we tested whether bicarbonate could stimulate Rap1 activation in PC12 cells. Assaying for GTP-loaded Rap1, we observed bicarbonate-induced Rap1 activation (Fig. 3C). These data indicate that sAC is present in PC12 cells and that sAC-generated cAMP is capable of activating Rap1.
sAC Uniquely Mediates the Activation of Rap1 Due to Depolarization-induced Calcium Influx
Depolarization-induced calcium influx has been shown to activate Rap1 via the cAMP target (cAMP-dependent protein kinase) in both PC12 cells and hippocampal neurons (40). Previously, calcium had been identified as an activator of sAC (20, 21, 28, 41). On the other hand, calcium is also known to regulate several types of tmACs, some of which are expressed in PC12 cells (31). We asked whether sAC, tmACs, or both contribute to the calcium influx-induced Rap1 activation.
To distinguish between the two sources of cAMP, we employed the tmAC-selective inhibitor 2′5′ddAdo and a small molecule sAC-specific inhibitor, KH7 (26). Importantly, KH7 is inert toward tmACs both in vitro and in whole cell contexts at concentrations up to 300 μm. As predicted, KCl depolarization-induced calcium influx resulted in a strong elevation in intracellular cAMP. This cAMP rise was partially blocked by 2′5′ddAdo as well as by KH7 (Fig. 4A), confirming the presence of both calcium-responsive tmACs and calcium-responsive sAC. Yet, examining the calcium influx-induced Rap1 activation, we found that it was completely inhibited by KH7 and unaffected by 2′5′ddAdo (Fig. 4B). Thus, sAC (but not tmACs) appeared to be involved in the activation of Rap1 due to calcium. This finding indicates the existence of at least three distinct pools of cAMP in PC12 cells, sAC-generated cAMP and PACAP-responsive tmAC-generated cAMP (both capable of activating Rap1) and calcium-responsive tmAC-generated cAMP (incapable of activating Rap1).
FIGURE 4. KCl depolarization-induced calcium influx raises cAMP via sAC and tmACs and activates Rap1 via sAC only.
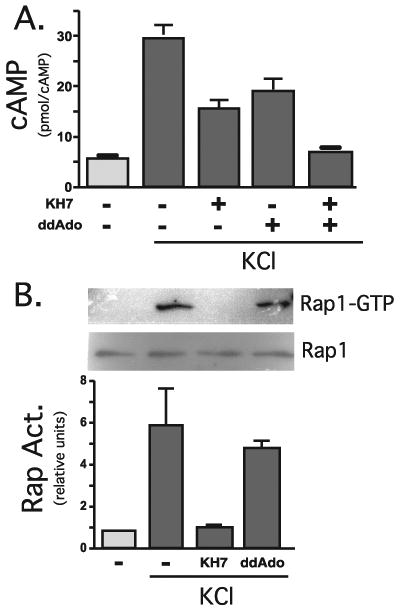
A, accumulated cAMP was measured in cells in the presence or absence of KCl (60 mm) for 2 min, in the presence of KH7 (50 μm), 2′5′dideoxyadenosine (50 μm), or Me2SO. Shown are averages of duplicate determinations of a representative experiment (repeated three times). B, PC12 cells pretreated for 15 min with KH7 (50 μm), 2′5′dideoxyadenosine (50 μm), or Me2SO were incubated for an additional 15 min in the presence or absence of KCl (60 mm). Rap1 activation was quantified by GST pull-down followed by Western blot. Western blot data from three separate experiments were quantified by spot densitometry (bar graph); activated Rap1 was calculated as the percentage of total Rap1 for each condition. The results were analyzed using Prism software.
sAC Is the Source of NGF-elicited cAMP
In contrast to depolarization-induced cAMP production, the elevation of cAMP elicited by NGF was completely blocked by KH7 (Fig. 5A). We confirmed sAC to be the source of cAMP generated in response to NGF using knockdown by siRNAs, which have been demonstrated to diminish sAC protein in neurons3 and cell lines.4 As seen with KH7, the NGF-induced cAMP increase was completely blocked by sAC-specific RNA interference (Fig. 5B). Finally, KH7 did not inhibit the cAMP increase due to PACAP (Fig. 5C), confirming that NGF and PACAP signal via distinct sources of cAMP.
FIGURE 5. NGF elevates cAMP in PC12 cells via sAC.

A, accumulated cAMP was measured in cells in the presence or absence of NGF (100 ng/ml) for 2 min and in the presence or absence of KH7 (50 μm). Shown are averages of quadruplicate determinations of a representative experiment (repeated three times). B, basal and NGF (2 min)-stimulated cAMP levels were measured in cells transfected with sAC-specific RNA interference or scrambled control. Shown are averages of duplicate determinations of a representative experiment (repeated three times). C, accumulated cAMP was measured in cells in the presence or absence of PACAP (200 nm) for 5 min, in the presence or absence of 2′5′dideoxyadenosine (50 μm), or KH7 (50 μm). Shown are averages of triplicate determinations of a representative experiment (repeated five times).
sAC Is Essential for the Sustained NGF-induced Activation of Rap1
Because NGF elicits cAMP elevation via sAC (Fig. 5) and sAC-generated cAMP can activate Rap1 (Fig. 3C), we asked whether sAC mediates NGF-induced sustained Rap1 activation. Indeed, the sustained NGF activation of Rap1 was abolished by the sAC-specific inhibitor KH7 (Fig. 6A). KH7 inhibition was dose-dependent (displaying an IC50 value of ∼25 μm (Fig. 6B)) and specific; i.e. it was rescued by 8-bromo-cAMP (Fig. 6C). The involvement of sAC was confirmed using RNA interference; knockdown of sAC by a gene-specific siRNA inhibited NGF-induced Rap1 activation, whereas scrambled siRNA control had no effect (Fig. 6D).
FIGURE 6. NGF-induced sustained Rap1 activation is specifically inhibited by KH7.
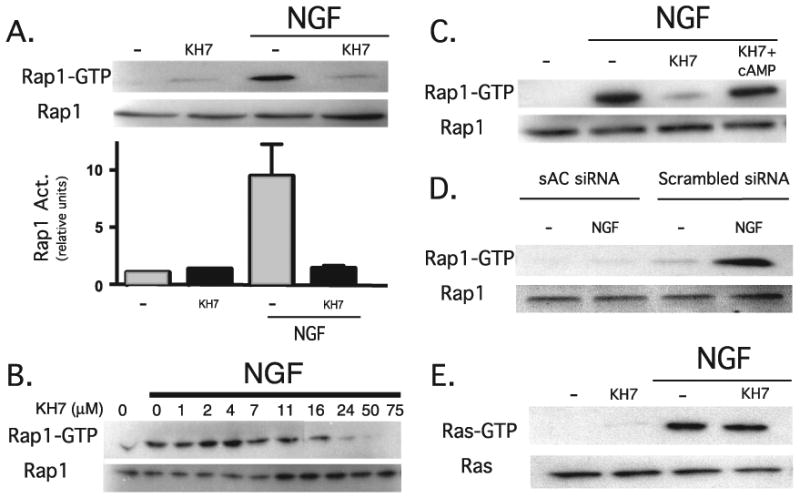
A, PC12 cells pretreated for 15 min with KH7 (50 μm) or Me2SO were incubated for an additional 15 min in the presence or absence of NGF (100 ng/ml). Rap1 activation was quantified by GST pull-down followed by Western blot. Western blot data from five separate experiments were quantified by spot densitometry (bar graph); activated Rap1 was calculated as a percentage of total Rap1 for each condition. The results were analyzed using Prism software. B, PC12 cells pretreated for 15 min with Me2SO or increasing doses of KH7 were incubated for an additional 15 min in the presence or absence of NGF (100 ng/ml). Rap1 activation was quantified by GST pull-down followed by Western blot. C, cells pretreated for 15 min with KH7 (50μm) or Me2SO were incubated for an additional 15 min in the presence of NGF (100 ng/ml) ± 8-bromo-cAMP (1 mm). Rap1 activation was quantified by GST pull-down followed by Western blot. D, 48 h after PC12 cells were transfected with sAC-specific RNA interference or scrambled control, the cells were incubated for 15 min in the presence or absence of NGF (100 ng/ml). Rap1 activation was quantified by GST pull-down followed by Western blot. E, cells pretreated for 15 min with KH7 (50 μm) or Me2SO were incubated for an additional 5 min in the presence or absence of NGF (100 ng/ml). Activated Ras, pulled down by GST-Raf (top), or total Ras (bottom) were detected by Western blot using Ras-specific antisera.
We used KH7 to examine the role of sAC in other NGF-induced effects, such as whether sAC was specifically involved in NGF-induced activation of Ras. In contrast to Rap1, NGF-induced activation of Ras was not inhibited by KH7 (Fig. 6E).
sAC Is Also Essential for the Early Phase NGF-induced Rap1 Activation
Having examined the effects of sAC inhibition on the sustained phase of NGF-induced Rap1 activation, we asked whether sAC is also required for the earlier phases of this process. In fact, we found that KH7 blocked the NGF-induced Rap1 activation at all of the times measured, including the earliest time point, 5 min after NGF addition (Fig. 7). Thus, sAC appears to be essential for all phases of Rap1 activation by NGF.
FIGURE 7. The early phase of NGF-induced Rap1 activation is also inhibited by KH7.
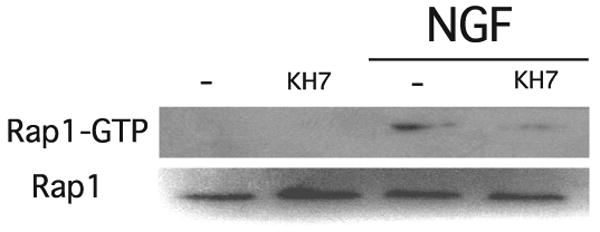
PC12 cells pretreated for 15 min with KH7 (50 μm) or Me2SO were incubated for an additional 5 min in the presence or absence of NGF (100 ng/ml). Rap1 activation was quantified by GST pull-down followed by Western blot.
Discussion
Use of selective small molecule inhibitors for sAC and tmAC enables us to differentiate between two distinct sources of cAMP. In the current study, it also provides evidence for a model that demonstrates two novel paradigms in signal transduction. First, two parallel signaling cassettes converge on the same target, the cAMP-activated GTPase Rap1, and they appear to differentially modulate Rap1 activity via distinct sources of cAMP. Our data reveal that PACAP activates Rap1 via tmAC-generated cAMP, whereas NGF activates Rap1 via sAC. Second, NGF is shown for the first time to signal directly through the second messenger cAMP via adenylyl cyclase activation. It was previously reported that NGF can elevate cAMP levels within 1 s in the membranes of PC12 cells (6). These investigators proposed cross-talk between the low affinity neurotrophin receptor p75NTR and a G protein-mediated cascade. However, our data show that NGF-induced Rap1 activation is insensitive to tmAC inhibition, revealing that NGF signaling does not involve heterotrimeric G proteins. A more recent study in PC12 cells indicates that NGF activation of Rap1 is dependent upon the high affinity receptor TrkA rather than p75NTR (11). Combining this finding with the data reported here yields a novel signaling axis that involves an adenylyl cyclase acting downstream of a receptor tyrosine kinase.
The actions of cAMP are mediated by multiple effector proteins, which are localized to different subcellular compartments, including the plasma membrane, cytosol, mitochondria, and the nucleus (22–24). Previously it was thought that cAMP is generated exclusively by tmACs at the plasma membrane and must, therefore, diffuse throughout the cell to reach its targets. Considering the multitude of intracellular processes that involve cAMP, the diffusion-dependent model fails to provide a mechanism for maintaining signal specificity and selectivity, particularly when signaling to distal effectors. Additionally, the actions of cAMP-catabolizing phosphodiesterases limit the propagation of cAMP signals throughout the cell. The discovery of sAC as a second source of cAMP in mammals and its localization to multiple subcellular compartments (22) provide the basis for a different model wherein cAMP, its source, and its effector form a single second messenger microdomain. Recent studies utilizing fluorescence resonance energy transfer to examine the dynamics of cAMP signaling in living cells, have shown local cAMP elevations to be far greater than the increases detected in whole cells (42), thus supporting the microdomain model. Our data reveal there to be three distinct pools of cAMP in PC12 cells, sAC-generated cAMP capable of activating Rap1, PACAP-responsive tmAC-generated cAMP capable of activating Rap1, and calcium-responsive tmAC-generated cAMP of unknown function. Clearly, these three microdomains are involved in different signal transduction pathways. Their individual roles and the question of whether cross-talk exists between them are subjects for future investigations.
Ultimately, the role of sAC-mediated signaling in neurite outgrowth-related phenomena must be examined. Recent studies in our laboratory (data not shown) suggest that sAC is necessary for the previously described NGF and phorbol ester-dependent fast neuronal migration in PC12 cells (43). Of more physiological interest, however, are the potential correlations with neuritogenesis in primary neuronal cultures. Neurotrophin-induced cAMP elevations have been shown to overcome the inhibition of axonal regeneration by myelin-associated glycoprotein in cerebellar neurons (9), and in Xenopus spinal neurons, cAMP has been implicated in the process of brain-derived neurotrophic factor-mediated axonal guidance (44, 45). Our development of selective inhibitors to differentiate between distinct sources of cAMP opens new avenues for future research in this quickly developing field.
Acknowledgments
We thank Drs. Lori Stohl and John Wagner for PC12 cells and Drs. Marie T. Filbin and Francis Lee for advice. We thank Chemical Diversity, Inc., Dr. Peter Meltzer, and Organix Inc. for synthesis of KH7. We also thank the Levin/Buck laboratory for critical reading of the manuscript.
Footnotes
This work was supported by Medical Scientist Training Program funding (to A. M. S. and J. H. Z.), National Institutes of Health Grants HD42060 and GM62328, the Ellison Medical Foundation (to J. B.), National Institutes of Health Grants HD38722 and AI64842, American Diabetes Association, and the Hirschl Weil-Caulier Trust (to L. R. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: NGF, nerve growth factor; cAMP, cyclic AMP; sAC, soluble adenylyl cyclase; tmAC, transmembrane adenylyl cyclase; PACAP, pituitary adenylyl cyclase-activating peptide; GST, glutathione S-transferase; siRNA, small interfering RNA; BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis.
K. Wu, M. Kamenetsky, J. Zippin, U. Hengst, J. Buck, L. R. Levin, and S. R. Jaffrey, submitted for publication.
M. Kamenetsky, L. R. Levin, J Buck, and J. H. Zippin, submitted for publication.
References
- 1.Dichter MA, Tischler AS, Greene LA. Nature. 1977;268:501–504. doi: 10.1038/268501a0. [DOI] [PubMed] [Google Scholar]
- 2.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ. Nature. 1998;392:622–626. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 3.Lu L, Anneren C, Reedquist KA, Bos JL, Welsh M. Exp Cell Res. 2000;259:370–377. doi: 10.1006/excr.2000.4984. [DOI] [PubMed] [Google Scholar]
- 4.Heidemann SR, Joshi HC, Schechter A, Fletcher JR, Bothwell M. J Cell Biol. 1985;100:916–927. doi: 10.1083/jcb.100.3.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vossler M, Yao H, York R, Pan M, Rim C, Stork P. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 6.Knipper M, Beck A, Rylett J, Breer H. FEBS Lett. 1993;324:147–152. doi: 10.1016/0014-5793(93)81382-a. [DOI] [PubMed] [Google Scholar]
- 7.Severin ES, Kondratyev AD. Adv Enzyme Regul. 1988;27:357–370. doi: 10.1016/0065-2571(88)90026-x. [DOI] [PubMed] [Google Scholar]
- 8.Schubert D, Whitlock C. Proc Natl Acad Sci U S A. 1977;74:4055–4058. doi: 10.1073/pnas.74.9.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Y, Nikulina E, Mellado W, Filbin MT. J Neurosci. 2003;23:11770–11777. doi: 10.1523/JNEUROSCI.23-37-11770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higuchi H, Yamashita T, Yoshikawa H, Tohyama M. EMBO J. 2003;22:1790–1800. doi: 10.1093/emboj/cdg177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obara Y, Labudda K, Dillon TJ, Stork PJ. J Cell Sci. 2004;117:6085–6094. doi: 10.1242/jcs.01527. [DOI] [PubMed] [Google Scholar]
- 12.Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, Journot L. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- 13.Barrie AP, Clohessy AM, Buensuceso CS, Rogers MV, Allen JM. J Biol Chem. 1997;272:19666–19671. doi: 10.1074/jbc.272.32.19666. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez A, Kimball B, Romanchuk G, Mulholland MW. Peptides (Elmsford) 1995;16:927–932. doi: 10.1016/0196-9781(95)00059-s. [DOI] [PubMed] [Google Scholar]
- 15.Okamura N, Onoe S, Kawakura K, Tajima Y, Sugita Y. Biochim Biophys Acta. 1990;1035:83–89. doi: 10.1016/0304-4165(90)90177-x. [DOI] [PubMed] [Google Scholar]
- 16.Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. Proc Natl Acad Sci U S A. 1999;96:79–84. doi: 10.1073/pnas.96.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, Pak CY. J Clin Endocrinol Metab. 2002;87:1476–1485. doi: 10.1210/jcem.87.4.8300. [DOI] [PubMed] [Google Scholar]
- 18.Sinclair ML, Wang XY, Mattia M, Conti M, Buck J, Wolgemuth DJ, Levin LR. Mol Reprod Dev. 2000;56:6–11. doi: 10.1002/(SICI)1098-2795(200005)56:1<6::AID-MRD2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Science. 2000;289:625–628. doi: 10.1126/science.289.5479.625. [DOI] [PubMed] [Google Scholar]
- 20.Jaiswal BS, Conti M. Proc Natl Acad Sci U S A. 2003;100:10676–10681. doi: 10.1073/pnas.1831008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. J Biol Chem. 2003;278:15922–15926. doi: 10.1074/jbc.M212475200. [DOI] [PubMed] [Google Scholar]
- 22.Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. FASEB J. 2003;17:82–84. doi: 10.1096/fj.02-0598fje. [DOI] [PubMed] [Google Scholar]
- 23.Bundey RA, Insel PA. Science's STKE 2004. 2004;231:pe19. doi: 10.1126/stke.2312004pe19. [DOI] [PubMed] [Google Scholar]
- 24.Zippin JH, Farrell J, Huron D, Kamenetsky M, Hess KC, Fischman DA, Levin LR, Buck J. J Cell Biol. 2004;164:527–534. doi: 10.1083/jcb.200311119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, Strik AM, Kuil C, Philipsen RL, Van Duin M, Conti M, Gossen JA. Proc Natl Acad Sci U S A. 2004;101:2993–2998. doi: 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess KC, Jones BH, Marquez B, Chen Y, Ord TS, Kamenetsky M, Miyamoto C, Zippin JH, Kopf GS, Suarez SS, Levin LR, Williams CJ, Buck J, Moss SB. Dev Cell. 2005;9:249–259. doi: 10.1016/j.devcel.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. J Biol Chem. 2003;278:49523–49529. doi: 10.1074/jbc.M309543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han H, Stessin A, Roberts J, Hess K, Gautam N, Kamenetsky M, Lou O, Hyde E, Nathan N, Muller WA, Buck J, Levin LR, Nathan C. J Exp Med. 2005;202:353–361. doi: 10.1084/jem.20050778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franke B, Akkerman JW, Bos JL. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai HL, Yang TH, Messing RO, Ching YH, Lin SC, Chern Y. J Biol Chem. 1997;272:4970–4977. doi: 10.1074/jbc.272.8.4970. [DOI] [PubMed] [Google Scholar]
- 31.Xia Z, Choi EJ, Wang F, Storm DR. Neurosci Lett. 1992;144:169–173. doi: 10.1016/0304-3940(92)90742-p. [DOI] [PubMed] [Google Scholar]
- 32.Johnson RA, Shoshani I. Methods Enzymol. 1994;238:56–71. doi: 10.1016/0076-6879(94)38006-6. [DOI] [PubMed] [Google Scholar]
- 33.Johnson RA, Yeung SM, Stubner D, Bushfield M, Shoshani I. Mol Pharmacol. 1989;35:681–688. [PubMed] [Google Scholar]
- 34.Gille A, Lushington GH, Mou TC, Doughty MB, Johnson RA, Seifert R. J Biol Chem. 2004;279:19955–19969. doi: 10.1074/jbc.M312560200. [DOI] [PubMed] [Google Scholar]
- 35.Geng W, Wang Z, Zhang J, Reed BY, Pak CY, Moe OW. Am J Physiol. 2005;288:C1305–C1316. doi: 10.1152/ajpcell.00584.2004. [DOI] [PubMed] [Google Scholar]
- 36.Jaiswal BS, Conti M. J Biol Chem. 2001;276:31698–31708. doi: 10.1074/jbc.M011698200. [DOI] [PubMed] [Google Scholar]
- 37.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 38.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 39.Braun T. Proc Soc Exp Biol Med. 1990;194:58–63. doi: 10.3181/00379727-194-43055. [DOI] [PubMed] [Google Scholar]
- 40.Horgan AM, York RD, Withers GS, Banker GA, Stork PJ. J Biol Chem. 2000;275:3722–3728. doi: 10.1074/jbc.275.5.3722. [DOI] [PubMed] [Google Scholar]
- 41.Steegborn C, Litvin TN, Hess KC, Capper AB, Taussig R, Buck J, Levin LR, Wu H. J Biol Chem. 2005;280:31754–31759. doi: 10.1074/jbc.M507144200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaccolo M, Pozzan T. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 43.Glowacka D, Ginty DD, Wagner JA. J Neurosci Res. 1992;31:263–272. doi: 10.1002/jnr.490310207. [DOI] [PubMed] [Google Scholar]
- 44.Song HJ, Ming GL, Poo MM. Nature. 1997;388:275–279. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- 45.Ming GL, Song HJ, Berninger B, Holt CE, Tessier-Lavigne M, Poo MM. Neuron. 1997;19:1225–1235. doi: 10.1016/s0896-6273(00)80414-6. [DOI] [PubMed] [Google Scholar]