

Abstract
With aging, multiple Ca2+-associated electrophysiological processes exhibit increased magnitude in hippocampal pyramidal neurons, including the Ca2+-dependent slow afterhyperpolarization (sAHP), L-type voltage-gated Ca2+ channel (L-VGCC) activity, Ca2+-induced Ca2+ release (CICR) from ryanodine receptors (RyRs), and Ca2+ transients. This pattern of Ca2+ dysregulation correlates with reduced neuronal excitability/plasticity and impaired learning/memory and has been proposed to contribute to unhealthy brain aging and Alzheimer's disease. However, little is known about the underlying molecular mechanisms. In cardiomyocytes, FK506-binding protein 1b/12.6 (FKBP1b) binds and stabilizes RyR2 in the closed state, inhibiting RyR-mediated Ca2+ release. Moreover, we recently found that hippocampal Fkbp1b expression is downregulated, whereas Ryr2 and Frap1/Mtor (mammalian target of rapamycin) expression is upregulated with aging in rats. Here, we tested the hypothesis that disrupting FKBP1b function also destabilizes Ca2+ homeostasis in hippocampal neurons and is sufficient to induce the aging phenotype of Ca2+ dysregulation in young animals. Selective knockdown of Fkbp1b with interfering RNA in vitro (96 h) enhanced voltage-gated Ca2+ current in cultured neurons, whereas in vivo Fkbp1b knockdown by microinjection of viral vector (3–4 weeks) dramatically increased the sAHP in hippocampal slice neurons from young-adult rats. Rapamycin, which displaces FKBP1b from RyRs in myocytes, similarly enhanced VGCC current and the sAHP and also increased CICR. Moreover, FKBP1b knockdown in vivo was associated with upregulation of RyR2 and mTOR protein expression. Thus, disruption of FKBP1b recapitulated much of the Ca2+-dysregulation aging phenotype in young rat hippocampus, supporting a novel hypothesis that declining FKBP function plays a major role in unhealthy brain aging.
Introduction
Substantial evidence indicates that, with aging, multiple Ca2+-associated electrophysiological processes are increased in magnitude in hippocampal pyramidal neurons, including the Ca2+-dependent slow afterhyperpolarization (sAHP), the Ca2+ action potential (AP), L-type voltage-gated Ca2+ channel (L-VGCC) activity, Na+ action potential failure (accommodation), synaptic depression, Ca2+-induced Ca2+ release (CICR) by the Ca2+-sensing/releasing ryanodine receptors (RyRs) of the smooth endoplasmic reticulum, and voltage-activated Ca2+ transients (Landfield and Pitler, 1984; Moyer et al., 1992; Disterhoft et al., 1996; Thibault and Landfield, 1996; Norris et al., 1998; Thibault et al., 2001, 2007; Clodfelter et al., 2002; Hemond and Jaffe, 2005; Kumar and Foster, 2005; Gant et al., 2006). This pattern of aging-related Ca2+ dysregulation has been correlated with impaired plasticity and learning/memory (Disterhoft et al., 1996; Thibault and Landfield, 1996; Tombaugh et al., 2005; Tonkikh et al., 2006; Disterhoft and Oh, 2007). Furthermore, increased neuronal RyR activity also appears to develop in transgenic mouse models of Alzheimer's disease (AD) (Stutzmann et al., 2007; Bezprozvanny and Mattson, 2008). Conversely, hippocampal long-term potentiation (LTP), which depends more on Ca2+ influx via NMDA receptors than VGCCs, is generally decreased or unchanged in aged animals (Watabe and O'Dell, 2003; Kumar and Foster, 2004; Burke and Barnes, 2006; Lynch et al., 2006; Magnusson et al., 2006; Smith et al., 2009). Altered Ca2+ regulation with aging, although of different patterns, is also found in nonpyramidal neurons (Michaelis et al., 1984; Gibson and Peterson, 1987; Reynolds and Carlen, 1989; Kirischuk et al., 1996; Murchison and Griffith, 2007). Together, these and other findings in aged animals have suggested the hypothesis that Ca2+ dysregulation is a major factor in unhealthy brain aging and AD (Gibson and Peterson, 1987; Landfield, 1987; Khachaturian, 1989; Disterhoft et al., 1996; Toescu and Verkhratsky, 2003; Disterhoft and Oh, 2007; Murchison and Griffith, 2007; Thibault et al., 2007).
Nevertheless, little is known about the molecular mechanisms that underlie aging changes in Ca2+ regulation in pyramidal neurons. In myocytes, the immunophilins FK506-binding protein 1a and 1b (FKBP1a and FKBP1b, also called FKBP12 and FKBP12.6) normally bind and stabilize RyR channel complexes in the closed state, thereby inhibiting intracellular Ca2+ release (Lehnart et al., 2003; Zalk et al., 2007). In particular, FKBP1b binds with high affinity to RyR2, the most abundant RyR isoform in brain and heart. In recent microarray studies, we found that expression levels of Fkbp1a and Fkbp1b are downregulated with aging in hippocampus of rats (Kadish et al., 2009, their Table 3). Thus, we tested here whether disrupting FKBP1b function in hippocampal neurons of young animals might be sufficient to recapitulate substantial aspects of the aging phenotype of Ca2+ dysregulation (Gant et al., 2006).
Table 3.
Basic experimental properties for all recorded CA1 neurons
| Control (n = 16) | Rapamycin (n = 21) | |
|---|---|---|
| Input resistance (MΩ) | 53.85 ± 6.53 | 50.31 ± 3.14 |
| Resting membrane potential (mV) | −61.67 ± 0.71 | −62.83 ± 0.70 |
| Action potential amplitude (mV) | 81.43 ± 1.79 | 82.03 ± 1.53 |
| Electrode resistance (MΩ) | 116.11 ± 6.38 | 112.50 ± 5.45 |
| RSS hyperpolarization (mV) | −5.4 ± 0.77 | −4.89 ± 0.42 |
| RSS first EPSP (mV) | 5.95 ± 1.10 | 4.55 ± 0.46* |
No differences were found except in baseline EPSP amplitude.
*p < 0.05; means ± SEM.
To disrupt FKBP1b function, we used in vitro and in vivo selective knockdown of Fkbp1b expression with small interfering RNA (siRNA) and treatment with rapamycin, an immunosuppressant drug that binds and displaces FKBP1b from RyR2 in myocytes, increasing intracellular Ca2+ release (Marks, 1997; Long et al., 2007; Zalk et al., 2007). Our results support a major role for FKBPs in the hippocampal aging phenotype.
Materials and Methods
Subjects.
Studies on adult animals used 3- to 4-month-old male Fischer 344 rats obtained from Harlan. All protocols and procedures were performed in accordance with institutional guidelines and were approved by the Animal Care and Use Committee.
FKBP1b knockdown in vitro.
Mixed neuronal–astrocyte cultures were prepared as described previously (Porter et al., 1997; Clodfelter et al., 2002; Norris et al., 2002) and transfected with 8 nm small interfering RNA against Fkbp1b mRNA (siFkbp1b) (58950, FKBP1b siGENOME SMARTpool; Dharmacon), 8 nm nontargeted siRNA (ON-TARGETplus Non-targeting siRNA #1; Dharmacon), or siRNA dilution buffer on 7 or 8 d in vitro (DIV). Using the SilenceMag protocol of MagnetoFection technology (OZ Biosciences), 8 nm of either siFkbp1b or nontargeting siRNA was mixed with 4 μl of SilenceMag beads in 200 μl of serum-free supplemented MEM at room temperature for 20 min and then added to each well of six-well hippocampal cell culture plates to a final volume of 2 ml. The cell cultures were then placed on a magnetic plate designed for magnetic transfection (OZ Biosciences) for 15 min at room temperature, after which they were placed in a 37°C cell culture incubator until analyses. Cultures were treated with siFkbp1b, nontargeting siRNA, or siRNA buffer for a period of ∼4 d, followed by recording and quantitative PCR (qPCR) analysis, performed on cells plated on “micro-islands” (Blalock et al., 1999).
FKBP1b knockdown in vivo with adeno-associated virus vectors.
To achieve selective knockdown in vivo, an adeno-associated virus (AAV) vector was used to express a short hairpin RNA (shRNA) targeting the rat Fkbp1b mRNA (GenBank accession no. NM_022675). The construction of this vector (shFkbp1b) was performed at the University of Pennsylvania Vector Core by first cloning a double-stranded fragment generated by annealing two oligonucleotides (top strand, 5′-CGGAAGGACATTCCCTAAG_TTCAAGAGA_CTTAGGGAATGTCCTTCCGC-3′; bottom strand, 3′-GCCTTCCTGTAAGGGATTC_AAGTTCTCT_GAATCCCTTACAGGAAGGCG-5′) into the AAV pZacf–U6–ZsGreen plasmid (provided by the University of Pennsylvania Vector Core) at the BamH1–EcoR1 restriction enzyme sites. The recombinant clone was then used to produce the AAV1 serotype shFkbp1b vector (AAV1–shFkbp1b). This design places the shRNA under the control of the human U6 promoter, which is followed by a downstream cytomegalovirus promoter-driven ZsGreen fluorescence marker. When cleaved, the shRNA was designed to generate siRNA targeting the CGGAAGGACATTCCCTAAG region of rat Fkbp1b mRNA. Purification and determination of AAV1–shFkbp1b vector titers were performed by the University of Pennsylvania Vector Core.
In recent years, considerable progress has been made in developing the techniques for viral-vector-mediated gene transfer (Fitzsimons et al., 2002; Klein et al., 2002, 2008; Zaiss and Muruve, 2005; Cearley and Wolfe, 2006; Mouravlev et al., 2006). Choice of the AAV1 serotype for specific transduction of targeted pyramidal neurons in the hippocampus in the present study was based on the previous work and on a series of pilot studies in which we compared distribution of neurons expressing green fluorescent protein (GFP) after injection of GFP-expressing AAV serotypes 1, 8, and 9. Under isoflurane anesthesia, rats were placed in a David Kopf Instruments stereotaxic frame. Small holes were drilled in the skull either unilaterally or bilaterally (at points from bregma: posterior, −4.5 mm; lateral, 3.0 mm), and the dura was pierced to allow for microinjection. A Hamilton microsyringe was lowered 1.6 mm into the dorsal hippocampus, a region critical for spatial memory (Moser and Moser, 1998). At the target injection site (CA1 stratum oriens at the peak of the curve of stratum pyramidale), 2 μl of AAV-containing vehicle was delivered at a rate of 0.2 μl/min using a Stoelting QSI microinjection pump. After injection, the syringe was left in place for 5 min, then the small holes in the skull were filled with bone wax, and the incision was sutured. Two to 4 weeks after unilateral microinjections of AAV, rats were killed, and the brains prepared for sectioning and microscopy. GFP expression showed that all serotypes successfully transduced hippocampal CA1–CA3 pyramidal neurons and that expression of GFP was primarily limited to the hippocampus proper. Serotypes 1 and 9 but not 8 exhibited longitudinal spread sufficient to transduce neurons throughout the dorsal regions of the ipsilateral hippocampus and did not spread substantially across either the hippocampal fissure into the dentate gyrus or the midline into the contralateral hippocampus. Serotype 1 appeared slightly more specific for the somal layer and was selected as the preferred serotype for additional studies. For this serotype, we estimated from hematoxylin–eosin-counterstained sections that 30–60% of CA1–CA3 pyramidal neurons in ipsilateral dorsal hippocampus expressed GFP.
For the electrophysiological experiments, one hippocampus (ipsilateral) of 3-month-old male F344 rats was microinjected with 2 μl of AAV [titer, 2.2E13 GC (genome copies per milliliter)] expressing short hairpin RNA against Fkbp1b (shFkbp1b), whereas the control hippocampus of each animal (contralateral) was either microinjected with 2 μl of empty-vector control AAV (AAV titer, 3.56E12 GC) or received no treatment. Three to 4 weeks after injection, intracellular recordings of the sAHP were obtained from CA1 pyramidal neurons in acute hippocampal slices from both ipsilateral and contralateral sides of nine shFkbp1b-injected animals. Subsequently, qPCR of Fkbp1b was performed on tissue from the dorsal tips and one slice each from both hippocampi of eight of the nine animals studied electrophysiologically. In addition, brains of two other animals unilaterally injected in hippocampus with shFkbp1b were prepared for immunohistochemical studies (see below).
qPCR.
For mRNA quantification, one-step real-time reverse transcription (RT)-PCR was used. RT-PCR amplification was performed as described previously (Chen et al., 2000) using an ABI prism 7700 sequence detection system (Applied Biosystems) and TaqMan One Step RT-PCR reagents (Applied Biosystems). All samples were run in duplicate in a final volume of 50 μl containing 50–100 ng of cellular RNA and a TaqMan probe (200 μm) and primers (300 nm each) with an amplicon spanning the rat Fkbp1b cDNA region from nucleotides 155 to 259. Cycling parameters for all assays were as follows: 30 min at 48°C, 10 min at 95°C followed by 40 cycles of 15 s at 95°C, and 1 min at 60°C. Primers were designed using Primer Express software (version 1.5; Applied Biosystems) and chemically synthesized by Applied Biosystems (forward primer, 5′-GCAAGCAGGAAGTCATCAAAGG-3′; reverse primer, 5′-CAGTAGCTCCATATGCCACATCA-3′; TaqMan probe, 5′-AGCTCATCTGGGCAGCGCCTTCTT-3′). The RNA levels of glyceraldehyde-3-phosphate dehydrogenase were used as normalization controls for RNA quantification.
Immunohistochemistry.
Immunohistochemistry (IHC) methods were similar to those described previously (Kadish and Van Groen, 2002). Rats were anesthetized and perfused transcardially with cold 0.9% NaCl, followed by perfusion with 4% paraformaldehyde in PBS; thereafter, they were stored overnight in the 4% paraformaldehyde/PBS solution and then transferred to 30% sucrose/PBS solution. Coronal sections (30 μm) were cut on a freezing sliding microtome. The following primary antibodies were used for overnight incubation: FKBP12.6/1b (1:500; sc-98742; Santa Cruz Biotechnology), mammalian target of rapamycin (mTOR) (1:500; ab32028; Abcam), and RyR2 (1:150; AB9080; Millipore Corporation). Sections were then rinsed and transferred to a solution containing appropriate biotinylated secondary antibody for 2 h, after which they were rinsed and transferred to a solution containing ExtraAvidin for 2 h. Sections were then incubated for 3 min with Ni-enhanced DAB solution. To obtain comparable staining across sections, all animals were processed simultaneously in the same staining tray. The FKBP1b/12.6-, mTOR-, and RyR2-stained sections of the dorsal hippocampus were digitized using an Olympus DP70 camera, and the resulting images were analyzed using the ScionImage (NIH Image) program. The DP70 camera settings were maintained throughout acquisition of all photomicrographs for statistical comparison. For densitometric analysis of FKBP1b, mTOR, and RyR2 immunohistochemistry, the CA1 pyramidal cell soma layer (stratum pyramidale) of the ipsilateral and contralateral hippocampus on two sections from each of two animals were outlined and quantified.
Drug application.
Rapamycin (A.G. Scientific) was dissolved in DMSO (0.1% final DMSO) and added to the culture dish (whole-cell recording) or holding chamber (intracellular recording in slices) to a final concentration of 5 μm. Rapamycin exposure was maintained for 1, 24, or 96 h in cultures and for 2–6 h in slices before recording began. Nimodipine was dissolved in DMSO and added to cultures at a final concentration of 10 μm (0.1% final DMSO dilution). Ryanodine (Alomone Labs) was dissolved in water and perfused into the recording chamber at a rate 1000 times slower than the oxygenated artificial CSF (ACSF) perfusate. Final concentration of ryanodine in the recording chamber was estimated at 20 μm.
Patch-clamp recording of Ca2+ channel current in cultured neurons.
Whole-cell recordings of pharmacologically isolated Ca2+ channel currents, using Ba2+ as the charge carrier, were obtained from neurons in primary hippocampal cell cultures at 9–12 DIV, as described previously (Porter et al., 1997; Blalock et al., 1999; Brewer et al., 2001; Norris et al., 2002). Electrophysiological data were acquired at 5–20 kHz using a patch-clamp amplifier (Axopatch 200A), DigiData 1320 digital input/output board and pClamp 7 (Molecular Devices). Cell capacitance measures, membrane resistance, access resistance, and holding current were obtained from the membrane properties function in pClamp7. All cells were held at −70 mV unless stated otherwise. Current–voltage (I–V) relationships were determined by stepping the voltage in increments of 10 mV from 70 to +60 mV for 150 ms. Current was averaged over five traces for each measurement point, and current density was determined by dividing current by cell membrane capacitance (picoamperes per picofarad).
Current-clamp intracellular recording in hippocampal slice pyramidal neurons.
Young-adult male F344 rats were anesthetized in a CO2 chamber before rapid decapitation. Our sharp-electrode electrophysiological methods have been described previously (Thibault et al., 2001; Gant et al., 2006). Briefly, intracellular recordings were obtained from CA1 pyramidal neurons in hippocampal slices (350-μm-thick) maintained in oxygenated ACSF (in mm: 128 NaCl, 1.25 KH2PO4, 10 glucose, 26 NaHCO3, 3 KCl, 2 CaCl2, and 2 MgCl2), using sharp glass pipettes filled with HEPES (10 mm), KMeSO4 (2 m), and Calcium Orange tetrapotassium salt (5 mm; Invitrogen). Electrophysiological data were acquired and analyzed using pClamp 8, a sharp-electrode amplifier (Axoclamp 2B), and a DigiData 1320 board (Molecular Devices). Voltage records were digitized at 2–20 kHz and low-pass-filtered at 1 kHz. Input resistance and AHP measures were obtained in current-clamp mode. AHP duration and sAHP amplitude were measured after four Na+ APs triggered by an intracellular depolarizing pulse. The 5 mm concentration of Ca2+ indicator used is in a range commonly used in slice imaging experiments (Jaffe et al., 1992; Brown and Jaffe, 1994; Jaffe and Brown, 1994) and apparently does not distort AHP waveforms or obscure aging differences in the AHP (Thibault et al., 2001; Gant et al., 2006).
Ca2+ imaging.
Methods used were similar to those described previously by several groups (Miyakawa et al., 1992; Brown and Jaffe, 1994; Jaffe and Brown, 1994; Magee and Johnston, 1995; Thibault et al., 2001; Gant et al., 2006). Individual CA1 neurons impaled with sharp pipettes and loaded with the Ca2+ indicator Calcium Orange were imaged on the stage of a Nikon E600 microscope equipped with a 40× water-immersion objective and a CCD camera (Roper Scientific/Princeton Instruments). Calcium Orange was allowed to diffuse into the cell for at least 10 min before Ca2+ fluorescence measures were performed. Calcium Orange was excited using a wavelength switcher (Sutter Instruments Lambda DG-4) and software control (Axon Imaging Workbench; version 2.2.1.54; Molecular Devices). The 575 nm wavelength was monitored through a dichroic mirror centered at 570 nm during excitation with the 550 nm wavelength. The intracellular Ca2+ response was elicited by 10 s of 7 Hz repetitive synaptic stimulation (RSS) delivered to the Schaffer collaterals via bipolar stimulation electrode. Stimulation intensity was set at AP threshold and generated an action potential on essentially each pulse (Thibault et al., 2001). Ca2+ responses were measured in a region of interest corresponding to the visible outline of the cell soma, excluding surrounding low-intensity diffracted light. Although the central region of the neuron appears brighter (see Fig. 6, inset), this may be related in part to greater indicator loading in this thicker central region of the cell (Tsien, 1988; Nicotera et al., 1994). After background subtraction from an area devoid of cells adjacent to the recorded cell, percentage change in fluorescence was determined relative to baseline (%ΔF/F). Optical sampling rate averaged ∼5 Hz, which appeared sufficiently sensitive to detect significant changes in the Ca2+ response. Excitation of Calcium Orange was essentially continuous during a 10 s train of RSS, but control runs showed that bleaching was minimal (−0.21 ± 0.09% at 5 s RSS; −0.69 ± 0.35% at 10 s RSS; n = 7 cells). The ryanodine-sensitive component (CICR component) of the Ca2+ response was determined by subtracting the post-ryanodine Ca2+ response from the pre-ryanodine response [(pre-ryanodine %ΔF/F) − (post-ryanodine %ΔF/F) = ryanodine-sensitive component].
Figure 6.

Rapamycin enhanced the ryanodine-sensitive component (CICR) of stimulated Ca2+ transients in hippocampal slice neurons. Representative traces showing Ca2+ indicator fluorescence responses during 7 Hz RSS in a control (A1) and a rapamycin-treated (A2) neuron, before and after ryanodine treatment. The before–after subtracted ryanodine-sensitive components (CICR) are shown at bottom of the traces. Note that the rapamycin-treated neuron exhibited increased CICR. Inset shows a representative grayscale image of the soma (region of interest) of an untreated pyramidal neuron at rest and during RSS. Brighter gray values reflect increased Ca2+ levels during stimulation but do not distinguish between nuclear and cytoplasmic signals (see Materials and Methods). B, Mean peak value (NS), integrated AUC during the first 5 s of RSS (p = 0.05), and rising time constant (p = 0.07) for the overall Ca2+ fluorescence transients of control (Ctr) versus rapamycin (Rap)-treated neurons before ryanodine treatment. C, Mean AUC values for only the ryanodine-sensitive component of the Ca2+ transient (A1, A2, bottom) in control and rapamycin-treated neurons during a 10 s train of RSS (p = 0.05). Neurons per group: n = 6 for control, n = 7 for rapamycin. *p < 0.05 versus control. Means ± SEM.
Statistical analyses.
Data analysis was performed with Clampfit8 (Molecular Devices), and statistical analysis was performed with StatView (SAS Institute). Variables were analyzed using ANOVA across all groups with means and SEs reported. Fisher's protected least significant difference (PLSD) test was used for post hoc group comparisons. p values <0.05 were considered significant.
Results
Disruption of FKBP1b function in vitro: effects on VGCC current
Because increased L-VGCC activity is a primary component of aging-related Ca2+ dysregulation (Campbell et al., 1996; Thibault et al., 2001, 2007), we tested the effects of prolonged exposure to small interfering RNA against Fkbp1b (siFkbp1b) or to rapamycin on pharmacologically isolated VGCC currents recorded in whole-cell patch-clamp mode from neurons in primary hippocampal cell cultures. Whole-cell recording data were analyzed from a total of 149 neurons across nine treatment groups, including untreated (control) cells and cells treated with siFkbp1b (8 nm, 96 h), nontarget control siRNA (8 nm, 96 h), siRNA buffer vehicle (96 h), rapamycin 5 μm (1, 24, and 96 h), rapamycin (5 μm, 96 h) plus the selective blocker of L-type VGCCs, nimodipine (10 μm), and nimodipine alone (10 μm). Neurons in all the different treatment conditions were recorded alternately and treatment groups were run concurrently over the length of the study, to control for any drift in experimental conditions. Table 1 provides values for basic electrophysiological properties and parameters, which revealed no significant differences across treatments on any of these variables.
Table 1.
Basic electrode and membrane properties from whole-cell recordings
| Treatment group | Electrode (MΩ) | Access resistance (MΩ) | Capacitance (pF) | Holding current (pA) | n |
|---|---|---|---|---|---|
| Control | 2.42 ± 0.06 | 10.01 ± 0.54 | 60.84 ± 4.79 | 147.91 ± 11.92 | 18 |
| Rapamycin, 1 h | 2.45 ± 0.10 | 9.73 ± 0.57 | 51.37 ± 3.62 | 113.80 ± 12.18 | 4 |
| Rapamycin, 24 h | 2.45 ± 0.08 | 10.44 ± 0.66 | 60.15 ± 3.69 | 133.26 ± 7.50 | 20 |
| Rapamycin, 4 d | 2.61 ± 0.07 | 10.04 ± 0.77 | 54.47 ± 2.86 | 113.74 ± 11.09 | 20 |
| Nimodipine | 2.55 ± 0.08 | 9.13 ± 0.33 | 53.07 ± 1.02 | 103.93 ± 17.42 | 12 |
| Rapamycin (4 d) + Nimodipine | 2.47 ± 0.08 | 8.97 ± 0.44 | 53.69 ± 1.75 | 107.77 ± 19.18 | 10 |
| siRNA FKBP | 2.62 ± 0.06 | 9.03 ± 0.45 | 57.47 ± 2.63 | 131.63 ± 12.02 | 23 |
| siRNA nontargeted | 2.52 ± 0.04 | 9.25 ± 0.060 | 57.86 ± 3.16 | 134.66 ± 16.58 | 19 |
| siRNA vehicle | 2.43 ± 0.05 | 9.05 ± 0.053 | 57.88 ± 2.90 | 128.20 ± 21.63 | 23 |
No differences in basic properties were observed across conditions; means ± SEM.
Figure 1 shows representative current traces, I–V curves, and mean peak current density for treatment groups in these experiments. An ANOVA across all groups revealed a main effect of treatment (F(8,140) = 18.49, p < 0.0001) on peak Ca2+ current density (Fig. 1C). Fisher's PLSD post hoc test showed that, after 96 h incubation in siFkbp1b, VGCC current density was significantly enhanced versus controls (p < 0.0001). Cultured neurons treated 96 h with either nontarget siRNA or siRNA vehicle exhibited no effects on VGCC current density (Fig. 1C, right bars). Real-time qPCR showed that the siFkbp1b transfection treatment achieved significant knockdown of cell culture Fkbp1b mRNA (mean ± SEM for siFkbp1b-treated vs control, 0.91 ± 0.05 vs 1.15 ± 0.05, p < 0.01).
Figure 1.

Knockdown of FKBP1b or treatment with rapamycin (Rap) increased Ca2+ channel current in cultured hippocampal neurons. A, Representative whole-cell patch-clamp current traces from each experimental condition. B, Mean I–V relationships for six of the nine experimental conditions. The I--V curves for Rap for 1 h, siRNA vehicle, and siRNA nontarget were not different from the control condition and are omitted for illustrative clarity. Exposure to siFkbp1b for 96 h or to rapamycin for 24 or 96 h induced enhancement of VGCC current. Treatment with siFkbp1b or with rapamycin altered the amplitude but not the voltage dependence of Ca2+ current, whereas nimodipine (Nim) shifted peak current to more positive voltage. C, Means ± SEM of peak Ca2+ current density for the nine conditions. *p < 0.05 and **p < 0.0001, significant differences from the control condition. n = 18 for control (Ctr), n = 4 for Rap for 1 h, n = 20 for Rap for 24 h, n = 20 for Rap for 96 h, n = 10 for Rap + Nim, n = 12 for Nim, n = 23 for siRNA vehicle (Veh), n = 19 for siRNA nontargets (Non-T), n = 23 for siFkbp1b.
Rapamycin exposure for 1 h had no effect on VGCC current density compared with controls, consistent with a previous study showing little effect of acute rapamycin exposure (Norris et al., 2002). However, after 24 or 96 h incubation with rapamycin, neurons exhibited substantially larger VGCC currents (p < 0.001, 24 or 96 h incubation duration vs controls) (Fig. 1C, left bars). Despite significant effects on current density, neither siFkbp1b nor rapamycin altered current inactivation during the voltage step (Fig. 1A) or shifted the I–V relationship (Fig. 1B). To determine whether the effect of rapamycin was mediated by actions specific for L-VGCCs, we added the specific L-VGCC blocker nimodipine (10 μm) to some cultures. Notably, nimodipine fully blocked the enhancing effect of rapamycin on total VGCC current (p < 0.0001), reducing current density levels in rapamycin-treated cells to those seen in untreated controls (Fig. 1C, middle bars). Furthermore, in cells treated with nimodipine alone, VGCC current density was significantly reduced by ∼30–35% compared with controls (p < 0.05), consistent with values previously reported for the L-type current fraction in hippocampal neurons (Blalock et al., 1999). As described previously, although slowly inactivating L-VGCCs are high voltage activated, nimodipine blockade shifts the peak of the I–V curve to more positive potentials (Fig. 1B) (Tsien et al., 1988; Regan et al., 1991; Porter et al., 1997; Blalock et al., 1999). Thus, most of the enhancing effect of rapamycin on VGCCs appeared to be accounted for by actions specific to L-type channels. Moreover, the similarities in the inactivation patterns and I–V curves of siFkbp1b-treated and rapamycin-treated neurons (Fig. 1A,B) indicate that these two treatments acted primarily on the same type (L-VGCC) of VGCC.
Disruption of FKBP1b function in vivo: effects on the sAHP
In these studies, we tested whether FKBP1b knockdown in adult neurons in vivo could increase sAHP magnitude because a larger sAHP is one of the most consistent electrophysiological markers of aging-related hippocampal Ca2+ dysregulation in pyramidal cells (Disterhoft and Oh, 2007; Thibault et al., 2007). AHP measures were obtained from nine young rats 3–4 weeks after unilateral hippocampal microinjection of shFkbp1b. The AHP was recorded intrasomatically from 17 CA1 neurons in slices from the hippocampus ipsilateral to the FKBP1b knockdown injection and from 17 CA1 neurons of the control (contralateral) hippocampus (Fig. 2, top). Fluorescence microscopy indicated that, 3–4 weeks after injection, the viral vector had infected neurons throughout much of CA1–C3 of the ipsilateral dorsal hippocampus. The AAV vector did not appear to spread across the midline to the contralateral hippocampus, although faint fluorescence was often seen in the contralateral CA1 stratum oriens and dentate gyrus (Fig. 2, middle), possibly reflecting transport via commissural fibers from the ipsilateral pyramidal neurons or uptake of AAV particles leaked into the ventricle. Nevertheless, neuronal somata were not generally infected in the contralateral hippocampus. Statistical analysis revealed that mean sAHP amplitude and duration approximately doubled in neurons from the FKBP1b knockdown side (Fig. 2). In addition, 9 of 17 knockdown side neurons showed sAHP amplitude or duration values above the highest control value. The sAHP magnitudes of control neurons were similar to those observed in young rats, whereas values from knockdown side neurons were similar to those observed in aged rats in previous studies (Gant et al., 2006).
Figure 2.
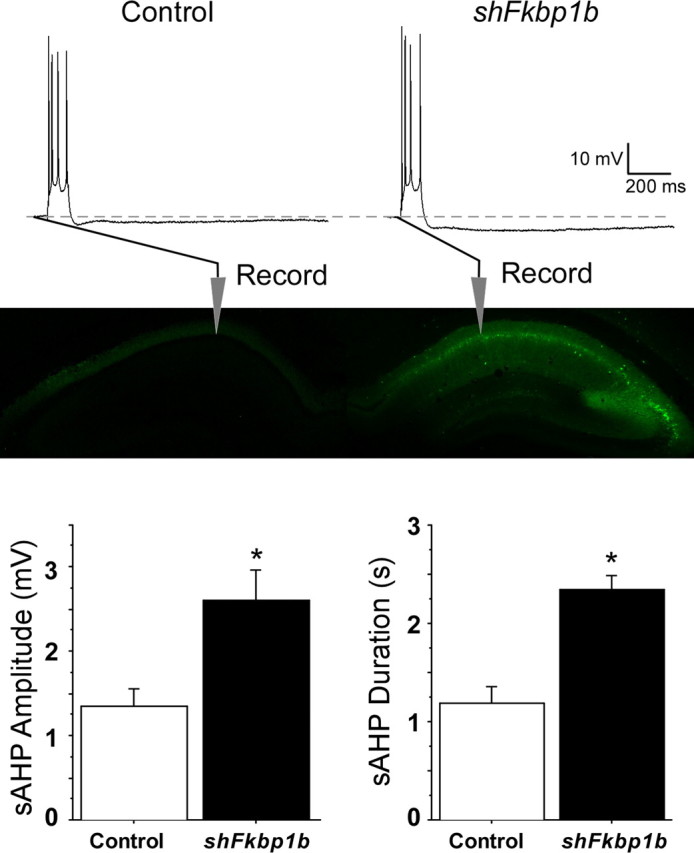
Knockdown of FKBP1b in vivo enhanced the slow AHP. Top, Representative sharp electrode intracellular recordings showing four triggered action potentials, followed by an AHP (dashed line indicates baseline) in CA1 neurons from slices of a control (left) and an AAV–shFkbp1b-injected (right) hippocampus. Middle, Fluorescent imaging in an animal receiving a similar AAV injection, showing strong expression in the injected, but not the contralateral, hippocampus and also illustrating general placement of recording pipettes in the pyramidal neuron somal layer (stratum pyramidale) of field CA1. Bottom, sAHP amplitude (left) and duration (right) were significantly increased by FKBP1b knockdown (shFkbp1b injection) (*p < 0.0025 for either variable, t test; n = 17 neurons per group).
qPCR (Fig. 3) and IHC densitometry (Fig. 4, top; Table 2) results from this knockdown study clearly showed that FKBP1b knockdown was achieved at both the mRNA (25–35%) and protein (Table 2) levels in the ipsilateral hippocampus. Moreover, the results revealed apparent upregulation of protein levels for two targets of FKBP inhibition, RyR2 and mTOR (see Discussion), on the FKBP1b knockdown side (Fig. 4; Table 2). Therefore, the present data suggest that FKBP1b may repress RyR2 and mTOR expression through an as yet unknown signaling pathway.
Figure 3.

qPCR of Fkbp1b mRNA after 3–4 weeks of in vivo knockdown. Degree of knockdown induced by injection of shFkbp1b was assessed in two hippocampal regions, the dorsal tip and a transverse 350-μm-thick slice from the more caudal dorsal hippocampal areas used for recording. Although the slice exhibited relatively higher Fkbp1b concentration, shFkbp1b injection was associated with a significant knockdown of Fkbp1b expression in both regions (*p < 0.05; main effects of region and treatment; two-way repeated measures ANOVA; n = 8 hippocampi per group), confirming considerable ipsilateral spread of the AAV vector. Gapdh, Glyceraldehyde-3-phosphate dehydrogenase.
Figure 4.

Immunohistochemical profile of protein expression after shFkbp1b-mediated knockdown of FKBP1b. Low-power photomicrographs of contralateral and ipsilateral dorsal hippocampus from the same coronal sections of a single rat ∼4 weeks after ipsilateral shFkbp1b knockdown of the FKBP1b/12.6 protein. Note marked reduction of FKBP1b staining in CA1 and other areas of the injected hippocampus, with associated ipsilateral upregulation of mTOR in CA1 and dentate gyrus and upregulation of RyR2 in the stratum oriens, stratum pyramidale (pyramidal neuron somal layer), and stratum radiatum of CA1 and the molecular layer of the dentate gyrus. Insets show higher-magnification views of stratum pyramidale (sp) and adjacent strata in CA1. Arrow in top right indicates injection target site, and small rectangle illustrates approximate region from where insets were taken (for data, see Table 2). Scale bars: low power, 1 mm; inset, 250 μm.
Table 2.
IHC densitometry data from AAV-injected animals
| contra | ipsi | p value | |
|---|---|---|---|
| Fkbp12.6 | 138.5 ± 3.5 | 115.3 ± 0.9 | 0.0068 |
| mTOR | 83.5 ± 1.9 | 96.0 ± 2.0 | 0.0292 |
| RyR2 | 110.5 ± 2.4 | 131.0 ± 5.9 | 0.0306 |
shFkbp1b injection resulted after 3–4 weeks in a decrease in FKBP1b and an increase in mTOR and RyR2 immunostaining in the ipsilateral hippocampus. n = 4 sections; paired t test; means ± SEM. contra, Contralateral; ipsi, ipsilateral.
Rapamycin also enhanced the sAHP
In other experiments on acute hippocampal slices of young-adult F344 rats, we tested whether rapamycin also could induce aging-like effects on the sAHP. For these studies, we again used intracellular sharp electrode recording techniques in individual CA1 pyramidal neurons. Slices were incubated in either control medium or rapamycin (5 μm) for 2–6 h before recording. After baseline recording, slices were perfused with control medium or ryanodine (20 μm), a selective ryanodine receptor blocker that is well established to inhibit CICR. Table 3 shows there were no effects of rapamycin on basic resting membrane parameters and measured potentials. These results are consistent with previous studies in hippocampal neurons in which no effect of rapamycin was found on voltage-gated Na+ and K+ currents or action potentials (Norris et al., 2002; Rüegg et al., 2007). However, baseline EPSP amplitude was reduced in rapamycin-treated cells (Table 3), an effect similar to that found in aged animals (Thibault et al., 2001; Burke and Barnes, 2006).
As seen in Figure 5, a significant enhancing effect of rapamycin was found on AHP measures. Rapamycin increased sAHP amplitude (p < 0.0001) and sAHP duration (p < 0.0001). Furthermore, a correlation analysis between incubation time and AHP measures revealed that this effect was time dependent because sAHP magnitude increased as a function of rapamycin incubation duration up to 6 h (sAHP amplitude vs incubation time, r = 0.74; p < 0.01). Treatment with ryanodine fully blocked the effect of rapamycin on AHP measures, indicating that the enhancing effect of rapamycin on AHP magnitude resulted primarily from elevated Ca2+ release from RyRs, as is also the case for the aging-related increase in the sAHP (Gant et al., 2006). Moreover, as reported previously (Kumar and Foster, 2005; Gant et al., 2006), ryanodine alone significantly reduced sAHP amplitude (p < 0.0001) and duration (p < 0.0001) compared with controls. A previous study reported a stimulatory effect of rapamycin on Ca2+-dependent K+ currents (Terashima et al., 1998). However, given the present data, that was likely an indirect effect resulting from FKBP1b disruption and increased CICR. Thus, our results indicate that rapamycin substantially increased sAHP magnitude by enhancing CICR in young-adult rat neurons, mimicking the effects of aging or Fkbp1b knockdown.
Figure 5.
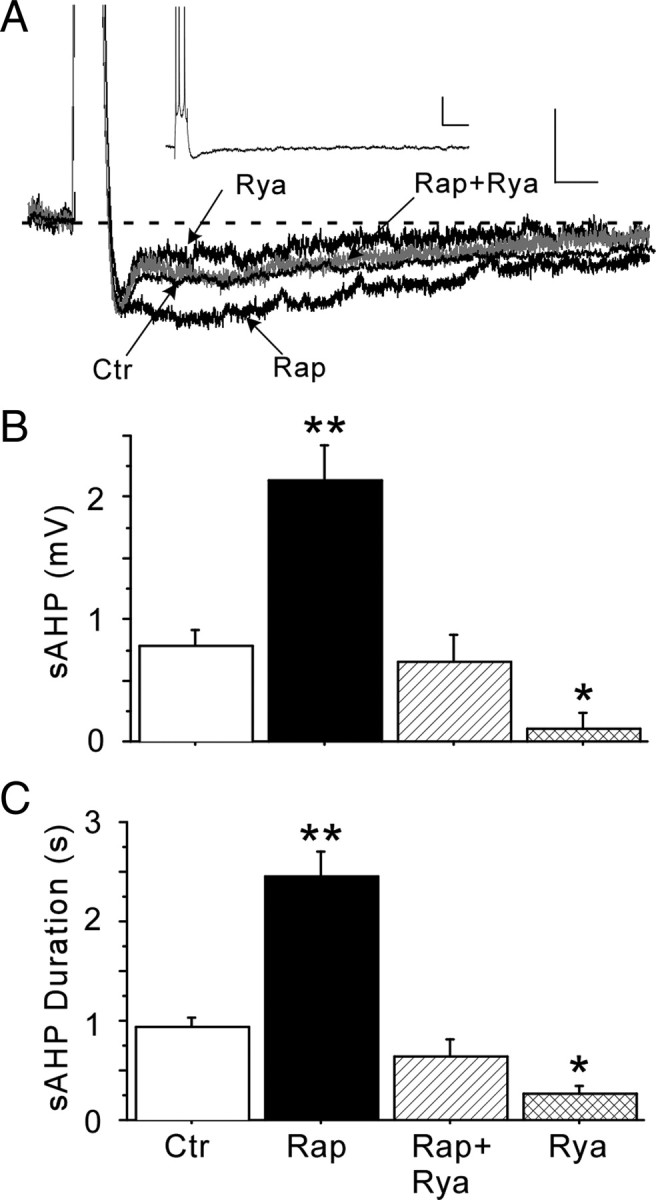
Rapamycin (Rap) enhanced the ryanodine (Rya)-sensitive AHP in hippocampal slices. A, Representative intracellular recording traces of AHPs, shown at high magnification to illustrate the enhancing effect of rapamycin on the AHP and its reversal by ryanodine, which blocks CICR. Calibration: 2 mV, 200 ms. The control (Ctr) and Rap + Rya traces partially overlap and obscure each other. Inset shows overview of a full AHP after a burst of four action potentials. Calibration: 3 mV, 150 ms. B, Rapamycin increased sAHP amplitude approximately twofold, an effect reversed by ryanodine. As described previously, ryanodine alone significantly reduced sAHP amplitude in control neurons. C, AHP duration was also enhanced by rapamycin and reversed by ryanodine. Neurons per group: n = 16 for control, n = 21 for rapamycin, n = 8 for rapamycin + ryanodine, n = 4 for ryanodine. *p < 0.05 and **p < 0.0001, significant difference between treatment and non-treated controls. Means ± SEM.
Rapamycin enhancement of intracellular Ca2+ transients and CICR
To more directly test the actions of rapamycin on CICR, we analyzed voltage-activated Ca2+ responses in a subset of the slice neurons in which the AHP was assessed. In previous studies of pyramidal neurons, it has been found that voltage-activated [Ca2+]i transients are increased with aging (Thibault et al., 2001; Hemond and Jaffe, 2005; Gant et al., 2006), apparently because of enhanced CICR (Gant et al., 2006). Ratiometric Ca2+ indicators were used in those previous studies to allow measures of absolute concentrations. Although single wavelength indicators do not generally provide information on absolute Ca2+ concentration values, they are useful for assessing Ca2+ responses relative to baseline (Jaffe et al., 1992; Miyakawa et al., 1992; Brown and Jaffe, 1994). Here, we used the single wavelength high-affinity fluorescent Ca2+ indicator Calcium Orange for improved signal/noise ratio and sampling speed (Fig. 6). To confirm comparable indicator loading, gray values were measured at baseline before obtaining stimulated fluorescence values. The baseline and post-ryanodine baseline gray values for nontreated control neurons were 206.9 ± 47.7 (mean ± SEM) and 209.3 ± 36.7 and for rapamycin-treated neurons were 232.4 ± 54.6 and 242.4 ± 58.7, indicating that there were no significant differences within or between treatment groups in indicator loading or baseline Ca2+. Postsynaptic intracellular Ca2+ transients were induced by 10 s trains of repetitive (7 Hz) synaptic stimulation at threshold for action potential generation (Fig. 6A1,A2), as described previously (Thibault et al., 2001; Gant et al., 2006). Rapamycin did not affect the RSS-activated peak fluorescence transient but increased the area under the curve (AUC) of the transient during the first 5 s of RSS (p = 0.05), primarily because of a near-significant decrease in rise time (p = 0.07) (Fig. 6B). The ryanodine-sensitive component (Fig. 6A1,A2) accounted for most of the enhancing effect of rapamycin on the AUC of the overall synaptically stimulated Ca2+ response, as shown by a significant increase specifically in the AUC of the ryanodine-sensitive component (p < 0.05) (Fig. 6C). Furthermore, the blocking effect of ryanodine indicates that the enhancement by rapamycin of Ca2+ transients, as of the AHP noted above, resulted primarily from enhancement by rapamycin of RyR-mediated CICR.
Discussion
The results of these studies show that disruption of FKBP1b/12.6 function, using interfering RNA or rapamycin, induced key components of the aging phenotype of Ca2+ dysregulation in young hippocampal pyramidal neurons. Notably, both FKBP1b knockdown and rapamycin treatment increased VGCC activity in vitro and enhanced the sAHP in vivo. Thus, both approaches to disrupting FKBP1b function induced two of the hallmark biomarkers of aging-related Ca2+ dysregulation in young rat hippocampal pyramidal neurons. Moreover, additional studies showed that the enhancement by rapamycin of the sAHP and the Ca2+ transient was mediated primarily by increasing the ryanodine-sensitive (i.e., CICR) component of those responses, a mode of action highly analogous to that seen with aging (Gant et al., 2006).
This is the first study to implicate declining function of FKBPs as a candidate molecular mechanism in hippocampal Ca2+ dysregulation. It therefore supports the novel hypothesis that an aging-related decline in hippocampal FKBP1b/1a function plays a key role in unhealthy brain aging (Fig. 7). In addition, the present study provides the first direct evidence that selective FKBP1b disruption destabilizes Ca2+ responses in brain neurons.
Figure 7.
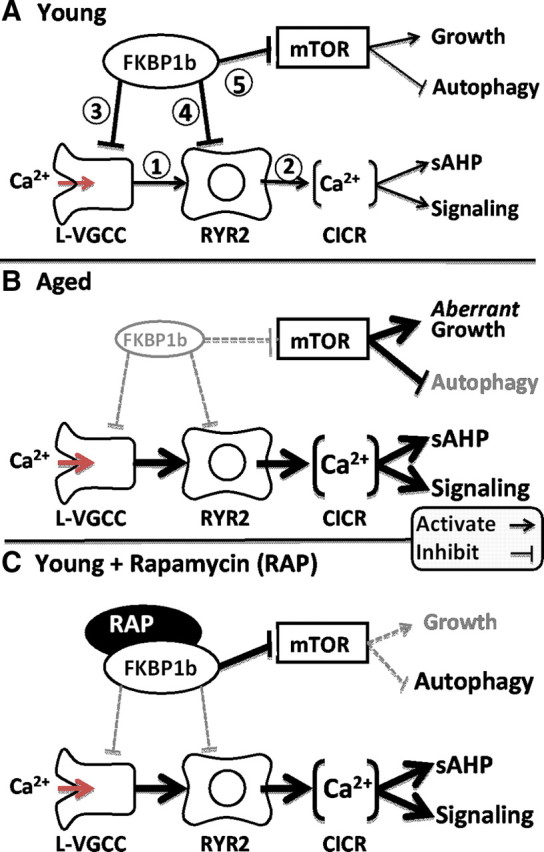
Working model of the role of FKBP1b in aging-related Ca2+ dyshomeostasis. A, In young subjects, neuronal FKBP1b exerts strong tonic inhibitory effects on two pathways: the Ca2+ regulatory pathway and the mTOR signaling pathway. In the Ca2+ regulatory pathway, FKBP1b inhibits cytosolic Ca2+ rises generated by extracellular influx via L-VGCCs (1) and intracellular release from RYRs (2), by inhibiting both channel types directly (3, 4). In the mTOR pathway, FKBP inhibition of mTOR (5) helps maintain the balance between growth-stimulating and autophagy-suppressing effects of mTOR. B, With aging (mimicked by FKBP1b knockdown), a decline of FKBP1b expression/function leads to weakened inhibition of both pathways and concomitant increases in both Ca2+ and mTOR signaling. Increased Ca2+ signaling results in dampened neuronal excitability and function, whereas mTOR disinhibition leads to aberrant increases in growth signaling as well as decreased autophagy. C, Rapamycin in young subjects exerts opposite effects on FKBP inhibition in the two pathways, mimicking the effect of aging of weakened FKBP inhibition in the Ca2+ pathway (B) but paradoxically augmenting FKBP inhibition of the mTOR pathway (C).
Interactions among L-VGCCs, RyR2, and FKBP1b in the brain
Several other intriguing observations emerge from these studies. For one, the present data show that, in addition to their established effects on Ca2+ release from RyRs in cardiomyocytes (Zalk et al., 2007), FKBPs in neurons appear to regulate Ca2+ influx via VGCCs (Fig. 1). The VGCC lies upstream of the RyR in the VGCC–RyR circuit that amplifies Ca2+ responses through CICR (Berridge, 1997; Verkhratsky, 2004), and it is feasible that FKBPs regulate Ca2+ currents through their actions on RyRs and secondary retrograde effects on VGCCs. However, a more parsimonious explanation may be that FKBPs regulate VGCC activity directly.
Furthermore, RyRs are closely juxtaposed to L-VGCCS in cardiomyocytes (Sedarat et al., 2000; Protasi, 2002) and neurons (Chavis et al., 1996; Kim et al., 2007; Berrout and Isokawa, 2009), an organization that enables L-VGCCs to serve as a preferred source of Ca2+ for activating RyR-mediated CICR. Thus, considering the evidence that FKBP1b may directly modulate plasmalemmal VGCCs (Fig. 1) as well as RyRs, we suggest that, in neurons, FKBP molecules may function together with L-VGCCs and RyRs in a multimeric Ca2+ regulating complex in which the FKBP directly stabilizes both the RyR and L-VGCC via separate interactions (see model in Fig. 7).
Another striking observation in these studies is that the expression of FKBPs and of their inhibitory targets, RyR2 and mTOR (also called FRAP1, for FK506–rapamycin-associated protein) may be inversely related. That is, knockdown of FKBP1b appeared associated with upregulation of mTOR/FRAP1 and RyR2 (Fig. 4; Table 2), two inhibitory targets of FKBPs (see below). An inverse relationship could account for why we found downregulated expression of Fkbp1b and Fkbp1a in conjunction with upregulated Ryr2 and Frap1/Mtor in gene microarray studies of the hippocampus in aging rats (Kadish et al., 2009, their Table 3 and supplemental material). Additionally, there is some evidence of aging-related upregulation of the CaV1.3 pore-forming subunit of the L-VGCC (Herman et al., 1998; Chen et al., 2000; Veng and Browning, 2002), which might also be an FKBP inhibitory target (Figs. 1, 7). If additional research confirms this putative inverse relationship, it would suggest the interesting conclusion that FKBP1b and FKBP1a may inhibit several targets not only by physical interactions but by repression of gene/protein expression as well.
The relatively slow activation of Ca2+-related processes by FKBP knockdown or rapamycin (hours) is consistent with derepression of gene expression by FKBP1b disruption but could also reflect gradual dissociation of FKBPs from target molecules, among other possibilities. In contrast, FK506, another immunosuppressant drug, exerts inhibitory actions on VGCCs in minutes, apparently by inhibiting calcineurin (Norris et al., 2002).
Other FKBP/rapamycin pathways: mammalian target of rapamycin
FKBP1a and FKBP1b are members of the FKBP family of immunophilins, which bind the immunosuppressant drugs FK506 and rapamycin. Many immunophilins exhibit peptidyl-prolyl isomerase activity and function as chaperones and stabilizers (Marks, 1997; Eitoku et al., 2008; Kang et al., 2008; Jakob et al., 2009). FKBP1b and FKBP1a interact with multiple pathways and exert pleiotropic effects, including inhibition of the mTOR/FRAP1 pathway that plays an important role in cell growth and plasticity in the brain (Jacinto and Hall, 2003; Hoeffer et al., 2008). Thus, FKBPs bind and inhibit both RyRs and mTOR, albeit via separate pathways (Fig. 7A). Somewhat paradoxically, however, the FKBP ligand rapamycin has opposite effects on these two inhibitory pathways. Although binding of rapamycin weakens FKBP-mediated inhibition of Ca2+ release pathways in myocytes (Marks, 1997), rapamycin binding strengthens FKBP-associated inhibition of mTOR. Recently, such rapamycin-enhanced mTOR inhibition has been reported to extend lifespan in mice and counteract some aging markers and AD-model brain pathology, possibly because of suppression of aberrant growth or of insulin signaling and tumorigenesis (Harrison et al., 2009; Caccamo et al., 2010). Nevertheless, in some studies, rapamycin has impaired LTP and/or cognitive function (Tang et al., 2002; Cracco et al., 2005; Parsons et al., 2006; Blundell et al., 2008), and the results of the present study also raise a clear caveat regarding the potential use of rapamycin as an “anti-aging” therapy. In any event, the increased inhibitory actions of rapamycin on mTOR appear essentially unrelated to its opposite disinhibitory effects on Ca2+ regulation, as the latter mimic the effects of aging. Therefore, with regard to Ca2+ regulation, knockdown of Fkbp1b and rapamycin appear to provide complementary approaches for selective disruption of FKBP1b function.
Possible mechanisms underlying age-related decline of FKBP1b function
The finding that disrupting hippocampal FKBP function can induce the Ca2+ dysregulation phenotype raises the question of whether similar disruption may occur with aging and, if so, what factors might underlie an initial aging-related decline in FKBP function. One possibility is that a decrease in the biosynthesis of energy-expensive proteins, notably including FKBPs, might result from a metabolic shift that appears to develop in neurons and glia of the hippocampus during aging (Rowe et al., 2007; Brinton, 2008; Kadish et al., 2009; Blalock et al., 2010). In addition, adrenal stress hormones (glucocorticoids), which have long been linked to brain aging (Porter and Landfield, 1998) and may exhibit altered impact with aging (Holmes and Seckl, 2006; Landfield et al., 2007; Blalock et al., 2010), enhance the Ca2+-dependent sAHP in the hippocampus (Joëls and de Kloet, 1989; Kerr et al., 1989). Similarly, mineralocorticoids have been shown to decrease FKBP1b expression and enhance RyR-mediated Ca2+ release (Gómez et al., 2009). Another possibility is that gradual vascular insufficiency might result in focally decreased FKBP protein (Kato et al., 2000). Alternatively, decreased FKBP function might be mediated by posttranslational events as well as by altered expression. In myocytes, hyperphosphorylation of RyR2 by protein kinase A (PKA) results in dissociation from FKBP1b and enhanced Ca2+ release by RyR2 (Marx et al., 2000). With aging, gene expression for PKA-pathway molecules appears increased in the hippocampus (Rowe et al., 2007; Kadish et al., 2009). However, there appear to be other pathways operating in the hippocampus because PKA activation does not increase but rather reduces the sAHP in hippocampal neurons (Oh et al., 2009). Clearly, substantial additional work will be needed to resolve these questions.
Implications of virally mediated manipulation of hippocampal FKBP1b
Viral-vector-mediated manipulation of gene expression in the adult CNS is a relatively new and powerful experimental tool that allows selective targeting of specific molecular pathways and cellular populations while circumventing some of the pitfalls of conventional genetic and pharmacological interventions (Fitzsimons et al., 2002; Klein et al., 2002, 2008; Zaiss and Muruve, 2005; Cearley and Wolfe, 2006; Mouravlev et al., 2006). In particular, it appears to provide an important complementary approach for gene array techniques because it can facilitate rapid and direct testing of hypotheses generated by genome-wide analyses of expression. AAV vector-mediated genomic intervention also appears to have substantial clinical potential, based on its persisting effects and its as yet relatively low pathogenic profile. In fact, several preclinical and clinical studies using virally mediated gene delivery for the treatment of neurodegenerative disorders have been conducted (Crystal et al., 2004; McPhee et al., 2006; Kaplitt et al., 2007; Johnston et al., 2009; Su et al., 2009; Kells et al., 2010). Thus, in concert with previous findings showing that FKBP1b expression in the hippocampus of incipient AD subjects is decreased and correlates with impaired cognition (Blalock et al., 2004), the present results on virally mediated manipulation of FKBP function may have future therapeutic implications.
In summary, we found that disrupting hippocampal FKBP1b function or expression was sufficient to induce aging-like Ca2+ dysregulation. Together, the results support a novel hypothesis of unhealthy brain aging, shown in Figure 7. In this model, an aging-related decline in hippocampal FKBP1b function, which can be mimicked by FKBP knockdown or rapamycin, leads to destabilization of both L-VGCCs and RyRs, resulting in elevated Ca2+ influx, greater CICR, larger sAHPs, and impaired excitability/plasticity and memory. If additional studies support this model, FKBP-mediated Ca2+-regulatory pathways may become important new therapeutic targets for combating aging-related cognitive impairment and neurodegenerative disease.
Footnotes
This work was supported by National Institute on Aging Grants AG004542 and AG010836 and National Center for Research Resources Grant P20-RR15592.
References
- Berridge MJ. Elementary and global aspects of calcium signalling. J Physiol. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrout J, Isokawa M. Homeostatic and stimulus-induced coupling of the L-type Ca2+ channel to the ryanodine receptor in the hippocampal neuron in slices. Cell Calcium. 2009;46:30–38. doi: 10.1016/j.ceca.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Porter NM, Landfield PW. Decreased G-protein-mediated regulation and shift in calcium channel types with age in hippocampal cultures. J Neurosci. 1999;19:8674–8684. doi: 10.1523/JNEUROSCI.19-19-08674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Grondin R, Chen KC, Thibault O, Thibault V, Pandya JD, Dowling A, Zhang Z, Sullivan P, Porter NM, Landfield PW. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci. 2010;30:6058–6071. doi: 10.1523/JNEUROSCI.3956-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Kouser M, Powell CM. Systemic inhibition of mammalian target of rapamycin inhibits fear memory reconsolidation. Neurobiol Learn Mem. 2008;90:28–35. doi: 10.1016/j.nlm.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21:98–108. doi: 10.1523/JNEUROSCI.21-01-00098.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer's disease. Adv Drug Deliv Rev. 2008;60:1504–1511. doi: 10.1016/j.addr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TH, Jaffe DB. Calcium imaging in hippocampal neurons using confocal microscopy. Ann N Y Acad Sci. 1994;747:313–324. doi: 10.1111/j.1749-6632.1994.tb44419.x. [DOI] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mTOR, Aβ and tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LW, Hao SY, Thibault O, Blalock EM, Landfield PW. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J Neurosci. 1996;16:6286–6295. doi: 10.1523/JNEUROSCI.16-19-06286.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Chen KC, Blalock EM, Thibault O, Kaminker P, Landfield PW. Expression of alpha 1D subunit mRNA is correlated with L-type Ca2+ channel activity in single neurons of hippocampal “zipper” slices. Proc Natl Acad Sci U S A. 2000;97:4357–4362. doi: 10.1073/pnas.070056097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clodfelter GV, Porter NM, Landfield PW, Thibault O. Sustained Ca2+-induced Ca2+-release underlies the post-glutamate lethal Ca2+ plateau in older cultured hippocampal neurons. Eur J Pharmacol. 2002;447:189–200. doi: 10.1016/s0014-2999(02)01843-5. [DOI] [PubMed] [Google Scholar]
- Cracco JB, Serrano P, Moskowitz SI, Bergold PJ, Sacktor TC. Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus. 2005;15:551–556. doi: 10.1002/hipo.20078. [DOI] [PubMed] [Google Scholar]
- Crystal RG, Sondhi D, Hackett NR, Kaminsky SM, Worgall S, Stieg P, Souweidane M, Hosain S, Heier L, Ballon D, Dinner M, Wisniewski K, Kaplitt M, Greenwald BM, Howell JD, Strybing K, Dyke J, Voss H. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum Gene Ther. 2004;15:1131–1154. doi: 10.1089/hum.2004.15.1131. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Alterations in intrinsic neuronal excitability during normal aging. Aging Cell. 2007;6:327–336. doi: 10.1111/j.1474-9726.2007.00297.x. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 1996;59:413–420. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- Eitoku M, Sato L, Senda T, Horikoshi M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell Mol Life Sci. 2008;65:414–444. doi: 10.1007/s00018-007-7305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons HL, Bland RJ, During MJ. Promoters and regulatory elements that improve adeno-associated virus transgene expression in the brain. Methods. 2002;28:227–236. doi: 10.1016/s1046-2023(02)00227-x. [DOI] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–3490. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, Peterson C. Calcium and the aging nervous system. Neurobiol Aging. 1987;8:329–343. doi: 10.1016/0197-4580(87)90072-8. [DOI] [PubMed] [Google Scholar]
- Gómez AM, Rueda A, Sainte-Marie Y, Pereira L, Zissimopoulos S, Zhu X, Schaub R, Perrier E, Perrier R, Latouche C, Richard S, Picot MC, Jaisser F, Lai FA, Valdivia HH, Benitah JP. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation. 2009;119:2179–2187. doi: 10.1161/CIRCULATIONAHA.108.805804. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemond P, Jaffe DB. Caloric restriction prevents aging-associated changes in spike-mediated Ca2+ accumulation and the slow afterhyperpolarization in hippocampal CA1 pyramidal neurons. Neuroscience. 2005;135:413–420. doi: 10.1016/j.neuroscience.2005.05.044. [DOI] [PubMed] [Google Scholar]
- Herman JP, Chen KC, Booze R, Landfield PW. Up-regulation of alpha1D Ca2+ channel subunit mRNA expression in the hippocampus of aged F344 rats. Neurobiol Aging. 1998;19:581–587. doi: 10.1016/s0197-4580(98)00099-2. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MC, Seckl JR. The role of 11beta-hydroxysteroid dehydrogenases in the brain. Mol Cell Endocrinol. 2006;248:9–14. doi: 10.1016/j.mce.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Brown TH. Confocal imaging of dendritic Ca2+ transients in hippocampal brain slices during simultaneous current- and voltage-clamp recording. Microsc Res Tech. 1994;29:279–289. doi: 10.1002/jemt.1070290404. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- Jakob RP, Zoldák G, Aumüller T, Schmid FX. Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc Natl Acad Sci U S A. 2009;106:20282–20287. doi: 10.1073/pnas.0909544106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, de Kloet ER. Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science. 1989;245:1502–1505. doi: 10.1126/science.2781292. [DOI] [PubMed] [Google Scholar]
- Johnston LC, Eberling J, Pivirotto P, Hadaczek P, Federoff HJ, Forsayeth J, Bankiewicz KS. Clinically relevant effects of convection-enhanced delivery of AAV2-GDNF on the dopaminergic nigrostriatal pathway in aged rhesus monkeys. Hum Gene Ther. 2009;20:497–510. doi: 10.1089/hum.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I, Van Groen T. Low levels of estrogen significantly diminish axonal sprouting after entorhinal cortex lesions in the mouse. J Neurosci. 2002;22:4095–4102. doi: 10.1523/JNEUROSCI.22-10-04095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29:1805–1816. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16:318–325. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, During MJ. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- Kato H, Oikawa T, Otsuka K, Takahashi A, Itoyama Y. Postischemic changes in the immunophilin FKBP12 in the rat brain. Brain Res Mol Brain Res. 2000;84:58–66. doi: 10.1016/s0169-328x(00)00210-2. [DOI] [PubMed] [Google Scholar]
- Kells AP, Eberling J, Su X, Pivirotto P, Bringas J, Hadaczek P, Narrow WC, Bowers WJ, Federoff HJ, Forsayeth J, Bankiewicz KS. Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J Neurosci. 2010;30:9567–9577. doi: 10.1523/JNEUROSCI.0942-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DS, Campbell LW, Hao SY, Landfield PW. Corticosteroid modulation of hippocampal potentials: increased effect with aging. Science. 1989;245:1505–1509. doi: 10.1126/science.2781293. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging (Milano) [Erratum (1989) 1:II] 1989;1:17–34. doi: 10.1007/BF03323872. [DOI] [PubMed] [Google Scholar]
- Kim S, Yun HM, Baik JH, Chung KC, Nah SY, Rhim H. Functional interaction of neuronal Cav1.3 L-type calcium channel with ryanodine receptor type 2 in the rat hippocampus. J Biol Chem. 2007;282:32877–32889. doi: 10.1074/jbc.M701418200. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Voitenko N, Kostyuk P, Verkhratsky A. Age-associated changes of cytoplasmic calcium homeostasis in cerebellar granule neurons in situ: investigation on thin cerebellar slices. Exp Gerontol. 1996;31:475–487. doi: 10.1016/0531-5565(95)02070-5. [DOI] [PubMed] [Google Scholar]
- Klein RL, Hamby ME, Gong Y, Hirko AC, Wang S, Hughes JA, King MA, Meyer EM. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp Neurol. 2002;176:66–74. doi: 10.1006/exnr.2002.7942. [DOI] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol Ther. 2008;16:89–96. doi: 10.1038/sj.mt.6300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol. 2004;91:2437–2444. doi: 10.1152/jn.01148.2003. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging. Brain Res. 2005;1031:125–128. doi: 10.1016/j.brainres.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Landfield PW. “Increased calcium-current” hypothesis of brain aging. Neurobiol Aging. 1987;8:346–347. doi: 10.1016/0197-4580(87)90074-1. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226:1089–1092. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Blalock EM, Chen KC, Porter NM. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer's disease. Curr Alzheimer Res. 2007;4:205–212. doi: 10.2174/156720507780362083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Huang F, Marx SO, Marks AR. Immunophilins and coupled gating of ryanodine receptors. Curr Top Med Chem. 2003;3:1383–1391. doi: 10.2174/1568026033451907. [DOI] [PubMed] [Google Scholar]
- Long C, Cook LG, Wu GY, Mitchell BM. Removal of FKBP12/12.6 from endothelial ryanodine receptors leads to an intracellular calcium leak and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1580–1586. doi: 10.1161/ATVBAHA.107.144808. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. Synaptic plasticity in early aging. Ageing Res Rev. 2006;5:255–280. doi: 10.1016/j.arr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol. 1995;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson KR, Kresge D, Supon J. Differential effects of aging on NMDA receptors in the intermediate versus the dorsal hippocampus. Neurobiol Aging. 2006;27:324–333. doi: 10.1016/j.neurobiolaging.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Marks AR. Intracellular calcium-release channels: regulators of cell life and death. Am J Physiol. 1997;272:H597–H605. doi: 10.1152/ajpheart.1997.272.2.H597. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- McPhee SW, Janson CG, Li C, Samulski RJ, Camp AS, Francis J, Shera D, Lioutermann L, Feely M, Freese A, Leone P. Immune responses to AAV in a phase I study for Canavan disease. J Gene Med. 2006;8:577–588. doi: 10.1002/jgm.885. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Johe K, Kitos TE. Age-dependent alterations in synaptic membrane systems for Ca2+ regulation. Mech Ageing Dev. 1984;25:215–225. doi: 10.1016/0047-6374(84)90142-8. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE, Johnston D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- Moser MB, Moser EI. Functional differentiation in the hippocampus. Hippocampus. 1998;8:608–619. doi: 10.1002/(SICI)1098-1063(1998)8:6<608::AID-HIPO3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Mouravlev A, Dunning J, Young D, During MJ. Somatic gene transfer of cAMP response element-binding protein attenuates memory impairment in aging rats. Proc Natl Acad Sci U S A. 2006;103:4705–4710. doi: 10.1073/pnas.0506137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol. 1992;68:2100–2109. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- Murchison D, Griffith WH. Calcium buffering systems and calcium signaling in aged rat basal forebrain neurons. Aging Cell. 2007;6:297–305. doi: 10.1111/j.1474-9726.2007.00293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotera P, Zhivotovsky B, Orrenius S. Nuclear calcium transport and the role of calcium in apoptosis. Cell Calcium. 1994;16:279–288. doi: 10.1016/0143-4160(94)90091-4. [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Landfield PW. Calcineurin enhances L-type Ca2+ channel activity in hippocampal neurons: increased effect with age in culture. Neuroscience. 2002;110:213–225. doi: 10.1016/s0306-4522(01)00574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, McKay BM, Power JM, Disterhoft JF. Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A. Proc Natl Acad Sci U S A. 2009;106:1620–1625. doi: 10.1073/pnas.0807708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Gafford GM, Helmstetter FJ. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci. 2006;26:12977–12983. doi: 10.1523/JNEUROSCI.4209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NM, Landfield PW. Stress hormones and brain aging: adding injury to insult? Nat Neurosci. 1998;1:3–4. doi: 10.1038/196. [DOI] [PubMed] [Google Scholar]
- Porter NM, Thibault O, Thibault V, Chen KC, Landfield PW. Calcium channel density and hippocampal cell death with age in long-term culture. J Neurosci. 1997;17:5629–5639. doi: 10.1523/JNEUROSCI.17-14-05629.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasi F. Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front Biosci. 2002;7:d650–d658. doi: 10.2741/A801. [DOI] [PubMed] [Google Scholar]
- Regan LJ, Sah DW, Bean BP. Ca2+ channels in rat central and peripheral neurons: high-threshold current resistant to dihydropyridine blockers and omega-conotoxin. Neuron. 1991;6:269–280. doi: 10.1016/0896-6273(91)90362-4. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Carlen PL. Diminished calcium currents in aged hippocampal dentate gyrus granule neurones. Brain Res. 1989;479:384–390. doi: 10.1016/0006-8993(89)91646-6. [DOI] [PubMed] [Google Scholar]
- Rowe WB, Blalock EM, Chen KC, Kadish I, Wang D, Barrett JE, Thibault O, Porter NM, Rose GM, Landfield PW. Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J Neurosci. 2007;27:3098–3110. doi: 10.1523/JNEUROSCI.4163-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedarat F, Xu L, Moore ED, Tibbits GF. Colocalization of dihydropyridine and ryanodine receptors in neonate rabbit heart using confocal microscopy. Am J Physiol Heart Circ Physiol. 2000;279:H202–H209. doi: 10.1152/ajpheart.2000.279.1.H202. [DOI] [PubMed] [Google Scholar]
- Smith CC, Vedder LC, McMahon LL. Estradiol and the relationship between dendritic spines, NR2B containing NMDA receptors, and the magnitude of long-term potentiation at hippocampal CA3-CA1 synapses. Psychoneuroendocrinology. 2009;34(Suppl 1):S130–S142. doi: 10.1016/j.psyneuen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann N Y Acad Sci. 2007;1097:265–277. doi: 10.1196/annals.1379.025. [DOI] [PubMed] [Google Scholar]
- Su X, Kells AP, Huang EJ, Lee HS, Hadaczek P, Beyer J, Bringas J, Pivirotto P, Penticuff J, Eberling J, Federoff HJ, Forsayeth J, Bankiewicz KS. Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum Gene Ther. 2009;20:1627–1640. doi: 10.1089/hum.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Nakai M, Hashimoto T, Kawamata T, Taniguchi T, Yasuda M, Maeda K, Tanaka C. Single-channel activity of the Ca2+-dependent K+ channel is modulated by FK506 and rapamycin. Brain Res. 1998;786:255–258. doi: 10.1016/s0006-8993(97)01435-2. [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007;6:307–317. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. Neuronal ageing from an intraneuronal perspective: roles of endoplasmic reticulum and mitochondria. Cell Calcium. 2003;34:311–323. doi: 10.1016/s0143-4160(03)00142-8. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM. The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25:2609–2616. doi: 10.1523/JNEUROSCI.5023-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkikh A, Janus C, El-Beheiry H, Pennefather PS, Samoilova M, McDonald P, Ouanounou A, Carlen PL. Calcium chelation improves spatial learning and synaptic plasticity in aged rats. Exp Neurol. 2006;197:291–300. doi: 10.1016/j.expneurol.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- Tsien RY. Fluorescence measurement and photochemical manipulation of cytosolic free calcium. Trends Neurosci. 1988;11:419–424. doi: 10.1016/0166-2236(88)90192-0. [DOI] [PubMed] [Google Scholar]
- Veng LM, Browning MD. Regionally selective alterations in expression of the alpha1D subunit (Cav1.3) of L-type calcium channels in the hippocampus of aged rats. Brain Res Mol Brain Res. 2002;107:120–127. doi: 10.1016/s0169-328x(02)00453-9. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Endoplasmic reticulum calcium signaling in nerve cells. Biol Res. 2004;37:693–699. doi: 10.4067/s0716-97602004000400027. [DOI] [PubMed] [Google Scholar]
- Watabe AM, O'Dell TJ. Age-related changes in theta frequency stimulation-induced long-term potentiation. Neurobiol Aging. 2003;24:267–272. doi: 10.1016/s0197-4580(02)00082-9. [DOI] [PubMed] [Google Scholar]
- Zaiss AK, Muruve DA. Immune responses to adeno-associated virus vectors. Curr Gene Ther. 2005;5:323–331. doi: 10.2174/1566523054065039. [DOI] [PubMed] [Google Scholar]
- Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]