

Abstract
PRDM9 has recently been identified as a likely trans-regulator of meiotic recombination hot spots in humans and mice1-3. The protein contains a zinc finger array that in humans can recognise a short sequence motif associated with hot spots4, with binding to this motif possibly triggering hot-spot activity via chromatin remodelling5. We now show that variation in the zinc finger array in humans has a profound effect on sperm hot-spot activity, even at hot spots lacking the sequence motif. Very subtle changes within the array can create hot-spot non-activating and enhancing alleles, and even trigger the appearance of a new hot spot. PRDM9 thus appears to be the preeminent global regulator of hot spots in humans. Variation at this locus also influences aspects of genome instability, specifically a megabase-scale rearrangement underlying two genomic disorders6 as well as minisatellite instability7, implicating PRDM9 as a risk factor for some pathological genome rearrangements.
PR domain-containing 9 (PRDM9) is a meiosis-specific histone H3 methyltransferase with a C-terminal tandem-repeat C2H2 zinc finger (ZnF) domain encoded by a minisatellite8. Mouse sub-species hybrids can show variation in recombination profiles and crossover hot-spot activity, depending on the Prdm9 alleles they carry3. In humans, evidence that PRDM9 influences recombination is less direct. First, ~40% of human hot spots contain the motif CCNCCNTNNCCNC that might serve as a binding site for a ZnF protein4,9 and bioinformatic analysis has identified PRDM9 as the human ZnF protein most likely to recognise this motif2. Second, the most common PRDM9 variant (allele A) binds this motif in vitro1. Third, individuals in a single pedigree carrying the variant PRDM9 allele I, coding a ZnF array that cannot bind this motif, show a genome-wide shift in hot-spot usage as shown by linkage analysis1.
We now test the influence of variation in the PRDM9 ZnF array on human recombination hot spots. The hypothesis is straightforward – hot spots containing a motif should show high activity in sperm from men carrying two motif-recognising PRDM9 alleles, and lower activity in men lacking such alleles. Conversely, hot spots lacking the motif will presumably be regulated by some other mechanism and should not be influenced by PRDM9 status. Limited PRDM9 variability in Europeans1,3 makes this test difficult. We therefore surveyed ZnF alleles in a panel of 74 African semen donors as well as 156 Europeans, revealing a diversity of alleles with 8–18 ZnFs, including known alleles A–E1 plus 24 novel alleles (L1–L24) (Fig. 1a, Supplementary Fig. 1). Allele I associated with a shift in hot-spot usage1 was not seen. Diversity in Africans was high3 (Fig. 1b) and classification of alleles according to their predicted ability to recognise the hot-spot motif (Fig. 1a, Supplementary Fig. 1) showed that nearly half should have impaired binding relative to the common A allele (Fig. 1b), thus providing a key resource for analysing PRDM9 effects.
Figure 1.
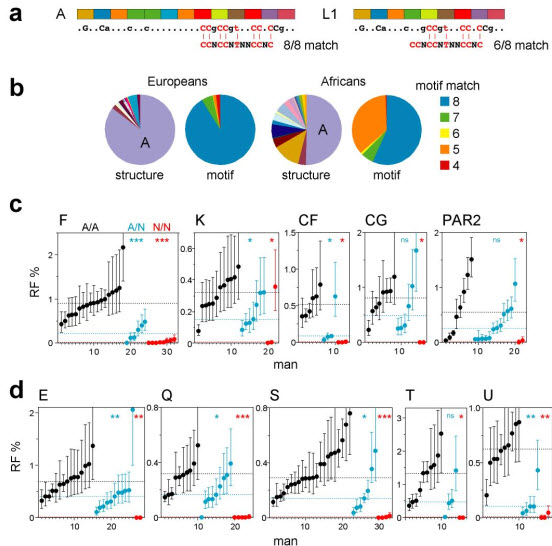
PRDM9 ZnF variants and crossover hot-spot activity in sperm. (a) Examples of tandem repeats encoding the ZnF array, with variant repeat units coloured differently. The predicted DNA binding sequence is shown below, with dots indicating weakly and uppercase the most strongly predicted bases, and aligned with the hot-spot motif CCNCCNTNNCCNC (ref. 4). The binding sequence for allele A matches all 8 specified bases in the motif, while allele L1 matches at best only 6 of the 8 bases. (b) PRDM9 ZnF array diversity in Europeans and Africans, with alleles classified either by structure or by the strength of the predicted match with the motif (details of alleles provided in Supplementary Fig. 1). (c) Variation between men in sperm crossover activity in hot spots, named above each panel, containing a central hot-spot motif. Different sets of men informative for SNPs required for crossover detection were analysed at each hot spot (Supplementary Table 1). Men carrying two PRDM9 A alleles (A/A, black), one A allele (A/N, blue) or two non-A alleles (N/N, red) were grouped separately in ascending order. Confidence intervals for each estimate of RF are shown, and median RFs within each group are indicated by dotted lines. Mann-Whitney test results for the significance of differences between the A/A group and the A/N or N/N groups are given at top right (ns P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001). (d) Corresponding analyses of hot spots lacking an obvious hot-spot motif (Supplementary Fig. 2).
We selected ten very active crossover hot spots for study, five with a central motif plus five lacking a clear motif10,11 (Supplementary Fig. 2). For each hot spot, we identified semen donors amenable to crossover detection12, and from these selected men carrying two common A alleles (A/A), plus men carrying one non-A allele (A/N) or two non-A alleles (N/N). Multiple men were analysed to smooth out variation caused by factors such as variable DNA quality and possible cis-acting variants11,13,14 that could influence recombination frequency (RF) estimates. Simple comparison of RFs in A/A, A/N and N/N men showed a dramatic effect, with almost all testable N/N men showing heavy reduction (>30-fold) at all hot spots, even at those that do not contain an obvious hot-spot motif (Fig. 1c, d, Supplementary Fig. 2). This association between N/N status and very low RF is highly significant. Thus, ranking of 112 men by RF over all hot spots tested (see Methods) showed that the 17 lowest-ranked individuals included 14 of the 15 N/N men (Supplementary Table 1) (P = 5×10−16). This association remained in a sub-group of men of Southern East African descent (11 N/N men all ranked lowest in 38 men tested, P = 2×10−9), ruling out differences between populations at other loci as significant confounding variables. PRDM9 is therefore a major regulator of human hot spots.
The strong RF suppression seen in most N/N men implies that most PRDM9 non-A alleles must be incapable of triggering activity at the hot spots tested. However, activating N alleles do exist as shown by one man fully active at hot-spot K (Fig. 1c) who carried two N alleles (L9, L10) both predicted to bind the motif (Supplementary Fig. 1), though it is impossible to determine whether both alleles are activating. As expected, A/N men tended to show significantly lower RFs than A/A men over all 10 hot spots (P = 7×10−18 for all hot spots combined) (Fig. 1c, d). Median RFs in A/N men were 41±16% of those in A/A men, consistent with a simple additive model whereby hot-spot activity is proportional to the number of activating alleles present.
Fourteen different PRDM9 alleles were identified as non-activators in heavily-suppressed N/N men, in each case failing to activate any testable hot spot (Fig. 2). Thus allele C, which is common in Africans and has major changes in the ZnF array, could not activate any of the nine testable hot spots. Surprisingly, there was no obvious relationship between the inability of an allele to activate and its predicted binding motif, with even subtle changes in the ZnF array creating hot-spot non-activating alleles. For example, allele L20 is closely related to the recombination-promoting allele A, differing only by a single Asn→His substitution in the 10th ZnF. This substitution is located at one of the four key DNA-contact residues in C2H2 ZnFs (position 3 of the −1, 2, 3 and 6 residues; Supplementary Fig. 1)1,2,15 and should alter DNA-binding characteristics. More remarkably, non-activating allele L13 is identical to allele A except for a Ser→Arg substitution at the non-contact position −2 in the 11th ZnF, a change not predicted to alter DNA binding15. This inability to activate hotspots might be due to allele L13 carrying an additional alteration that inactivates the gene, though such null alleles should be rare given that Prdm9 disruption in mice causes sterility8.
Figure 2.
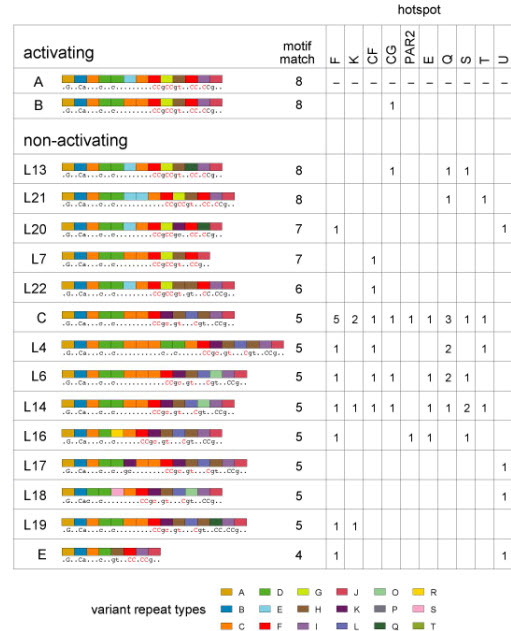
Activating and non-activating PRDM9 alleles. Non-activating alleles at each hot spot were defined as alleles present in N/N men who showed <5% of the median RF seen in A/A men. Allele structures are coded as in Supplementary Fig. 1, with predicted DNA binding sequences and best motif matches shown as in Fig. 1. Data for each hot spot give the number of specific N alleles detected in suppressed men; for example, 5 such men typed at hot-spot F carried the C allele. Evidence that the B allele is active is based on a B/L6 heterozygote assayable only at hot-spot CG who showed crossovers at 40% of the median frequency seen in A/A homozygotes. Since allele L6 is a non-activator at CG, this implies that B is similar in activity to A.
We extended the search for PRDM9 influences to a pair of hot spots (MSTM1a,b; Supplementary Fig. 2) previously shown to vary in sperm crossover activity independently of local DNA sequence16. MSTM1b showed 75-fold variation in activity (Fig. 3a), and the lowest-ranked man carried the non-A allele L2, an association of borderline significance (P = 0.04). Intriguingly, the four men with the highest RF all carried allele L20, a significant association (P = 0.0002) establishing that L20 is a recombination-enhancing allele for MSTM1b. Under a simple additive model, we estimate that this allele enhances recombination ~13-fold relative to allele A. Remarkably, L20 cannot activate hot-spots F and U (Fig. 2), indicating that PRDM9 alleles can have different influences on different hot spots.
Figure 3.

PRDM9 variation influences crossover activity at hot-spots MSTM1a and MSTM1b. (a) Variant alleles present in men previously typed for sperm crossovers at these hot spots16; all other men were A/A homozygotes. RFs at each hot spot are shown, together with 95% CIs. Note that alleles L9/L24 associate not only with MSTM1a activity but also apparently with elevated RF at MSTM1b (P = 0.035). (b) DNA sequence structures of these alleles, coloured as in Fig. 2, plus amino acid sequence changes relative to allele A, with locations given with respect to the main ZnF DNA-contact residues (−1, 2, 3 and 6)15.
The neighbouring hot-spot MSTM1a was active in only three of the 25 men surveyed16. These all carried the non-A alleles L9 or L24 (Fig. 3a) that differ from each other by a single synonymous base substitution and thus code for the same ZnF array. This association between L9/L24 and hot-spot activity is significant (P = 0.0004). Alleles L9/L24 differ from allele A by a single Lys→Glu replacement not predicted to affect DNA binding (Fig. 3b). Thus, a very subtle change in amino acid sequence can trigger the appearance of a new hot spot.
The hot-spot motif is also associated with genomic regions prone to rearrangement4. We therefore extended the PRDM9 survey to modes of meiotic genome instability detectable as de novo events in sperm. Small-pool PCR analysis of three highly-unstable minisatellites17-19, which show instability driven substantially by meiotic gene conversion-like events, revealed a major PRDM9 effect with 10–30-fold reduction of instability in N/N men as seen at crossover hot spots (Fig. 4). Rank-order analysis of men showed that low minisatellite instability and N/N status are significantly associated (P = 0.000015). PRDM9 clearly regulates minisatellite instability, though it is unclear whether this occurs by interaction of the ZnF array with hot-spot motifs present within the minisatellites themselves (Fig. 4a)4 or with a flanking recombination hot spot that in turn drives repeat turnover20,21.
Figure 4.

Influence of PRDM9 variation on minisatellite instability in sperm. (a) Minisatellite repeat units, with the best matches to the hot-spot motif indicated. Minisatellites MS1 and CEB1 both contain perfect matches. Two contiguous repeats are shown for MS1. (b) Representative small-pool PCR results for minisatellite CEB1, for a PRDM9 A/A homozygote and C/L16 heterozygote. (c) Mutation frequencies in A/A (black), A/N (blue) and N/N (red) men, with CIs plus median values per group indicated as in Fig. 1.
The genomic disorders Charcot-Marie-Tooth type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP) result from unequal meiotic exchanges between 1.5–Mb-separated CMT1A-REP sequences22, and de novo rearrangements in sperm can be detected by PCR amplification of the exchange junctions6. Exchanges cluster into a narrow hot spot that contains a hot-spot motif4, and which may also function in allelic crossover23. Analysis of the frequency of these rearrangements in sperm again showed a major PRDM9 effect, with ~25-fold reduction of unequal exchanges in N/N men and reduced instability in A/N men (Fig. 5).
Figure 5.

PRDM9 variation and de novo HNPP/CMT1A rearrangements in sperm DNA. (a) Detection of HNPP deletion junctions in sperm DNA from a PRDM9 A/A homozygote and a C/L14 heterozygote, with 40 ng DNA input per PCR reaction. (b) Rearrangement frequencies in A/A (black), A/N (blue) and N/N (red) men for HNPP deletions and CMT1A duplications, with CIs and median values per group shown as in Fig. 1. The same men were analysed for both rearrangements, but not all men were typed for duplications, which arise at a lower frequency than deletions6. Rearrangements in blood were rare6 (<2.2×10−6 for deletions, <1.3×10−6 for duplications, P > 0.95).
We also tested whether PRDM9 alleles influence the frequency of the recurrent translocation t(11;22)(q23;q11). Translocation junctions map within a short palindromic AT-rich repeat (PATRR), allowing de novo translocations to be detected by PCR24. As found previously24, we saw these translocations in sperm but not somatic DNA (Fig. 6a). Sperm frequencies varied widely between men (Fig. 6b), reflecting the sensitivity of the process to variation in the PATRRs25. However, comparison of PRDM9 A/A, A/N and N/N men showed no significant influence of PRDM9 on translocation frequency, with indistinguishable median frequencies, no evidence for suppression in N/N men and no indication of specific non-A alleles associated with unusual translocation frequencies. This suggests that, while these translocations are probably meiotic in origin24, they are not facilitated by PRDM9-mediated chromatin remodelling.
Figure 6.

PRDM9 variation and t(11;22) translocation frequencies. (a) Detection of de novo der(22) translocation junctions by nested PCR amplification29 of multiple 150 ng aliquots of blood and sperm DNA. Minor variation in junction size results from differences in translocation breakpoint locations within the PATRR24. (b) der(22) translocation frequencies, with CIs, in sperm DNA in PRDM9 A/A, A/N and N/N men, with identities of N alleles shown above. The median frequency per group, similar to previously-reported translocation frequencies24, is indicated by a dotted line. There is no significant difference in translocation frequency between groups (Kruskal-Wallis test, P = 0.98). No de novo translocations were seen in blood DNA tested from four different men (frequency < 5×10–7, P > 0.95).
Variation within the PRDM9 ZnF domain thus strongly influences hot-spot activity. The differential effects of allele L20 on different hot spots is consistent with PRDM9 directly interacting with hot spots. The independent variation seen at hot-spots MSTM1a/b, with centres only 2 kb apart, suggests an interaction highly local to the hot spot and could argue against extended chromatin domains being opened by PRDM9. It is surprising how sensitive hot spots can be to minor variation within the ZnF domain, with variant alleles differing by a single amino acid substitution that can enhance or fail to activate hot spots or even trigger the appearance of a new hot spot. This raises a fascinating conundrum – how can PRDM9 variants predicted not to influence DNA binding have a major impact on hot-spot activity, while hot-spot promoting alleles such as A can activate hot spots lacking an obvious hot-spot motif? If PRDM9 does indeed regulate hot spots by binding to the motif, as suggested by ZnF bioinformatic surveys2 and in vitro studies1, then the rules governing PRDM9/hot-spot interactions must be subtle and complex, and most likely only addressable by in vivo studies. In any event, these rules are evidently not well captured by consensus motifs established from large-scale comparisons of historical hot spots4, nor do such motifs provide clear predictions of hot-spot locations (Supplementary Fig. 2).
PRDM9 status is a major risk factor for de novo CMT1A/HNPP rearrangements, with non-A alleles being strongly protective in N/N individuals, at least in males. Substantial shifts in PRDM9 allele frequencies between populations imply corresponding shifts in the incidence of de novo rearrangements. They also suggest major changes in crossover hot spots used in different populations. If variant PRDM9 alleles trigger the appearance of new hot spots genome-wide (as implied by the L9/L24-specific appearance of hot-spot MSTM1a), then Africans are likely to use a relatively broad repertoire of hot spots, providing an explanation, in addition to greater population age, for the relatively short haplotype blocks and rapid decay of linkage disequilibrium (LD) seen in Africans26. One challenge will be to identify African-specific hot spots and to see whether they too are “tuned” to a specific subset of PRDM9 alleles. We note that almost all hot spots in the present study were initially identified from LD breakdown in Europeans10,11 and were therefore likely to be activated by PRDM9 allele A. The only exception was hot-spot MSTM1a, which has not left a mark on LD in Europeans16 and is not activated by this allele.
Are there other trans-regulators, equivalent to PRDM9, of meiotic recombination hot spots in humans? Even though all hot spots analysed to date are regulated by PRDM9, substantial variation in RF can remain between men of the same PRDM9 status (for example among A/A men at hot-spot S, Fig. 1d). Such variation might provide a lead to other trans-regulators such as loci already identified that associate with variation in linkage map lengths27,28. Finally, the influence of PRDM9 ZnF variants on minisatellite instability is intriguing, given that these variants themselves are encoded by a minisatellite. Could PRDM9 influence its own evolution, by generating alleles that affect instability at the PRDM9 minisatellite to promote the generation of new alleles? If so, then bursts of new PRDM9 alleles could be generated whenever an interacting allele appears in a population, implying a potentially chaotic mode of hot-spot evolution.
METHODS
Characterising PRDM9 ZnF alleles
Semen samples were collected with approval from the Leicestershire Health Authority Research Ethics Committee and with informed consent. The collection of African samples has been described previously30. DNAs were extracted and manipulated under conditions designed to minimise the risk of contamination20. PRDM9 minisatellite alleles were amplified from 230 men using primers PN0.6F and PN2.5R (see Supplementary Table 2) designed not to amplify the paralogous locus PRDM71. Alleles in men showing two different length minisatellite alleles were separated by agarose gel electrophoresis and sequenced with primers PN1.2F and PN2.4R (Supplementary Table 2) using BigDye Terminator v3.1 Cycle Sequencing (Applied Biosystems) on a 3730 DNA Analyzer (Applied Biosystems). All remaining length homozygotes who were subsequently subjected to crossover analysis (almost exclusively men carrying two 13-repeat alleles) were sequenced from diploid PCR products. In most instances, non-mixed sequence traces were obtained that matched PRDM9 allele A, indicating A/A homozygotes. All remaining cases of mixed traces could be fully explained by heterozygosity for allele A and another already-characterised allele, allowing allele status to be deduced. In total, 300 PRDM9 alleles were sequenced. The DNA binding sequence predicted for each allele was established as described by Baudat et al. (ref. 1) and simplified to show the most strongly predicted bases, in lowercase for >80% consensus for a specific base and uppercase for >95% consensus across >60 observations. Analysis of sperm counts estimated from DNA yields across all Africans showed no significant differences between A/A, A/N and N/N men (Kruskal-Wallis test, 2 df, P = 0.97), suggesting no obvious effect of PRDM9 ZnF variation on fertility.
Hot-spot analysis
A panel of donors including all 74 Africans plus 98 Europeans, including those with atypical-length PRDM9 alleles, was genotyped for SNPs across 10 different hot spots. Hot-spots E, F, K, Q and S have been described previously10,11. Hot-spot S is a doublet in some Europeans, with the second hot spot triggered by a single base substitution in cis11; however, this substitution was absent from all additional men tested in the present study who will therefore only contain a single hot spot. Details of the remaining five hot spots (T, U, CF, CG, PAR2) are provided in Supplementary Fig. 2. Men for crossover analysis were selected based on appropriate hot-spot genotypes and PRDM9 status. Crossovers were analysed as described previously12 using nested repulsion-phase allele-specific long PCR to selectively amplify crossover molecules from sperm DNA. Shifts in SNP frequencies in Africans at some hot spots forced the development of additional allele-specific primers to maximise the number of analysable men; a full list of primers is provided in Supplementary Table 3. For each hot spot and man, we typically analysed 4×12 aliquots of sperm DNA estimated from RF data to each contain 0.5, 1.0, 2.0 and 4.5 crossover molecules per aliquot; this strategy provided reliable estimates of RF in active men, and sufficient power to detect major (>30 fold) reduction in RF in inactive men. All DNA concentrations were quantified on a NanoDrop 1000 spectrophotometer, and a single DNA molecule PCR efficiency of 50% was assumed throughout10. A full listing of RFs in all men tested at each hot spot is provided in Supplementary Table 1. In rank-order analysis of RFs in groups of men, PRDM9 allele B was treated as an A allele given its similarity to A and its lack of major hot-spot–shifting effects in linkage maps1, plus evidence that it is fully active (Fig. 2); further evidence for B activity came from A/B heterozygotes, assayed at hot-spots F, CF, CG, S and U, who showed crossover activity at 80% of the median values seen in A/A homozygotes. To assess associations between PRDM9 status and RF, we normalised RFs at each hot spot in each man with respect to the median value seen in all A/A men. We then averaged these normalised RFs over all hot spots tested in a man. Men were then ranked using these mean RFs and the association between low RF and N/N status assessed by the Mann-Whitney test. This approach is conservative since RF suppression over multiple hot spots in a man was treated as a single observation.
Additional analyses of crossovers
Crossover locations already established for hot-spots MSTM1a/b (ref. 16), plus those analysed over men typed at hot-spot PAR2, showed no significant shifts in hot-spot location between men. PAR2 contains a central hot-spot motif with a motif-disrupting SNP (rs700442) that suppressed crossover activity by ~3-fold. This cis-acting influence was ignored in Fig. 1c, though similar conclusions about PRDM9 influence were drawn for men matched for allelic status at this SNP. A full description of this hot spot will be provided elsewhere. Possible cis-acting SNPs at hot-spots E and F were also checked by resequencing all analysed donors over a 1.5 kb interval spanning the centre of the hot spot; no obvious cis effects were detected.
Analysis of minisatellite instability
Men were typed for allele length at all three minisatellites17-19, and individuals carrying two relatively short (0.6–5.2 kb) alleles were chosen for mutation analysis to facilitate mutant resolution. De novo mutants were analysed by small-pool PCR of multiple aliquots of sperm DNA each containing 0.2–0.4 ng DNA (40–80 haploid genomes, assuming 60% single molecule PCR efficiency), followed by agarose gel electrophoresis and Southern blot hybridisation with a 32P-labelled minisatellite probe17-19. Mutation frequencies in all men tested are provided in Supplementary Table 1.
CMT1A/HNPP rearrangements
Exchange junctions within the CMT1A-REPs were the same as described by Turner et al. (ref. 6) but were detected using different paralogue-specific PCR primers (listed in Supplementary Table 3). Primer sites in both REPs were sequenced in all men selected for analysis, and individuals carrying a variant at any site were excluded. De novo exchanges were detected using nested paralogue-specific PCR on typically 72 PCR reactions per man each seeded with 40–80 ng sperm DNA (6,700–13,300 haploid genomes, assuming 50% single molecule PCR efficiency), in parallel with 12–24 PCR reactions seeded with 60–80 ng blood DNA as a negative control6. Cycling for HNPP deletions was at 96°C for 1 min, followed by 28/34 cycles (1°/2° PCR) at 96°C for 20 sec, 49°C/48°C (1°/2° PCR) for 30 sec, and 66°C for 2.5 min. Cycling for CMT1A duplications was the same but with annealing at 46°C/49°C (1°/2° PCR). Rearrangement frequencies in all men tested are provided in Supplementary Table 1.
Translocations
De novo der(22) junctions arising from t(11;22) translocations were detected by nested PCR using primers and amplification conditions described previously29, but with the PCR extension temperature reduced to 60°C to facilitate amplification across the AT-rich junction. Twenty-four PCR reactions, each seeded with 75–150 ng sperm DNA, were analysed per man, with additional assays at lower DNA inputs for men showing the highest translocation frequencies. Assays for the reciprocal der(11) junction29 performed on five men yielded translocation frequencies indistinguishable from der(22) frequencies. Translocation frequencies in all men tested are provided in Supplementary Table 1.
ACKNOWLEDGMENTS
We thank J. Blower and volunteers for providing semen samples, colleagues for helpful discussions, and the Medical Research Council, the Wellcome Trust (ref. 081227/Z/06/Z), the Boehringer Ingelheim Fonds, the Royal Society and the Louis-Jeantet Foundation for funding support.
Footnotes
PRDM9 SEQUENCES
Sequences of novel alleles L1–L24 have been deposited in GenBank, accession numbers HM210983–HM211006.
References
- 1.Baudat F, et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myers S, et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 5.Paigen K, Petkov P. Mammalian recombination hot spots: properties, control and evolution. Nat. Rev. Genet. 2010;11:221–233. doi: 10.1038/nrg2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeffreys AJ, et al. Complex gene conversion events in germline mutation at human minisatellites. Nat. Genet. 1994;6:136–145. doi: 10.1038/ng0294-136. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi K, Yoshida K, Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–378. doi: 10.1038/nature04112. [DOI] [PubMed] [Google Scholar]
- 9.The International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb AJ, Berg IL, Jeffreys A. Sperm cross-over activity in regions of the human genome showing extreme breakdown of marker association. Proc. Natl. Acad. Sci. USA. 2008;105:10471–10476. doi: 10.1073/pnas.0804933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeffreys AJ, Neumann R. The rise and fall of a human recombination hot spot. Nat. Genet. 2009;41:625–629. doi: 10.1038/ng.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeffreys AJ, Ritchie A, Neumann R. High resolution analysis of haplotype diversity and meiotic crossover in the human TAP2 recombination hotspot. Hum. Mol. Genet. 2000;9:725–733. doi: 10.1093/hmg/9.5.725. [DOI] [PubMed] [Google Scholar]
- 13.Jeffreys AJ, Neumann R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat. Genet. 2002;31:267–271. doi: 10.1038/ng910. [DOI] [PubMed] [Google Scholar]
- 14.Jeffreys AJ, Neumann R. Factors influencing recombination frequency and distribution in a human meiotic crossover hotspot. Hum. Mol. Genet. 2005;14:2277–2287. doi: 10.1093/hmg/ddi232. [DOI] [PubMed] [Google Scholar]
- 15.Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 1999;3:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 16.Neumann R, Jeffreys AJ. Polymorphism in the activity of human crossover hotspots independent of local DNA sequence variation. Hum. Mol. Genet. 2006;15:1401–1411. doi: 10.1093/hmg/ddl063. [DOI] [PubMed] [Google Scholar]
- 17.Buard J, Bourdet A, Yardley J, Dubrova Y, Jeffreys AJ. Influences of array size and homogeneity on minisatellite mutation. EMBO J. 1998;17:3495–3502. doi: 10.1093/emboj/17.12.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamaki K, May CA, Dubrova YE, Jeffreys AJ. Extremely complex repeat shuffling during germline mutation at human minisatellite B6.7. Hum. Mol. Genet. 1999;8:879–888. doi: 10.1093/hmg/8.5.879. [DOI] [PubMed] [Google Scholar]
- 19.Berg I, Neumann R, Cederberg H, Rannug U, Jeffreys AJ. Two modes of germline instability at human minisatellite MS1 (locus D1S7): complex rearrangements and paradoxical hyperdeletion. Am. J. Hum. Genet. 2003;72:1436–1447. doi: 10.1086/375629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffreys AJ, Murray J, Neumann R. High-resolution mapping of crossovers in human sperm defines a minisatellite-associated recombination hotspot. Mol. Cell. 1998;2:267–273. doi: 10.1016/s1097-2765(00)80138-0. [DOI] [PubMed] [Google Scholar]
- 21.Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat. Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- 23.Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am. J. Hum. Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurahashi H, Emanuel BS. Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat. Genet. 2001;29:139–140. doi: 10.1038/ng1001-139. [DOI] [PubMed] [Google Scholar]
- 25.Kato T, et al. Genetic variation affects de novo translocation frequency. Science. 2006;311:971. doi: 10.1126/science.1121452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reich DE, et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 27.Kong A, et al. Sequence variants in the RNF212 gene associate with genome-wide recombination rate. Science. 2008;319:1398–1401. doi: 10.1126/science.1152422. [DOI] [PubMed] [Google Scholar]
- 28.Chowdhury R, et al. Genetic analysis of variation in human meiotic recombination. PLoS Genet. 2009;5:e1000648. doi: 10.1371/journal.pgen.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurahashi H, et al. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22) Am. J. Hum. Genet. 2000;67:763–768. doi: 10.1086/303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monckton DG, et al. Minisatellite mutation rate variation associated with a flanking DNA sequence polymorphism. Nat. Genet. 1994;8:162–170. doi: 10.1038/ng1094-162. [DOI] [PubMed] [Google Scholar]