

Abstract
The JAK/STAT pathway has pleiotropic roles in animal development, and its aberrant activation is implicated in multiple human cancers1–3. JAK/STAT signaling effects have been attributed largely to direct transcriptional regulation by STAT of specific target genes that promote tumor cell proliferation or survival. We show here in a Drosophila melanogaster hematopoietic tumor model, however, that JAK overactivation globally disrupts heterochromatic gene silencing, an epigenetic tumor suppressive mechanism4. This disruption allows derepression of genes that are not direct targets of STAT, as evidenced by suppression of heterochromatin-mediated position effect variegation. Moreover, mutations in the genes encoding heterochromatin components heterochromatin protein 1 (HP1) and Su(var)3-9 enhance tumorigenesis induced by an oncogenic JAK kinase without affecting JAK/STAT signaling. Consistently, JAK loss of function enhances heterochromatic gene silencing, whereas overexpressing HP1 suppresses oncogenic JAK-induced tumors. These results demonstrate that the JAK/STAT pathway regulates cellular epigenetic status and that globally disrupting heterochromatin-mediated tumor suppression is essential for tumorigenesis induced by JAK overactivation.
The D. melanogaster genome contains a single JAK, named Hopscotch (Hop), and a single STAT, STAT92E, that are most similar to JAK2 and STAT5, respectively5–7. Hop and STAT92E function in a canonical JAK/STAT pathway, mediating cell proliferation, differentiation and migration in a variety of developmental processes7–9. Tumorous-lethal (Tum-l) is an oncogenic allele of hop and encodes a hyperactive JAK kinase due to a G341E substitution10–12. hopTum-l is associated with high incidence of hematopoietic tumors in heterozygous animals, a leukemia-like phenotype. These tumors manifest as melanotic masses of blood cell aggregates in the larval or adult body cavity, resulting from overproliferation and differentiation of particular blood cell types10,11. It has been shown that Tum-l induces hematopoietic tumors by overactivating STAT92E, as STAT92E is hyperphosphorylated by HopTum-l, and that reducing the gene dosage of STAT92E suppresses Tum-l tumorigenicity5,6,13. Classical genetic screens and genome-wide RNA interference (RNAi) analyses in D. melanogaster have led to the identification of many components involved in the JAK/STAT pathway or in its modulation5,6,13–15. However, as in human leukemias, the genes underlying tumorigenesis induced by overactivation of the JAK/STAT pathway remain unclear.
It has become increasingly clear that perturbation of epigenetic gene regulation is important in tumorigenesis16,17. Epigenetic gene regulation refers to stably heritable activation or repression of gene expression via covalent modifications of DNA or histones without changes in the DNA sequence of the gene18,19. Local hypermethylation of tumor-suppressor genes and global hypomethylation of genomic DNA are both commonly found in human cancers and can have a causal role in tumorigenesis16,17. Histone modifications represent another type of epigenetic gene regulation. For instance, methylation of histone H3 at Lys9 (H3mK9), catalyzed by methyltransferase Su(var)3-9 homologs, is associated with heterochromatin formation and heterochromatic gene silencing. H3mK9 provides binding sites for HP1, which in turn recruits Su(var)3-9 and associated proteins, leading to heterochromatin assembly and spreading20. Recently, it has been shown that induction of heterochromatin formation mediated by Su(var)3-9 can function as a tumor-suppressive mechanism that limits cancer development4.
In order to understand how overactivation of the JAK/STAT pathway causes hematopoietic tumors, we undertook a genetic screen to identify genes important for the tumorigenicity of hopTum-l. We first screened a collection of chromosomal deficiencies (Dfs), which altogether span over 70% of the autosomes, for dominant enhancers and suppressors of Tum-l tumorigenesis. We quantified Tum-l tumorigenicity by tumor index (see Methods and Fig. 1a). Among the 208 Df stocks tested, we selected 32 enhancers, termed E(Tum-l), and five suppressors, termed Su(Tum-l), whose tumor index values (in hopTum-l/+; Df/+ flies) differed by more than two standard deviations from the mean of the control crosses (Fig. 1b; see Methods). We then tested mutant alleles of genes disrupted by these Dfs. This allowed us to determine, for most of the Dfs, the genes responsible for the modification of hopTum-l tumorigenicity (see Methods and Supplementary Table 1 online).
Figure 1.
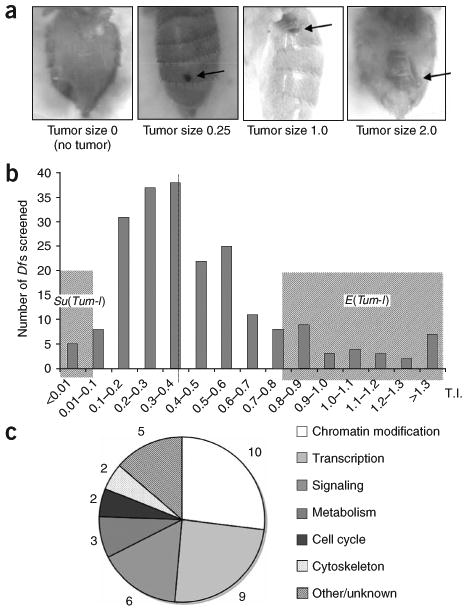
Genetic mutations influence hopTum-l hematopoietic tumorigenicity. (a) Examples of different sizes of hematopoietic tumors (arrow) associated with hopTum-l. Adult fly abdomens are shown. (b) Distribution of tumor indices (TI) of hopTum-l/+; Df/+ flies. The F1 progeny with the hopTum-l/+; Df/+ genotype from hopTum-l/+ females and different deficiency-carrying males were analyzed and TIs were calculated. Each bar represents the number of crosses in which the TIs fall in the indicated range. Dashed line indicates the mean TI (0.39 ± 0.11). Shaded regions indicate chromosomal deficiencies designated as E(Tum-l) or Su(Tum-l). (c) Distribution of hopTum-l modifiers in each category by number.
We then assigned these modifiers of hopTum-l to different categories based on their known molecular function (Supplementary Table 1 and Fig. 1c). Among the hopTum-l modifiers identified, several are known to interact with the JAK/STAT pathway or to be involved in blood or tumor formation. These include the human tumor suppressor nm23-H1 homolog abnormal wing discs (awd), Toll, PIAS, NURF301 and Serrate (Ser) (Supplementary Table 1)7,15,21–24, supporting the specificity of this screen.
A major class of E(Tum-l) genes identified in this screen encode proteins involved in chromatin modification. These include HP1, the histone H3-K9 methyltransferase Su(var)3-9, histone deacetylase (HDAC) Rpd3 and several other chromodomain-containing proteins (Fig. 1c and Supplementary Table 1). Animals transheterozygous for hopTum-l and either Su(var)205 (encoding HP1), Su(var)3-9 or Rpd3 demonstrated significantly increased numbers of melanotic tumors and larval blood cells (Fig. 2a,b). Animals heterozygous (Fig. 2a,b) or even hemizygous (data not shown) for Su(var)205, Su(var)3-9 or Rpd3 mutations alone, however, did not show melanotic tumors or increases in blood cell numbers. This suggests that a loss of either HP1, Su(var)3-9 or Rpd3 alone is not sufficient for tumorigenesis in the absence of hopTum-l and further suggests that these chromatin-modifying proteins may not modulate JAK/STAT signaling activity.
Figure 2.
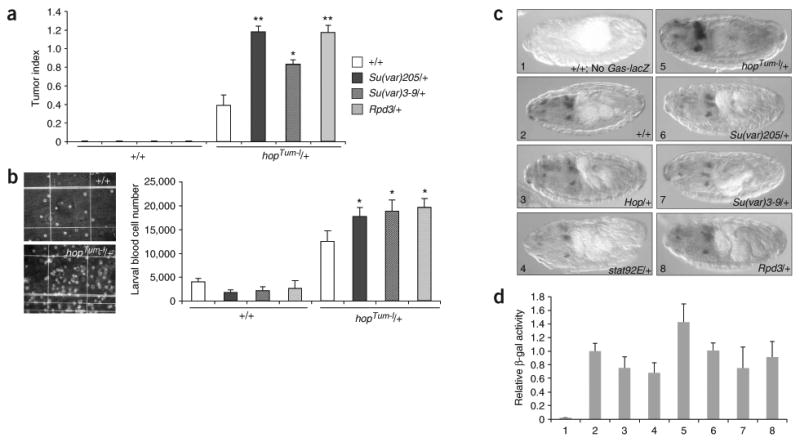
Su(var)205, Su(var)3-9 and Rpd3 mutations enhance hopTum-l tumorigenicity without affecting JAK/STAT signaling. (a) Mutations in Su(var)205, Su(var)3-9 and Rpd3 dominantly enhance hematopoietic tumor formation associated with hopTum-l. Tumor indices (TI) of adult flies of indicated genotypes are shown in histograms with s.d. Flies without the hopTum-l mutations (left) have no tumors (TI = 0.0). (b) Mutations in Su(var)205, Su(var)3-9 and Rpd3 cause a further increase in the larval blood cell number of hopTum-l heterozygotes. Images show larval blood cells from wild-type (top) and hopTum-l (bottom) hemolymph. Numbers of blood cells and standard deviations are shown at right for each genotype. *P < 0.01; **P < 0.001, Student's t-test. (c) Representative patterns of GAS-lacZ expression (detected by antibodies to β-gal (dark staining)) are shown for each indicated genotype. β-gal is detected in distinctive cells and tissues in the anterior region of stage 15 embryos. The levels of β-gal are moderately increased in hopTum-l/+ embryos, slightly decreased in hop+/− and STAT92E+/− embryos and relatively unchanged in Su(var)205, Su(var)3-9 and Rpd3 heterozygotes. (d) β-gal enzyme activity detected in embryo extracts of indicated genotypes. Error bars indicate s.d. Numbers correspond to genotypes as shown in c.
To confirm that mutations in Su(var)205, Su(var)3-9 or Rpd3 do not cause increased JAK/STAT signaling, we examined the effects of halving the dosage of these genes on the expression of a lacZ reporter transgene (GAS-lacZ) that is controlled by multiple STAT binding sites25 and the expression of which depends on Hop/STAT92E activity in vivo25. Indeed, embryos heterozygous for hopTum-l, STAT92E or hop mutations showed increased or decreased β-galactosidase (β-gal) activity, respectively, as measured by immunostaining (Fig. 2c) and β-gal activity assays (Fig. 2d). However, we found that reducing the dosage of Su(var)205, Su(var)3-9 and Rpd3 by half did not lead to increased levels of β-gal produced by GAS-lacZ (Fig. 2c,d), suggesting that the chromatin-modifying proteins HP1, Su(var)3-9 and Rpd3 do not normally repress JAK/STAT signaling.
This interpretation is consistent with the fact that the chromatin-modifying proteins identified in this study were not found in the genome-wide RNAi screens based on changes in STAT92E transcriptional activity13,14. Moreover, mutations in the gene encoding NURF301, the large nucleosome remodeling factor (NURF) subunit, have been shown to cause blood tumors without affecting JAK/STAT activity23. Thus, the chromatin-modifying proteins we identified may instead be involved in steps downstream of JAK/STAT signaling in hematopoietic tumorigenesis, such as in counteracting tumorigenesis as tumor suppressors.
HP1 and Su(var)3-9 are essential components of heterochromatin and are required for heterochromatin-mediated gene silencing20, which has been shown recently to function as a tumor suppressor mechanism4. JAK/STAT overactivation may compete with the HP1/Su(var)3-9-mediated silencing system for access to STAT target genes that promote tumorigenesis without affecting this silencing system per se. Alternatively, JAK/STAT overactivation may actively disrupt HP1- or Su(var)3-9-mediated heterochromatic gene silencing, allowing expression of normally repressed genes that are not necessarily STAT target genes.
We tested whether JAK overactivation could disrupt heterochromatin-mediated gene silencing. Heterochromatic gene silencing is responsible for position-effect variegation (PEV) and tandem repeat–induced variegation, and mutations in the genes encoding HP1 and Su(var)3-9 methyltransferase dominantly suppress PEV20. DX1 is a transgenic line containing seven tandem copies of the p[lacW] transgene (containing a mini-white+ and a lacZ gene) inserted at the cytogenetic location 50C in the euchromatic region26. Owing to sequence repetition, these tandem p[lacW] transgenes induce heterochromatin formation, resulting in repression of the mini-white+ gene and variegated red eye pigmentation in DX1 flies26 (Fig. 3a). We found that hopTum-l suppressed the variegating phenotype of DX1, as evidenced by derepression of the mini-white+ transgene and increased eye pigmentation (Fig. 3a). hopTum-l, however, did not affect the expression of the mini-white+ transgene from a single p[lacW] inserted at the same location (6-2 mini-w), which does not induce heterochromatin formation and served as a control26,27 (Fig. 3a). It is thus unlikely that the mini-white+ transgene itself is transcriptionally regulated by the JAK/STAT pathway. Consistent with these results, hopTum-l also increased the expression of the lacZ gene contained in the p[lacW] of DX1, without affecting lacZ expression from the single p[lacW] of 6-2 mini-w flies (Fig. 3b). These results indicate that hopTum-l counteracts heterochromatin-mediated silencing of genes that are not normally regulated by the JAK/STAT pathway.
Figure 3.
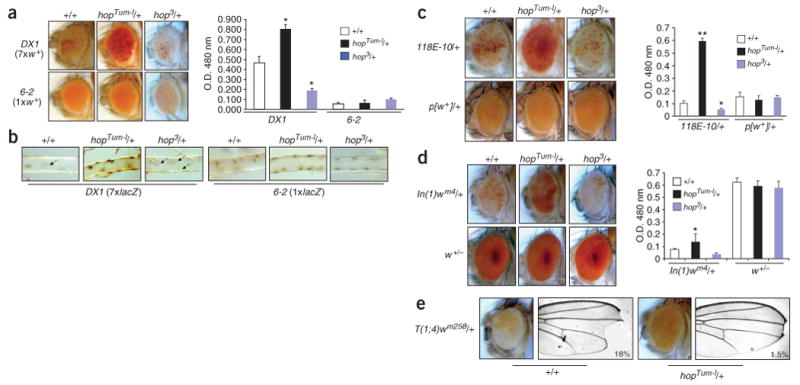
JAK gain of function suppresses and loss of function enhances heterochromatic gene silencing. (a) Mutations in hopTum-l (gain of function) and hop3 (loss of function) dominantly suppress and enhance, respectively, repeat-induced variegation, as shown by the expression of the heterochromatinized mini-w+ transgene (top) and levels of the red eye pigment (measured by optical density). Average values of more than three independent measurements are shown with s.d. *P < 0.01 compared with +/+ control (Student's t-test). (b) hopTum-l or hop3 increases or decreases, respectively, expression of the heterochromatinized lacZ transgene of DX1 (left) but not of mini-w 6-2 (right), as visualized by anti-β-gal immunostaining of third instar larval tracheal branches. Brown spots indicate β-gal–positive nuclei on the surface of the trachea. Arrows point to nuclei with no β-gal expression owing to heterochromatin-mediated repression. (c) hopTum-l and hop3 suppress and enhance, respectively, PEV associated with a single p[w+] transgene (118E-10) inserted in pericentric heterochromatin without affecting single p[w+] transgenes inserted in the euchromatin region (only one example shown). (d) hopTum-l and hop3 suppress and enhance, respectively, PEV associated with chromosomal inversion, In(1)wm4, in which the w+ gene is juxtaposed to heterochromatin without affecting expression of the w+ gene in its normal locus. (e) hopTum-l suppresses PEV associated with a translocation, T(1;4)wm258, in which the X chromosome is fused with heterochromatinized chromosome 4, resulting in silencing of the w+ and Notch+ genes. Representative images of eye and wing, and the percentage of flies having the defects, are shown (n > 200 for each genotype).
To confirm that JAK overactivation counteracts heterochromatic gene silencing in general, we tested additional independent variegating lines resulting from juxtaposition of euchromatic genes to heterochromatin. Indeed, we found that hopTum-l dominantly derepressed silenced transgenes inserted in pericentric heterochromatin (118E-10; Fig. 3c) as well as euchromatic genes that were brought into close proximity to heterochromatin by inversion (In(1)wm4; Fig. 3d) or translocation (T(1;4)wm258; Fig. 3e). Taken together, these results strongly suggest that overactivation of the JAK/STAT pathway globally disrupts heterochromatic gene silencing.
Consistent with the effects of hopTum-l, we found that hop loss-of-function mutations dominantly enhanced gene silencing in DX1, 118E-10 and In(1)wm4 flies (Fig. 3a–d). These results suggest that the JAK/STAT signaling pathway normally functions to counteract heterochromatic gene silencing during D. melanogaster development.
To investigate how JAK/STAT signaling counteracts heterochromatic gene silencing, we examined the effects of JAK gain or loss of function on heterochromatin. Hallmarks of heterochromatin include HP1 localization and histone H3 Lys9 dimethylation (H3mK9), which are most prominent in the centromeric region20. We found that in salivary gland cells of hopTum-l/+ third instar larvae, the levels of HP1 and H3mK9 normally seen as concentrated foci in the nucleus were much lower than in wild-type larvae (Fig. 4a,b). In addition, in hop loss-of-function heterozygotes, we detected increased areas of HP1 and H3mK9 concentration in the nucleus (Fig. 4a,b). Protein blot analysis of embryo extracts indicated that the total amount of H3mK9 was much reduced in hop gain-of-function and increased in loss-of-function animals (Fig. 4c). Notably, the total HP1 level was increased by 40% in flies heterozygous for hopTum-l (Fig. 4c), suggesting that the lack of HP1 foci in hopTum-l/+ nuclei is due to a failure of HP1 localization to the centromeric region. Thus, JAK gain or loss of function reduces or increases centromeric heterochromatin and HP1 localization, respectively.
Figure 4.
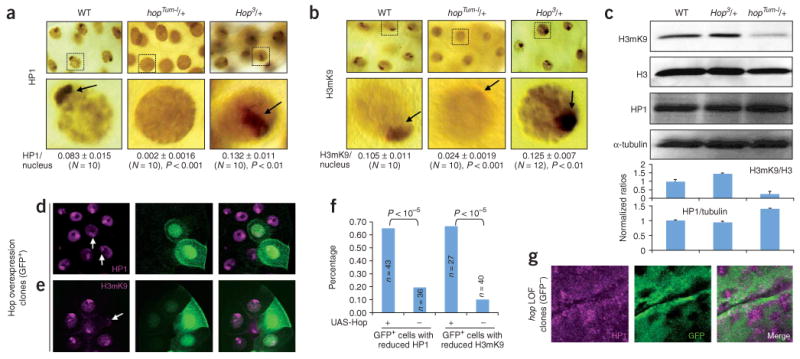
JAK gain of function decreases, and loss of function increases, HP1 and H3mK9 localization on heterochromatin. (a) Representative giant nuclei of salivary glands of indicated genotypes stained with anti-HP1. HP1 signals on heterochromatin (dark brown stain and arrow) are decreased in hopTum-l and increased in hop3 heterozygotes. Quantifications are shown as the ratio of the area of HP1-positive focus (centromeric heterochromatin) versus the area of the nucleus. We used ten represented nuclei from different stained salivary glands for quantification. (b) H3mK9 signals are higher in hopTum-l and lower in hop3 heterozygotes than in wild type. Quantification of H3mK9 staining was done as above. (c) Protein blots of total proteins extracted from 40 12- to 24-h-old embryos from females of indicated genotypes. The membrane was blotted with anti-H3mK9, stripped and sequentially blotted with anti-H3, anti-HP1 and anti-α-tubulin. More than three independent blots were performed and the gel bands were quantified; ratios (± s.d.) are shown at bottom. (d,e) Overexpression of Hop (JAK overactivation) in salivary gland cells (marked by GFP (green)) result in decreased levels of HP1 (d) and H3mK9 (e). (f) Reductions in HP1 or H3mK9 were quantified for all GFP+ cells found in third instar salivary glands with indicated transgene combination. Control animals did not contain UAS-Hop. The differences were analyzed by χ2 test. (g) Increased HP1 levels are detected in hop loss-of-function (LOF) homozygous clones (marked by a lack of GFP). Part of a third instar imaginal disc is shown.
To confirm that JAK/STAT signaling affects the levels of heterochromatin and HP1 localization, we examined JAK gain or loss of function by clonal analysis. First, we overexpressed Hop in random cells marked by green fluorescent protein (GFP) (see Methods). Overexpression of Hop and HopTum-l are equivalent in causing STAT92E phosphorylation and phenotypic consequences11. We found that salivary gland cells overexpressing Hop showed reduced levels of HP1 (Fig. 4d,f) and H3mK9 (Fig. 4e,f). In addition, in cells mutant for hop, we found increased levels of HP1 (Fig. 4g). These results demonstrate that JAK signaling is essential in regulating heterochromatin formation or stability, and its overactivation destabilizes heterochromatin.
Having established that JAK overactivation globally disrupts heterochromatic gene silencing and destabilizes heterochromatin, we investigated whether disrupting heterochromatic gene silencing is important for hopTum-l-induced hematopoietic tumorigenesis. To this end, we examined the effects of increasing HP1 levels via an hsp70-Su(var)205 transgene28 on hopTum-l tumorigenicity. To our surprise, we found that a moderate increase in the levels of HP1, provided by the basal-level expression of hsp70-Su(var)205, was able to completely suppress hopTum-l-induced hematopoietic tumor formation (Fig. 5a), such that we did not find any melanotic tumors in hopTum-l/+; hsp70-Su(var)205+/− flies (n > 800). Thus, increasing HP1 levels, which promotes heterochromatic silencing28, is sufficient to suppress hopTum-l induced hematopoietic tumor formation, suggesting that reducing HP1 localization on heterochromatin is essential for hematopoietic tumorigenesis induced by JAK gain of function.
Figure 5.
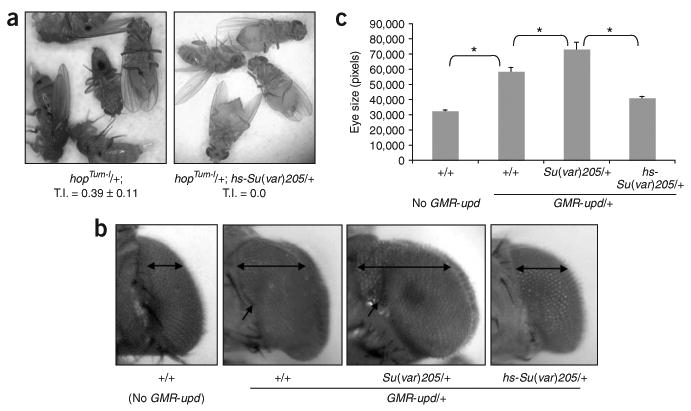
HP1 counteracts tumorigenesis induced by JAK/STAT overactivation. (a) A moderate increase in HP1 levels by the basal expression from an hsp70-Su(var)205 transgene completely suppresses hopTum-l-dependent hematopoietic tumors. Shown are representative flies of indicated genotypes and the tumor index calculated from 500 flies. (b) The enlarged eye phenotype, due to eye cell overproliferation induced GMR-upd (second from left), is enhanced by halving the dosage of HP1 (second from right) and suppressed by the basal expression of hsp70-Su(var)205 (far right). Double arrows indicate the widths of eyes of females of indicated genotypes viewed dorsally. Single arrows point to the furrow due to eye enlargement induced by GMR-upd, which is deepened when HP1 levels are reduced (second from right) and nearly absent when HP1 levels are increased (right). (c) Eye size was quantified by measuring the pixel numbers of more than five representative eye pictures (taken dorsally at the same magnification and distance) of indicated genotypes. * P < 0.01 (Student's t-test).
To investigate whether HP1 is important in general for tumorigenesis induced by JAK/STAT overactivation, we examined the effects of changing HP1 levels on overproliferation of eye cells caused by targeted expression of the JAK/STAT ligand Unpaired (Upd)15. Overexpression of Upd by an eye-specific promoter (GMR-upd) leads to eye cell overproliferation and eye enlargement (Fig. 5b; also see ref. 15). This effect is mediated by the JAK/STAT pathway, as reducing the dosage of Hop or STAT92E suppresses the large-eye phenotype13,15. We found that reducing or increasing HP1 levels significantly enhanced or suppressed the large-eye phenotype of GMR-upd flies, respectively (Fig. 5b,c). Changing HP1 levels as above had no effect on eye development in an otherwise wild-type genetic background (data not shown). These results suggest that HP1 levels affect the outcome of JAK/STAT overactivation-induced tumorigenesis not only in hematopoietic tissues but possibly also in general.
We have investigated the mechanism of how overactivation of the JAK/STAT pathway induces hematopoietic tumors in D. melanogaster. Taken together, our results demonstrate that (i) the levels of HP1 and the heterochromatic gene silencing machinery determine the outcome of hopTum-l tumorigenicity without affecting JAK/STAT signaling per se, (ii) JAK signaling has a previously unknown function in globally counteracting heterochromatin-mediated gene silencing during normal D. melanogaster development and (iii) disruption of heterochromatic gene silencing is essential for JAK overactivation–induced cell overproliferation and tumorigenesis.
Methods
Fly stocks and genetics
All crosses were carried out at 25 °C on standard cornmeal/agar medium. Fly stocks of hopTum-l, hop2, hop3, Stat92E6346, Rpd304556, Su(var)2055, Su(var)3-91, In(1)wm4, T(1;4)wm258 and the Bloomington Deficiency Kit Stocks were all from the Bloomington Drosophila Stock Center. Other stocks used in this study were generous gifts from other researchers. These include DX1 and 6-2 mini-white+ (J. Birchler), 118E-10 (S. Elgin), hsp70-Su(var)205 (L. Wallrath), GAS-lacZ (N. Reich), UAS-hop (N. Perrimon) and GMR-upd flies (E. Bach).
To screen for enhancers or suppressors of hopTum-l using autosomal Df stocks, hopTum-l/FM7 virgin females were crossed to males heterozygous for each individual Df (Df/Balancer) in vials. The F1 adult flies were scored for the presence of hematopoietic tumors, which manifest as melanotic masses most frequently found in the abdomen (Fig. 1a). Typically, more than 200 flies were scored for each cross. Potential modifiers were retested. To alleviate possible genetic background effects carried by individual Df stocks, the Df heterozygous males were outcrossed to a CyO/Sco (for chromosome 2 deficiencies) or Ly/TM3 (for chromosome 3 deficiencies) stock. Tumorigenicity was quantified by tumor index (TI), which is defined as the sum of tumor size multiplied by occurrence and divided by the total number of flies of a particular genotype (TI = Σ(tumor size × n)/N, where n is the number of occurrences of a particular tumor size and N is the total number of flies counted for a particular genotype). Tumor size 1 is defined as a tumor with a diameter equal to the width of an average abdominal segment (Fig. 1a). TI 1.0 is equivalent to all flies of a category each having a size 1.0 tumor.
Control crosses were carried out by crossing hopTum-l/FM7 females to males of wild-type or stocks bearing different ‘marker’ genes, such as Oregon R, Canton S, w1118 and y1. The mean TI of the hopTum-l/+ F1 progeny from these crosses was 0.39 ± 0.11. When mated with a collection of chromosomal deficiencies, the TI values of the hopTum-l/+; Df/+ F1 progeny flies ranged from 0 (no tumors) to > 1.0 in a quasi-normal distribution (Fig. 1b).
E(Tum-l) and Su(Tum-l) mutations were selected based on the TI values of the hopTum-l/+; Df/+ flies that fell beyond two (for Su(Tum-l)) or three (for E(Tum-l)) standard deviations from the mean of the control crosses. To determine which gene or genes were responsible for the modification of hopTum-l tumorigenicity for a particular Df, we tested more than five candidate genes on average for each modifier Df, and in most cases only one demonstrated a TI similar to the Df and was designated as a candidate gene.
Immunohistochemistry, β-gal assay and eye pigmentation measurement
Mouse antibodies to β-gal (Promega; 1:1000) and to HP1 (C1A9; Developmental Hybridoma Bank, Iowa; 1:50 to 1:500) and rabbit antibodies to histone H3 dimethyl K9 (Upstate Biotechnology) were used as primary antibodies for whole-mount immunostaining (1:200) and protein blots (1:1,000). Biotinylated secondary antibodies and the ABC Elite Kit (Vector Labs) or fluorescent secondary antibodies (Molecular Probes) were used to visualize signals. Stained tissues were photographed with an Axiophot compound microscope or a Leica confocal microscope.
To assay for β-gal activity produced by GAS-lacZ, females of appropriate genotypes were mated to males homozygous for a p[GAS-lacZ] insertion, and 200 embryos (4–20 h old) were collected and assayed with the β-Galactosidase Enzyme Assay System (Promega) according to the manufacturer's protocol.
To measure eye pigmentation, the heads of 40 female flies (2–3 d old; raised at 25 °C) of each genotype were homogenized in 1 ml of methanol (acidified with 0.1% HCl). Eye pigmentation was represented as the absorbance of the supernatant at 480 nm.
Supplementary Material
Acknowledgments
We thank K. Larson and D. Guo for technical assistance; J. Birchler, S. Elgin, L. Wallrath, N. Reich, N. Perrimon, E. Bach and the Bloomington Drosophila Stock Center for various Drosophila strains and L. Silver-Morse for helpful comments on the manuscript. J.L. was a recipient of the Wilmot Cancer Research Fellowship. H.C.C. was a trainee of the Post-Baccalaureate Research Education Program (PREP) of the US National Institutes of Health. This study was supported by grants from the US National Institutes of Health (R01GM65774 and R01GM077046) and an American Cancer Society Research Scholar Grant (RSG-06-196-01-TBE) to W.X.L.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Author Contributions: The initial Df screen was performed by H.C.C., J.L. and L.L.; identification of modifier genes and phenotype assessment were performed by S.S.; F.X. contributed to the protein blot analysis. W.X.L. designed the study and wrote the paper.
Competing Interests Statement: The authors declare that they have no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Bromberg JF. Activation of STAT proteins and growth control. Bioessays. 2001;23:161–169. doi: 10.1002/1521-1878(200102)23:2<161::AID-BIES1023>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 2.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 3.Yu H, Jove R. The STATs of cancer–new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 4.Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 5.Hou XS, Melnick MB, Perrimon N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell. 1996;84:411–419. doi: 10.1016/s0092-8674(00)81286-6. [DOI] [PubMed] [Google Scholar]
- 6.Yan R, Small S, Desplan C, Dearolf CR, Darnell JE., Jr Identification of a Stat gene that functions in Drosophila development. Cell. 1996;84:421–430. doi: 10.1016/s0092-8674(00)81287-8. [DOI] [PubMed] [Google Scholar]
- 7.Hou SX, Zheng Z, Chen X, Perrimon N. The Jak/STAT pathway in model organisms. Emerging roles in cell movement. Dev Cell. 2002;3:765–778. doi: 10.1016/s1534-5807(02)00376-3. [DOI] [PubMed] [Google Scholar]
- 8.Agaisse H, Perrimon N. The roles of JAK/STAT signaling in Drosophila immune responses. Immunol Rev. 2004;198:72–82. doi: 10.1111/j.0105-2896.2004.0133.x. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Xia F, Li WX. Coactivation of STAT and Ras is required for germ cell proliferation and invasive migration in Drosophila. Dev Cell. 2003;5:787–798. doi: 10.1016/s1534-5807(03)00328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanratty WP, Dearolf CR. The Drosophila Tumorous-lethal hematopoietic oncogene is a dominant mutation in the hopscotch locus. Mol Gen Genet. 1993;238:33–37. doi: 10.1007/BF00279527. [DOI] [PubMed] [Google Scholar]
- 11.Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 1995;14:2857–2865. doi: 10.1002/j.1460-2075.1995.tb07285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo H, Hanratty WP, Dearolf CR. An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J. 1995;14:1412–1420. doi: 10.1002/j.1460-2075.1995.tb07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller P, Kuttenkeuler D, Gesellchen V, Zeidler MP, Boutros M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature. 2005;436:871–875. doi: 10.1038/nature03869. [DOI] [PubMed] [Google Scholar]
- 14.Baeg GH, Zhou R, Perrimon N. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 2005;19:1861–1870. doi: 10.1101/gad.1320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bach EA, Vincent S, Zeidler MP, Perrimon N. A sensitized genetic screen to identify novel regulators and components of the Drosophila janus kinase/signal transducer and activator of transcription pathway. Genetics. 2003;165:1149–1166. doi: 10.1093/genetics/165.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 17.Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 18.Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 19.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 20.Grewal SI, Elgin SC. Heterochromatin: new possibilities for the inheritance of structure. Curr Opin Genet Dev. 2002;12:178–187. doi: 10.1016/s0959-437x(02)00284-8. [DOI] [PubMed] [Google Scholar]
- 21.Zinyk DL, McGonnigal BG, Dearolf CR. Drosophila awdK-pn, a homologue of the metastasis suppressor gene nm23, suppresses the Tum-1 haematopoietic oncogene. Nat Genet. 1993;4:195–201. doi: 10.1038/ng0693-195. [DOI] [PubMed] [Google Scholar]
- 22.Betz A, Lampen N, Martinek S, Young MW, Darnell JE., Jr A Drosophila PIAS homologue negatively regulates stat92E. Proc Natl Acad Sci USA. 2001;98:9563–9568. doi: 10.1073/pnas.171302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badenhorst P, Voas M, Rebay I, Wu C. Biological functions of the ISWI chromatin remodeling complex NURF. Genes Dev. 2002;16:3186–3198. doi: 10.1101/gad.1032202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lebestky T, Jung SH, Banerjee UA. Serrate-expressing signaling center controls Drosophila hematopoiesis. Genes Dev. 2003;17:348–353. doi: 10.1101/gad.1052803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert MM, Weaver BK, Gergen JP, Reich NC. A novel functional activator of the Drosophila JAK/STAT pathway, unpaired2, is revealed by an in vivo reporter of pathway activation. Mech Dev. 2005;122:939–948. doi: 10.1016/j.mod.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 26.Dorer DR, Henikoff S. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell. 1994;77:993–1002. doi: 10.1016/0092-8674(94)90439-1. [DOI] [PubMed] [Google Scholar]
- 27.Ronsseray S, Boivin A, Anxolabehere D. P-Element repression in Drosophila melanogaster by variegating clusters of P-lacZ-white transgenes. Genetics. 2001;159:1631–1642. doi: 10.1093/genetics/159.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Danzer JR, Alvarez P, Belmont AS, Wallrath LL. Effects of tethering HP1 to euchromatic regions of the Drosophila genome. Development. 2003;130:1817–1824. doi: 10.1242/dev.00405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.