

Abstract
The efficacy of ACTH, particularly in high doses, for rapid and complete elimination of infantile spasms (IS) has been demonstrated in prospective controlled studies. However, the mechanisms for this efficacy remain unknown. ACTH promotes the release of adrenal steroids (glucocorticoids), and most ACTH effects on the central nervous system have been attributed to activation of glucocorticoid receptors. The manner in which activation of these receptors improves IS and the basis for the enhanced therapeutic effects of ACTH — compared with steroids — for this disorder are the focus of this chapter. First, a possible “common excitatory pathway,” which is consistent with the many etiologies of IS and explains the confinement of this disorder to infancy, is proposed. This notion is based on the fact that all of the entities provoking IS activate the native “stress system” of the brain. This involves increased synthesis and release of the stress-activated neuropeptide, corticotropin-releasing hormone (CRH), in limbic, seizure-prone brain regions. CRH causes severe seizures in developing experimental animals, as well as limbic neuronal injury. Steroids, given as therapy or secreted from the adrenal gland upon treatment with ACTH, decrease the production and release of CRH in certain brain regions. Second, the hypothesis that ACTH directly influences limbic neurons via the recently characterized melanocortin receptors is considered, focusing on the effects of ACTH on the expression of CRH. Experimental data showing that ACTH potently reduces CRH expression in amygdala neurons is presented. This downregulation was not abolished by experimental elimination of steroids or by blocking their receptors and was reproduced by a centrally administered ACTH fragment that does not promote steroid release. Importantly, selective blocking of melanocortin receptors prevented ACTH-induced downregulation of CRH expression, providing direct evidence for the involvement of these receptors in the mechanisms by which ACTH exerts this effect. Thus, ACTH may reduce neuronal excitability in IS by two mechanisms of action: (1) by inducing steroid release and (2) by a direct, steroid-independent action on melanocortin receptors. These combined effects may explain the robust established clinical effects of ACTH in the therapy of IS.
I. How Do the Many Etiologies of Infantile Spasms (IS) Lead to Excitability and Seizures? The Scope of the Problem
IS is an age-specific disorder of brain excitability, with diverse genetic, teratogenic, perinatal, and postnatally acquired etiological factors. Two key elements characteristic of this entity are (1) the remarkable number and variability of the predisposing factors and (2) the fact that regardless of the time of onset of the underlying etiology (i.e., conception, intrauterine, prenatal, perinatal, or postnatal), IS commence at a distinct developmental age (typically the third to seventh postnatal month). These facts have suggested that (a) there must be a “final common pathway” for the etiologies, leading to IS, and (b) this final common pathway must be operative only during the state of brain maturation that occurs during infancy.
Mechanistic theories for the development of IS have included those invoking abnormal immune function, brain stem dysfunction (Hrachovy and Frost, 1989), developmental arrest (Riikonen, 1983), and cortical microdysplasia (Vinters et al., 1992; DallaBernadina and Dulac, 1994). The latter has gained in popularity with the advent of magnetic resonance imaging. However, foci of microdysgenesis have been described in autopsied brains of normal individuals (Lyon and Gastaut, 1985), casting doubt on the etiological role of these in IS. In addition, the majority of infants with symptomatic IS have etiologies, such as those of metabolic, chromosomal, or infectious nature, that do not involve cortical dysplasias or other structural anomalies.
Any theory for the pathogenesis of IS has to account for the unique features of this disorder. For example, how can a single entity have so many etiologies? Why do IS arise only in infancy, even when a known insult had occurred prenatally, and why do they disappear? Why are IS associated with lasting cognitive dysfunction, and why do these seizures — unlike most others — respond to ACTH [in 86–88% of cases (Baram et al., 1996)]. ACTH may accelerate central nervous system (CNS) myelination and dendritic formation, and thus may shorten a hypothetical period of vulnerability to IS (Riikonen, 1983). ACTH may also act as a direct anticonvulsant via GABAergic or other mechanisms (Holmes, 1987). However, the hormonal actions of ACTH (as opposed to other potential effects) have been shown to be necessary for efficacy, as analogs of ACTH without hormonal effects do not eliminate IS (Pentella et al., 1982; Willing and Lagenstein, 1982). Furthermore, the rapid (median response time of 2 days), all-or-none, and often permanent effects of ACTH on IS are not consistent with conventional anticonvulsant properties (Hrachovy and Frost, 1989; Baram, 1993; Baram et al., 1996, 1999).
II. Can the Seizure-Selective, Striking Efficacy of ACTH for IS Provide Insight into the Mechanisms of This Disorder?
A. The Known Hormonal Role of ACTH Is to Induce High Plasma and Brain Steroid Levels
The established hormonal role of ACTH in human physiology is to function in the neuroendocrine cascade of the responses to all stressful stimuli, including insults to the brain. Native ACTH is synthesized in the pituitary and functions to stimulate the adrenal cortex to release steroids into the bloodstream as part of the neuroendocrine stress response (Fig. 1). ACTH, whether released from the pituitary or given as therapy, functions via promoting the release of glucocorticoids (steroids, primarily Cortisol in the human and corticosterone in the rat) from the adrenal gland. These hormones have a multitude of effects throughout the body. Importantly, steroids cross the blood–brain barrier readily and bind to receptors that are widely expressed throughout the brain. Activation of glucocorticoid receptors has little direct anticonvulsant effects (Karst et al., 1994). However, glucocorticoids modulate the expression and release of a number of neurotransmitters and neuromodulators, including the proconvulsant neuropeptide corticotropin-releasing hormone (CRH).
Fig. 1.
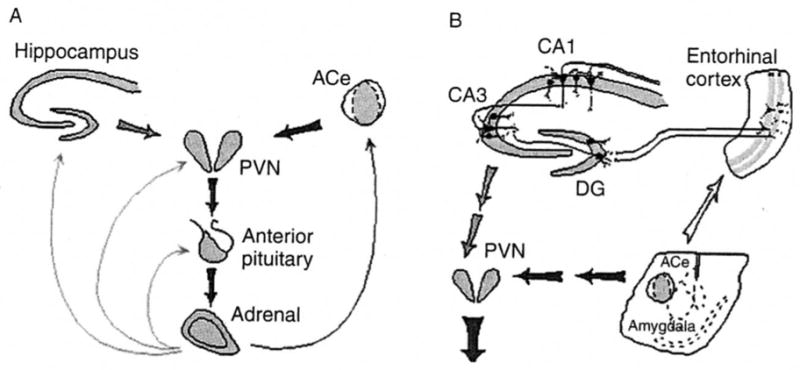
Neuroendocrine (A) and limbic (B) stress-activated corticotropin-releasing hormone (CRH) loops. (A) Stress-conveying signals rapidly activate immediate early genes in CRH expressing neurons of the central nucleus of the amygdala (ACe). Rapid CRH release in the ACe activates CRH expression in the hypothalamic paraventricular nucleus (PVN) to secrete CRH into the hypothalamo-pituitary portal system, inducing ACTH and glucocorticoid secretion from the pituitary and adrenal, respectively. Glucocorticoids exert a negative feedback on the PVN (directly and via the hippocampus), yet activate CRH gene expression in the amygdala, potentially promoting further CRH release in this region. (B) CRH-expressing GABAergic interneurons (small round cells) in the principal cell layers of the hippocampal CA1, CA3, and the dentate gyrus (DG) are positioned to control excitability of the pyramidal and granule cells, respectively. These neurons may be influenced by the stress-evoked release of CRH from the ACe via connections in the entorhinal cortex. Dark and light arrows denote established or putative potentiating and inhibitory actions, respectively. Arrows do not imply monosynaptic connections.
B. The Many Etiologies of IS Activate the Corticotropin-Releasing Hormone (CRH) –ACTH–Steroid Cascade: Human and Animal Data
IS, at least the symptomatic cases, occur in the context of an insulted and stressed developing brain (Baram, 1993). The many etiologies of IS all lead to activation of the stress response, including the stress neurohormone CRH. CRH has been shown, in infant animal models, to cause severe seizures and death of neurons in areas involved with learning and memory (Baram et al., 1992a; Brunson et al., 2001a). These effects of CRH are largely restricted to the infancy period because the receptors for CRH, which mediate its actions on neurons, are most abundant during this developmental period (Avishai-Eliner et al., 1996; Baram and Hatalski, 1998). The mechanisms by which CRH increases excitability of limbic neurons have been examined in vitro (e.g., Aldenhoff et al., 1983; Hollrigel et al., 1998) and involve a suppression of afterhyperpolarization and a potentiation of glutamatergic neurotransmission. Thus, the possibility that IS result from “excessive” levels of CRH in limbic synapses, which are a result of activation of the production and secretion of CRH as part of the CNS stress response to the etiologies of IS, has been suggested (Baram, 1993; Baram and Hatalski, 1998; Baram et al., 1999).
Data from human infants with IS support a disruption of the CRH–ACTH stress cascade in the brains of these infants. High brain CRH levels are expected to reduce cerebrospinal fluid (CSF) levels of ACTH and of steroids [because chronic activation of CRH receptors leads to their desensitization (Hauger et al., 1993) and decreased ACTH release]. Indeed, several groups have independently reported reduced ACTH levels in IS patients compared with age-matched controls (Nalin et al., 1985; Baram et al., 1992b; 1995; Heiskala, 1997). Thus, these data are consistent with enhanced levels of endogenous CRH in brains of youngsters with IS. Based on animal data described later, increased CRH at limbic synapses should promote increased excitability, potentially leading to the chronically abnormal neuronal activity (hypsarrhythmia) and spasms characteristic of IS.
C. Excess CRH Provokes Limbic Seizures Involving the Amygdala and Hippocampus
In an immature rat, particularly during the stage of brain development that is comparable to that of human infants, administration of minute amounts of CRH directly into the CSF causes prolonged and severe seizures (Baram and Schultz, 1991; Baram et al., 1992a; Baram and Hatalski, 1998). These seizures involve activation of CRH receptors in the amygdala and hippocampus. Therefore, it is reasonable to assume that increased levels of endogenous CRH in the amygdala and hippocampus would also increase excitability in the amygdala–hippocampal limbic circuit. Interestingly, CRH is highly expressed in both immature and adult central nucleus of the amygdala (Gray and Bingaman, 1996; Hatalski et al., 1998). A robust expression of CRH in hippocampal neurons has been demonstrated as well (Yan et al., 1998), although levels of CRH in mature hippocampus are rather low (Swanson et al., 1983). In addition, recurrent stress has been shown to increase CRH levels in the amygdala of the infant rat (Hatalski et al., 1998), and some stresses also increase CRH expression in the immature hippocampus (Hatalski et al., 2000). Therefore, the following scenario may be considered: The many etiologies of IS share the common denominator of being “stressful” to the immature brain in the sense of activating the intrinsic stress response. While stress does not increase CRH synthesis in the neonate (Yi and Baram, 1994), increased CRH levels in the amygdala, and potentially in the hippocampus, does occur in the infant. This would lead to excess activation of CRH receptors, abundant during infancy in amygdala and hippocampus (see earlier discussion), and to hyperexcitability and seizures. Drastic reduction in the abundance of CRH receptors, occurring with maturation, would be responsible for the reduction in this peptide-mediated hyperexcitability later in life (Baram and Hatalski, 1998).
D. ACTH, Acting via Steroids, Modulates Excitability via Altering CRH Expression
Earlier studies have focused on the hormonal action of ACTH, specifically of its induction of steroid release, as the key mechanism of its efficacy for IS. This view was supported by several facts. First, steroids by themselves are effective in a significant portion of infants with IS. In addition, ACTH fragments that did not release steroids were not effective for IS. (It is now known, however, that these fragments also do not activate ACTH/melanocortin receptors in the brain, see later.) While the full spectrum of the mechanisms of action of steroids in ameliorating IS is unknown (see, e.g., Karst et al., 1994; Pavlides et al., 1995), these hormones reduce CRH expression in some brain regions, notably in the brain stem (Imaki et al., 1991), a region suspected to be involved in IS (Hrachovy and Frost, 1989). However, steroids do not reduce CRH levels in limbic regions such as the amygdala and hippocampus.
III. Direct Actions of ACTH May Modulate Excitability Independent of Steroids by Downregulating CRH Expression in Limbic Regions
A. Distribution and Roles of Native, Endogenous ACTH
Neurons containing ACTH have been localized to the CNS, particularly the hypothalamus, and ACTH-immunoreactive cell bodies or fibers have also been described in the amygdala, cerebral cortex, brain stem, and cerebellum (Pilcher and Joseph, 1984). In contrast to pituitary ACTH, the functions of CNS–ACTH have not been well defined. Evidence from both human and animal studies has suggested that CNS–ACTH may function as a neurotransmitter or neuromodulator (Pranzatelli, 1994; de Wied, 1977). Indeed, central physiological roles for ACTH, including modulation of learning and memory processes and facilitation of arousal states, have been suggested, but the mechanisms for these actions of ACTH have remained unclear (Pranzatelli, 1994; de Wied, 1977). Characterization of the melanocortin receptor family, consisting of several members possessing binding affinity for ACTH (Mountjoy et al., 1992; Adan and Gispen, 1997), has yielded insight regarding the likely sites and mechanisms of ACTH effects. However, the downstream effects of activation of these receptors and the specific molecular changes that can lead to the clinical effects of ACTH on CNS function have not been elucidated.
As mentioned earlier, pituitary ACTH synthesis and secretion are regulated by CRH, and both hormones participate in the neuroendocrine response to stress (Fig. 1). Conversely, ACTH reduces CRH gene expression in the hypothalamus as part of the neuroendocrine feedback loop of the stress response (Sawchenko and Arias, 1995). ACTH and CRH are found in close proximity in excitable, seizure-prone brain regions such as the amygdala, and complex interactions between ACTH- and CRH-expressing neuronal systems have been suggested (Wu et al., 1997; Hauger et al., 1993; Joseph et al., 1985). For example, ACTH-immunoreactive fibers have been demonstrated in the central nucleus of the amygdala, a major limbic locus of CRH-expressing neurons. As mentioned earlier, CRH production in the central nucleus is activated when diverse stressors or brain insults induce the neuroendocrine stress cascade. In view of these facts, could suppression of CRH expression in the central nucleus of the amygdala and the resulting reduced excitability (Baram et al., 1992a) account for some of the actions of ACTH on IS?
B. Experimental Evidence for Direct Reduction of CRH Expression in the Central Nucleus of the Amygdala by ACTH
ACTH, in the form typically used for therapy of IS in the United States (ACTHARGEL, Rhone-Poulenc Rorer, Collegeville, PA), was administered into the peritoneal cavity (ip) of infant rats at a high dose (80 IU/kg) because of the limited blood–brain barrier penetration of the peptide (Nicholson et al., 1978; Mezey et al., 1878). ACTH4–10, an analog that binds melanocortin receptors but does not induce steroid secretion from the adrenal, was also tested and infused directly into the cerebral ventricles (Brunson et al., 2001b). Steroid receptors were blocked using RU 38486 (Roussel UCLAF, Romainville, France), whereas melanocortin receptors were blocked using SHU9119 (courtesy Dr. K. Yagaloff, Roche, New Jersey).
The effects of ACTH in the presence and absence of endogenous steroids, and with blockade of steroid or ACTH/melanocortin receptors, were examined in infant rats. CRH expression in the central nucleus of the amygdala was determined using semiquantitative in situ hybridization histochemistry (ISH; Eghbal-Ahmadi et al., 1999; Brunson et al., 2001). Analysis of the CRH–mRNA ISH signal was performed as described in these publications, including unbiased sampling of sections, and performance of all analyses without knowledge of treatment group (“blindly”). C14 standards were used to ascertain that analyzed signals fell in the linear range, and values were expressed as relative optical density units. Differences among groups were determined using a one-way ANOVA or student’s t test, as appropriate (Prism GraphPad, San Diego, CA), and significance levels were set at p < 0.05. Further analysis used Tukey’s multiple comparison posthoc tests to determine the source of the detected significance in the ANOVAs.
Administration of ACTH downregulated CRH expression in the central nucleus by 35%, considered highly significant physiologically (for review, see Hatalski et al., 1998). Glucocorticoid receptor activation was not required for the ACTH-induced downregulation of CRH expression because this downregulation was also observed in the absence of endogenous steroids: in adrenalectomized rats — as in intact littermates — ACTH resulted in a significant (33%) suppression of CRH expression. It should be noted that adrenalectomy — eliminating steroids and the consequent negative feedback on ACTH secretion — resulted in high levels of intrinsic, endogenous ACTH. These ACTH levels, by themselves, were sufficient to significantly downregulate CRH expression (34% reduction from sham-operated controls).
The effect of ACTH on CRH expression in amygdala was central: Similar to systemically administered ACTH, the analog ACTH4–10 infused into the CSF resulted in a significant downregulation of CRH expression (45% reduction from vehicle-infused control levels). The dose of the ACTH4–10 analog was relatively small and was therefore unlikely to reach the adrenal when given centrally. In addition, this ACTH analog does not stimulate corticosterone secretion from the adrenal and did not raise plasma steroid levels in the experiments described here. These data suggest that ACTH4–10 (and ACTH) reduced CRH expression in amygdala via a central rather than a steroid-mediated peripheral mechanism.
A specific blockade of melanocortin receptors or of glucocorticoid receptor occupancy demonstrated that occupancy and activation of the former were required for the action of ACTH on amygdala CRH: SHU9119, a blocker of melanocortin receptors, blocked ACTH-induced reduction of CRH expression. In contrast, when RU 38486, a glucocorticoid receptor blocker, was given to intact animals together with ACTH, it failed to block the effects of ACTH administration on CRH expression in amygdala (a 63% reduction from ip injected controls). These data indicate that activation of melanocortin receptors, but not of glucocorticoid receptors, is required for the actions of ACTH on CRH expression in the central nucleus of the amygdala.
IV. Implications of Steroid-Independent Effects of ACTH for the Therapy of IS
The studies described here demonstrate direct actions of systemically or centrally administered ACTH on limbic neurons that are independent of adrenal steroids. ACTH-induced reduction of CRH expression in the amygdala may provide a mechanism for the efficacy of ACTH in IS and, potentially, in other human disorders. Importantly, these effects of ACTH involve activation of specific melanocortin receptors, which may provide new and important therapeutic targets.
ACTH, particularly in high doses, has been shown to provide efficacy for IS (86–88%), which is greater than that of steroids. Indeed, controlled prospective and blinded studies demonstrated a clear superiority of high doses of ACTH over steroids (Baram et al., 1996, but see Hrachovy et al., 1983). These clinical data raise several issues. First, if ACTH acts via steroids, then maximal doses of both agents should have similar effects. Second, once doses of ACTH that suffice to maximally release endogenous steroids are used, then higher doses should not have additional benefits. The enhanced potency of ACTH compared with steroids and the superiority of extremely high doses of ACTH compared with lower ones are consistent with direct, steroid-independent actions of ACTH within the CNS. In addition, the poor penetration of systemically given ACTH through the blood–brain barrier (Nicholson et al., 1978; Mezey et al., 1978) may underlie the requirement for large systemic doses.
The studies described here support the notion of direct CNS actions of ACTH. Using doses analogous to those used clinically, systemic ACTH influenced the expression of CRH in specific brain regions regardless of the presence or absence of adrenal steroids. These effects were generated by ACTH given both systemically or directly into the cerebral ventricles and persisted when glucocorticoid receptors were blocked, but not when a subtype of ACTH receptors (melanocortin receptors) was antagonized, showing that activation of these latter receptors was required and sufficient for these actions of ACTH.
Melanocortin receptors, a family of transmembrane G-protein-coupled receptors, have been demonstrated in regions subserving ACTH-mediated functions (e.g., substantia nigra for grooming and amygdala for learning and memory). In addition, activation of central melanocortin receptors plays a role in appetitive and grooming behavior (Adan and Gispen, 1997; Fisher et al., 1999). Data reviewed in this chapter demonstrate that ACTH may activate melanocortin receptor type 4 and are consistent with a scenario in which ACTH, released from fiber terminals in the central nucleus of the amygdala, acts locally on melanocortin receptors located on CRH-expressing neurons. ACTH, given in clinical settings, should mimic these effects.
Interestingly, the (4–10) fragment of ACTH that does not release adrenal steroids also led to the depression of CRH gene expression. Earlier clinical studies have shown that shorter fragments (4–9) of ACTH, which failed to release adrenal steroids, were not efficacious for infantile spasms (Willig and Lagenstein, 1982; Pentella et al., 1982). Currently available information explains the apparent discrepancy: new information about the binding of ACTH fragments to melanocortin receptors shows that while the 4–10 fragment used here activates these receptors, the 4–9 fragment does not (Adan and Gispen, 1997; Gantz et al., 1993). Again, these data are consistent with activation of melanocortin receptors as the mechanism for the observed effects of ACTH on amygdala neurons.
In summary, data discussed here provide two potential mechanisms for the efficacy of ACTH for developmental seizure disorders and specifically for IS. The first is the “conventional” activation of steroid release and the second involves the direct modulation of amygdala neurons leading to decreased production of the proconvulsant peptide CRH. Importantly, data reviewed here highlight components of the ACTH–CRH system as potential targets for the development of new therapeutic agents for IS.
Acknowledgments
Supported by NIH RO1 NS28912 and R41 HD34975 (TZB) and by a system-wide University of California Biotechnology-oriented predoctoral award (KLB). The authors thank M. Hinojosa for excellent editorial assistance.
References
- Adan RAH, Gispen WH. Brain melanocortin receptors: From cloning to function. Peptides. 1997;8:1279–1287. doi: 10.1016/s0196-9781(97)00078-8. [DOI] [PubMed] [Google Scholar]
- Aldenhoff JB, Gruol DL, Rivier J, Vale W, Siggins GR. Corticotropin releasing factor decreases postburst hyperpolarizations and excites hippocampal neurons. Science. 1983;221:875–877. doi: 10.1126/science.6603658. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Yi SJ, Baram TZ. Developmental profile of messenger RNA for the corticotropin-releasing hormone receptor in the rat limbic system. Dev Brain Res. 1996;91:59–163. doi: 10.1016/0165-3806(95)00158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ. Pathophysiology of massive infantile spasms: Perspective on the putative role of the brain adrenal axis. Ann Neurol. 1993;33:231–236. doi: 10.1002/ana.410330302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: A key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Hirsch E, Snead OC, III, Schultz L. Corticotropin-releasing hormone-induced seizures in infant rats originate in the amygdala. Ann Neurol. 1992a;31:488–494. doi: 10.1002/ana.410310505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Brunson K, Haden E. Infantile spasms: Hypothesis-driven therapy and pilot human infant experiments using corticotropin-releasing hormone receptor antagonists. Dev Neurosci. 1999;21:281–289. doi: 10.1159/000017407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Snead OC, II, Horton EJ. Corticotropin and Cortisol are increased in the cerebrospinal fluid of infants with massive infantile spasms. Pediatr Neurol. 1995;13:108–110. doi: 10.1016/0887-8994(95)00121-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Snead OC, III, Horton EJ, Saito M. Brain adrenal axis hormones are altered in the CSF of infants with massive infantile spasms. Neurology. 1992b;42:1171–1175. doi: 10.1212/wnl.42.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Tournay A, et al. High-dose corticotropin (ACTH) versus prednisone for infantile spasms: A prospective, randomized, blinded study. Pediatrics. 1996;97:375–379. [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Schultz L. Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Dev Brain Res. 1991;61:97–101. doi: 10.1016/0165-3806(91)90118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, Bender R, Chen Y, Baram TZ. Progressive hippocampal cell loss and long-term cognitive dysfunction induced by early-life administration of corticotropin releasing hormone reproduce the effects of early-life stress. Proc Nat Acad Sci USA. 2001a;98:8856–8861. doi: 10.1073/pnas.151224898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Khan N, Eghbal-Ahmadi M, Baram TZ. ACTH acts directly on amygdala neurons to down-regulate corticotropin releasing hormone gene expression. Ann Neurol. 2001b;49:304–312. [PMC free article] [PubMed] [Google Scholar]
- DallaBernadina B, Dulac O. Introduction to etiology. In: Dulac O, Chugani H, Dalla Bernadina B, editors. Infantile Spasms and West Syndrome. Saunders; London: 1994. [Google Scholar]
- de Wied D. Behavioral effects of neuropeptides related to ACTH, MSH, and βLPH. Ann NY Acad Sci. 1977;297:263–275. doi: 10.1111/j.1749-6632.1977.tb41859.x. [DOI] [PubMed] [Google Scholar]
- Eghbal-Ahmadi M, Avishai-Eliner S, Hatalski CG, Baram TZ. Differential regulation of the expression of corticotropin-releasing factor receptor type 2 (CRF2) in hypothalamus and amygdala of the immature rat by sensory input and food intake. J Neurosci. 1999;19:3982–3991. doi: 10.1523/JNEUROSCI.19-10-03982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SL, Yagaloff KA, Burn P. Melanocortin and leptin signaling systems: Central regulation of catabolic energy balance. J Recept Signal Transduct Res. 1999;19:203–216. doi: 10.3109/10799899909036646. [DOI] [PubMed] [Google Scholar]
- Gantz I, Miwa H, Konda Y, et al. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- Gray TS, Bingaman EW. The amygdala: Corticotropin-releasing factor, steroids, and stress. Crit Rev Neurobiol. 1996;10:155–168. doi: 10.1615/critrevneurobiol.v10.i2.10. [DOI] [PubMed] [Google Scholar]
- Hatalski CG, Guirguis C, Baram TZ. Corticotropin releasing factor mRNA expression in the hypothalamic paraventricular nucleus and the central nucleus of the amygdala is modulated by repeated acute stress in the immature rat. J Neuroendocrinal. 1998;10:663–669. doi: 10.1046/j.1365-2826.1998.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauger RL, Irwin MR, Lorang M, et al. High intracerebral levels of CRH result in CRH receptor downregulation in the amygdala and neuroimmune desensitization. Brain Res. 1993;616:283–292. doi: 10.1016/0006-8993(93)90219-d. [DOI] [PubMed] [Google Scholar]
- Heiskala H. CSF ACTH and beta-endorphin in infants with West syndrome and ACTH therapy. Brain Dev. 1997;5:339–342. doi: 10.1016/s0387-7604(97)00026-0. [DOI] [PubMed] [Google Scholar]
- Hollrigel GS, Chen K, Baram TZ, Soltesz I. The pro-convulsant actions of corticotropin-releasing hormone in the hippocampus of infant rats. Neuroscience. 1998;84:78–84. doi: 10.1016/s0306-4522(97)00499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL, Weber DA. Effects of ACTH on seizure susceptibility in the developing brain. Ann Neurol. 1986;1:82–88. doi: 10.1002/ana.410200114. [DOI] [PubMed] [Google Scholar]
- Hrachovy RA, Frost JD. Infantile spasms. Pediatr Clin North Am. 1989;36:311–329. doi: 10.1016/s0031-3955(16)36651-2. [DOI] [PubMed] [Google Scholar]
- Hrachovy RA, Frost JD, Glaze DG. High-dose long-duration versus low-dose short duration corticotropin therapy for infantile spasms. J Pediatr. 1994;124:803. doi: 10.1016/s0022-3476(05)81379-4. [DOI] [PubMed] [Google Scholar]
- Hrachovy RA, Frost JD, Kellaway P, Zion TE. Double-blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103:641–645. doi: 10.1016/s0022-3476(83)80606-4. [DOI] [PubMed] [Google Scholar]
- Imaki T, Nahan JL, Rivier C, Sawchenko PE, Vale W. Differential regulation of corticotropin-releasing factor mRNA in rat brain regions by glucocorticoids and stress. J Neurosci. 1991;11:585–599. doi: 10.1523/JNEUROSCI.11-03-00585.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SA, Pilcher WH, Knigge KM. Anatomy of the corticotropin-releasing factor and opiomelanocortin systems of the brain. Federation Proc. 1985;44:100–107. [PubMed] [Google Scholar]
- Karst H, Wadman WJ, Joels M. Corticosteroid receptor-dependent modulation of calcium currents in rat hippocampal CA1 neurons. Brain Res. 1994;649:234–242. doi: 10.1016/0006-8993(94)91069-3. [DOI] [PubMed] [Google Scholar]
- Lyon G, Gastaut H. Considerations on the significance attributed to unusual cerebral histological findings recently described in eight patients with primary generalized epilepsy. Epilepsia. 1985;26:365–367. doi: 10.1111/j.1528-1157.1985.tb05664.x. [DOI] [PubMed] [Google Scholar]
- Mezey E, Palkovitz M, de Kloet ER, et al. Evidence for pituitary-brain transport of a behaviorally potent ACTH analog. Life Sci. 1978;22:831–838. doi: 10.1016/0024-3205(78)90606-9. [DOI] [PubMed] [Google Scholar]
- Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:543–546. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- Nalin A, Facchinetti F, Galli V, et al. Reduced ACTH content in cerebrospinal fluid of children affected by cryptogenic infantile spasms with hypsarrhythmia. Epilepsia. 1985;26:446–449. doi: 10.1111/j.1528-1157.1985.tb05678.x. [DOI] [PubMed] [Google Scholar]
- Nicholson WE, Liddle RA, Puett D, Liddle GW. Adrenocorticotropic hormone biotransformation, clearance, and catabolism. Endocrinology. 1978;103:1344–351. doi: 10.1210/endo-103-4-1344. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Kimura A, Magarinos AM, McEwen BS. Hippocampal homosynaptic long-term depression/depotentiation induced by adrenal steroids. Neuroscience. 1995;68:379–385. doi: 10.1016/0306-4522(95)94332-s. [DOI] [PubMed] [Google Scholar]
- Pentella K, Bachman DS, Sandman CA. Trial of an ACTH 49 analog in children with intractable seizures. Neuropediatrics. 1982;13:59–62. doi: 10.1055/s-2008-1059598. [DOI] [PubMed] [Google Scholar]
- Pilcher WH, Joseph SA. Co-localization of CRF-ir perikarya and ACTH-ir fibers in rat brain. Brain Res. 1984;299:91–102. doi: 10.1016/0006-8993(84)90791-1. [DOI] [PubMed] [Google Scholar]
- Pranzatelli MR. On the molecular mechanism of adrenocorticotrophic hormone in the CNS: Neurotransmitters and receptors. Exp Neurol. 1994;125:142–161. doi: 10.1006/exnr.1994.1018. [DOI] [PubMed] [Google Scholar]
- Riikonen R. Infantile spasms: Some new theoretical aspects. Epilepsia. 1983;24:159–168. doi: 10.1111/j.1528-1157.1983.tb04875.x. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Shibasaki T, Lederis K. Corticotropin releasing factor-like immunoreactivity in the rat brain as revealed by a modified cobalt-glucose oxidase-diaminobenzidine. J Comp Neurol. 1987;260:256–298. doi: 10.1002/cne.902600209. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Arias C. Evidence for short-loop feedback effects of ACTH on CRF and vasopressin expression in parvocellular neurosecretory neurons. J Neuroendocrinal. 1995;7:721–731. doi: 10.1111/j.1365-2826.1995.tb00814.x. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: An immuno-histochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Vinters HV, Fisher RS, Cornford ME. Morphological substrates of infantile spasms: Studies based on surgically resected cerebral tissue. Child Nerv Sys. 1992;8:8–17. doi: 10.1007/BF00316556. [DOI] [PubMed] [Google Scholar]
- Watson SJ, Richard CW, III, Barchas JD. Adrenocorticotropin in rat brain: Immuno-cytochemical localization in cells and axons. Science. 200:1180–1182. doi: 10.1126/science.206967. [DOI] [PubMed] [Google Scholar]
- Willig RP, Lagenstein I. Use of ACTH fragments in children with infantile spasms. Neuro-pediatrics. 1982;13:55–58. doi: 10.1055/s-2008-1059597. [DOI] [PubMed] [Google Scholar]
- Wu HC, Chen KY, Lee WY, Lee EHY. Antisense oligonucleotides to corticotropin-releasing factor impair memory retention and increase exploration in rats. Neuroscience. 1997;78:147–153. doi: 10.1016/s0306-4522(96)00533-7. [DOI] [PubMed] [Google Scholar]
- Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: Light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SJ, Baram TZ. Corticotropin-releasing hormone mediates the response to cold stress in the neonatal rat without compensatory enhancement of the peptide’s gene expression. Endocrinology. 1994;135:2364–2368. doi: 10.1210/endo.135.6.7988418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SJ, Masters JN, 7Baram TZ. The effect of a specific glucocorticoid receptor antagonist on CRH gene expression in the neonatal rat hypothalamus. Dev Brain Res. 1993;73:253–259. doi: 10.1016/0165-3806(93)90145-z. [DOI] [PubMed] [Google Scholar]