

Abstract
Independent of the aetiology, AD (Alzheimer's disease) neurofibrillary degeneration of abnormally hyperphosphorylated tau, a hallmark of AD and related tauopathies, is apparently required for the clinical expression of the disease and hence is a major therapeutic target for drug development. However, AD is multifactorial and heterogeneous and probably involves several different aetiopathogenic mechanisms. On the basis of CSF (cerebrospinal fluid) levels of Aβ1-42 (where Aβ is amyloid β-peptide), tau and ubiquitin, five different subgroups, each with its own clinical profile, have been identified. A successful development of rational therapeutic disease-modifying drugs for AD will require understanding of the different aetiopathogenic mechanisms involved and stratification of AD patients by different disease subgroups in clinical trials. We have identified a novel aetiopathogenic mechanism of AD which is initiated by the cleavage of SET, also known as inhibitor-2 (I2PP2A) of PP2A (protein phosphatase 2A) at Asn175 into N-terminal (I2NTF) and C-terminal (I2CTF) halves and their translocation from neuronal nucleus to the cytoplasm. AAV1 (adeno-associated virus 1)-induced expression of I2CTF in rat brain induces inhibition of PP2A activity, abnormal hyperphosphorylation of tau, neurodegeneration and cognitive impairment in rats. Restoration of PP2A activity by inhibition of the cleavage and of I2PP2A/SET activity offers a promising therapeutic opportunity in AD with this aetiopathogenic mechanism.
Keywords: abnormally hyperphosphorylated tau, Alzheimer's disease, microtubule-associated protein, neurofibrillary pathology, protein phosphatase 2A (PP2A), tauopathy
Introduction
AD (Alzheimer's disease), the single major cause of dementia in middle- and old-age individuals, is multifactorial and heterogeneous. Less than 1% of the AD cases are caused by a mutation in APP (β-amyloid precursor protein) or PS (presenilin) 1 or PS2 [1]. The aetiology of the remaining, i.e. over 99%, AD cases, commonly referred to as the sporadic form of the disease, is at present not established. Independent of the aetiology, AD is histopathologically characterized by the co-occurrence of β-amyloidosis and neurofibrillary degeneration of the brain. The former is seen as β-amyloid plaques and congophilic angiopathy. Neurofibrillary degeneration is seen as intraneuronal NFTs (neurofibrillary tangles) of PHFs (paired helical filaments) admixed with SFs (straight filaments) in the cell soma, in the neuropil as neuropil threads, and in dystrophic neurites surrounding the plaque core β-amyloid. β-Amyloid is made up of the APP metabolite Aβ (amyloid β-peptide), mostly Aβ1-40 and Aβ1-42 [2,3]. The major protein subunit of PHFs/SFs is the microtubule-associated protein tau in an abnormally hyperphosphorylated state [4–6]. Studies on clinicopathological correlation have repeatedly demonstrated that neurofibrillary pathology and not β-amyloidosis correlate with the presence of dementia in humans [7–9]. Although, according to the amyloid cascade hypothesis, β-amyloidosis is upstream of neurofibrillary degeneration, an increasing number of studies suggest that the latter is pivotal for at least the clinical expression of AD. Neurofibrillary degeneration of the AD type is made up of the abnormally hyperphosphorylated tau which apparently involves several different aetiopathogenic mechanisms. The present article reviews the pivotal role of the neurofibrillary pathology of the abnormally hyperphosphorylated tau and the multifactorial nature of this lesion.
Pivotal role of tau pathology in neurodegeneration and dementia
Whereas neurofibrillary degeneration and β-amyloidosis are two required histopathological features of AD, each of these two lesions are seen in the absence of the other in different human conditions. In a significant number of healthy aged individuals, there is as much β-amyloid plaque burden in the brain as in typical cases of AD, except that, in the former case, plaques lack dystrophic neurites with neurofibrillary pathology surrounding the β-amyloid cores [8,10–12]. On the other hand, neurofibrillary degeneration of the AD type, but in the absence of β-amyloid, is seen in several tauopathies such as Guam parkinsonism dementia complex, dementia pugilistica, corticobasal degeneration, Pick's disease and FTDP-17 tau (frontotemporal dementia with parkinsonism linked with chromosome 17 and tau mutations) and progressive supranuclear palsy. All of these tauopathies with neocortical lesions are clinically characterized by dementia; in progressive supranuclear palsy, the neurofibrillary degeneration is limited to the brain stem and is associated with motor impairment.
In healthy aged individuals, neurofibrillary pathology is seen in the entorhinal cortex. In AD, neurofibrillary degeneration spreads from the entorhinal cortex first to the hippocampus and then to the rest of the neocortex [13]. Apparently neurofibrillary degeneration in the neocortex is required for the dementia; neither β-amyloidosis of the brain in the absence of neurofibrillary degeneration nor the presence of the neurofibrillary pathology in the entorhinal cortex alone are sufficient for the clinical expression of the disease.
In the case of the inherited cases of FTDP-17, almost equal numbers are caused by a mutation in the tau gene (FTDP-17 tau) as in the TDP (TAR-DNA-binding protein)-43 gene. In FTDP-17 tau, certain missense mutations in the tau gene, including those that affect the alternative splicing of its pre-mRNA, favouring the 4R (four microtubule-binding repeat) tau isoforms, co-segregate with the disease [14–16]. These mutated taus and the 4R taus are respectively more favourable substrates for abnormal hyperphosphorylation than wild-type tau and 3R taus [17]. Inclusions of hyperphosphorylated tau have also been observed in small numbers in glial cells in the white matter, especially in frontolobal dementias [18,19].
In AD-affected brain, all of the six tau isoforms are hyperphosphorylated and aggregated into PHFs/SFs [4–6,20,21]. Although conformational changes [22–24] and truncation of tau [25–27] following its hyperphosphorylation [28] have been reported in AD, the most established and compelling cause of neurofibrillary degeneration in AD and related tauopathies is the abnormal hyperphosphorylation of this protein [5,29,30].
Two major known functions of tau are its ability to promote assembly and to maintain structure of microtubules [31]. These functions of tau are regulated by its degree of phosphorylation [29,32–34]. An increase in phosphorylation beyond the normal brain level of 2–3 mol of phosphate depresses the biological activity of tau.
As much as 40 % of the abnormally hyperphosphorylated tau in AD-affected brain is present in the cytosol and not polymerized into PHFs/NFTs [30,35,36]. Unlike normal tau, the cytosolic AD P-tau (AD abnormally hyperphosphorylated tau) does not bind to tubulin and promote microtubule assembly, but instead it inhibits assembly and disrupts microtubules [29,30,37,38]. This inhibitory property of the AD P-tau involves the sequestration of normal tau, MAP (microtubule-associated protein) 1, and MAP2 by this diseased protein [29,39,40]. This toxic behaviour of the AD P-tau appears to be solely due to its abnormal hyperphosphorylation because the dephosphorylation of the diseased tau by protein phosphatase converts it into a normal-like protein [29,37,38]. Hyperphosphorylation of tau induced by intracerebroventricular injection of forskolin, a PKA (protein kinase A) activator, in rats caused cognitive impairment and on co-administration of Rp-cAMPS (Rp isomer of adenosine 3′,5′-monophosphothioate), a PKA inhibitor, both the hyperphosphorylation and cognitive impairment were reversed [41]. The inhibitory activity of the cytosolic AD P-tau has also been confirmed in tau-transfected yeast [42,43], tau-transgenic Drosophila [44] and a P301L transgenic mouse model [45]. On self-assembly into PHFs/NFTs, the AD P-tau loses its ability to sequester normal MAPs and inhibit or disrupt microtubules [32,33,46].
Tau mutations, which cause FTDP-17 tau, result either in an increase in the 4R/3R tau ratio or in missense mutations. Both 4R and mutated taus are more easily abnormally hyperphosphorylated than the normal wild-type protein [17,47]. Opposite to FTDP-17 tau, in Pick's disease and Down's syndrome, the 3R/4R ratio is increased [48–50]. Since the activity of 3R tau is lesser than that of 4R tau in binding to tubulin/microtubules, the unbound 3R tau becomes abnormally hyperphosphorylated because free tau is a more favourable substrate than tau on microtubules for phosphorylation [51]. Thus it appears that a loss of the normal balance of 4R/3R taus can promote tau pathology.
Multifactorial nature of neurofibrillary degeneration
Neurofibrillary degeneration of AD P-tau, a histopathological hallmark of AD and related tauopathies, is caused by multiple factors. These multiple causes include not only mutations in the tau gene, APP gene, PS1 gene and PS2 gene mentioned above, but also metabolic abnormalities and environmental factors.
Tau is a substrate for several protein kinases [52,53]. Among these kinases, GSK3 (glycogen synthase kinase 3), Cdk5 (cyclin-dependent protein kinase 5), PKA, CaMKII (Ca2+/calmodulin-dependent protein kinase II), CK1 (casein kinase I), MAPK (mitogen-activated protein kinase) ERK1/2 (extracellular-signal-regulated kinase 1/2) and SAPKs (stress-activated protein kinases) have been most implicated in the abnormal hyperphosphorylation of tau [54,55].
The state of phosphorylation of a phosphoprotein is a function of the balance between the activities of the protein kinases and the protein phosphatases that regulate its phosphorylation. The activity of PP2A (protein phosphatase 2A), which accounts for over 70% of tau phosphatase activity in human brain [56–58] is compromised in AD–affected brain [59,60] and has been strongly implicated as a cause of abnormal hyperphosphorylation of tau [55,61,62]. On dephosphorylation with PP2A, the AD P-tau loses both its ability to inhibit microtubule assembly and self-assemble into PHFs/SFs [63]. Interestingly, this PP2A-dephosphorylated tau can be converted back into AD P-tau by more than one combination of tau kinases, suggesting that AD neurofibrillary degeneration can involve several different aetiopathogenic mechanisms [63].
PP2A activity is regulated by two heat-stable proteins, inhibitor-1 (I1PP2A) and inhibitor-2 (I2PP2A) [64–67]. The mRNAs and protein expression of both of these PP2A inhibitors are up-regulated in AD-affected brain [68]. I2PP2A, also called SET, a primarily nuclear protein, is selectively cleaved into an N-terminal half (I2NTF) and a C-terminal half (I2CTF), and is translocated from the neuronal nucleus to the cytoplasm and co-localizes with NFTs in AD-affected brain [68]. Expression of I2CTF in the brain causes abnormal hyperphosphorylation of tau and reference memory impairment in rats [69], suggesting a novel aetiopathogenic mechanism of neurofibrillary degeneration involving cleavage of I2PP2A and generation of I2CTF.
Virtually all cases of Down's syndrome, which is caused by partial or complete trisomy 21, develop AD histopathology when they reach the fourth decade of life. In addition to APP, another important gene within the chromosome 21, Down's syndrome-critical region is Dyrk1A, which encodes a serine/threonine protein kinase DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A) [70]. Recent studies suggest that overexpression of DYRK1A may cause neurofibrillary degeneration, both by leading to abnormal hyperphosphorylation of tau by priming it for further phosphorylation by GSK3β and by promoting exclusion of microtubule-binding repeat 2 through phosphorylation of ASF (alternative splicing factor) [50,71].
Phosphorylation of tau is also regulated by its degree of O-GlcNAcylation which involves serine/threonine residues [72,73]. O-GlcNAcylation, including that of tau, is down-regulated in AD-affected brain [74]. This is probably due to a decrease in brain glucose metabolism caused by a decrease in the level of the glucose transporters Glut1 and Glut3 [75,76]; the brain level of Glut3 is also decreased in diabetes and in cases of AD with diabetes, providing an explanation of diabetes as a risk factor and a metabolic cause of AD. All of these studies taken together suggest that the sporadic AD is multifactorial. Understanding of these different aetiopathogenic mechanisms involved is required for rational development of potent disease-modifying drugs for the treatment of AD.
Acknowledgments
We are grateful to Ms Janet Murphy for secretarial assistance. Dr Ezzat El-Akkad helped to prepare Figure 1.
Figure 1. Multifactorial nature, pivotal role, and mechanism of AD neurofibrillary degeneration.
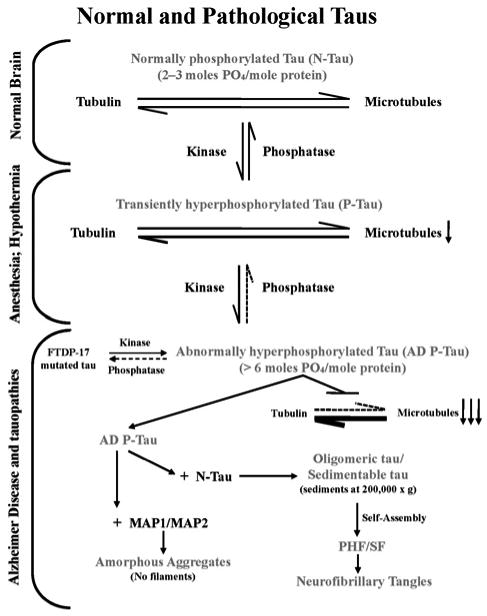
Neurofibrillary degeneration of the AD type is a product of multiple aetiological factors. Independent of the aetiology, neurofibrillary degeneration in AD and tauopathies involves abnormal hyperphosphorylation of tau and its occurrence in the neocortex is associated with dementia; neurofibrillary degeneration in the brainstem in progressive supranuclear palsy is associated with motor impairment. A cause of hyperphosphorylation of tau is a decrease in the activity of PP2A, a major tau phosphatase. Unlike normal tau, AD P-tau, instead of interacting with tubulin and promoting its assembly into microtubules, sequesters normal tau, MAP1 and MAP2, forming 200 000 g sedimentable oligomeric tau. The sequestration of normal MAPs disrupts microtubules and compromises axonal transport and consequently retrograde degeneration and dementia. The AD P-tau polymerizes into tangles of PHFs/SFs, which, unlike the cytosolic/oligomeric AD P-tau, interacts neither with tubulin nor with normal MAPs, but eventually obstructs further the microtubule network and axoplasmic flow as a space-occupying lesion.
Funding: Studies in our laboratories were supported in part by the New York State Office of Mental Retardation and Developmental Disabilities, National Institutes of Health [grant numbers AG019158, AG028538 and AG27429] and Alzheimer's Association (Chicago, IL) [grant numbers IIRG-00-2002, HRG-05-13095 and NIRG-08-91126].
Abbreviations used
- Aβ
amyloid β-peptide
- AD
Alzheimer's disease
- AD P-tau
AD abnormally hyperphosphorylated tau
- APP
β-amyloid precursor protein
- DYRK1A
dual-specificity tyrosine-phosphorylated and -regulated kinase 1A
- FTDP-17 tau
frontotemporal dementia with parkinsonism linked with chromosome 17 and tau mutations
- Glut
glucose transporter
- GSK3
glycogen synthase kinase 3
- MAP
microtubule-associated protein
- NFT
neurofibrillary tangle
- PHF
paired helical filament
- PKA
protein kinase A
- PP2A
protein phosphatase 2A
- I2PP2A
PP2A inhibitor-2
- I2CTF
I2PP2A C-terminal half
- PS
presenilin
- R
microtubule-binding repeat
- SF
straight filament
References
- 1.Bird TD. Genetic aspects of Alzheimer disease. Genet Med. 2008;10:231–239. doi: 10.1097/GIM.0b013e31816b64dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 3.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau: a component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 5.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iqbal K, Grundke-Iqbal I, Smith AJ, George L, Tung YC, Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:5646–5650. doi: 10.1073/pnas.86.14.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–242. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- 8.Alafuzoff I, Iqbal K, Friden H, Adolfsson R, Winblad B. Histopathological criteria for progressive dementia disorders: clinical–pathological correlation and classification by multivariate data analysis. Acta Neuropathol. 1987;74:209–225. doi: 10.1007/BF00688184. [DOI] [PubMed] [Google Scholar]
- 9.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 10.Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer's disease: a double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- 11.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 12.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 13.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 14.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 15.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 16.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alonso AD, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–34881. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 18.Kumaran R, Kingsbury A, Coulter I, Lashley T, Williams D, de Silva R, Mann D, Revesz T, Lees A, Bandopadhyay R. DJ-1 (PARK7) is associated with 3R and 4R tau neuronal and glial inclusions in neurodegenerative disorders. Neurobiol Dis. 2007;28:122–132. doi: 10.1016/j.nbd.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, Fraser G, Crowther RA, Goedert M, Budka H, Ghetti B. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008;67:963–975. doi: 10.1097/NEN.0b013e318187a80f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 21.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 22.Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res. 1999;55:713–723. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 23.Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 24.Jicha GA, Rockwood JM, Berenfeld B, Hutton M, Davies P. Altered conformation of recombinant frontotemporal dementia-17 mutant tau proteins. Neurosci Lett. 1999;260:153–156. doi: 10.1016/s0304-3940(98)00980-x. [DOI] [PubMed] [Google Scholar]
- 25.Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. 2005;64:104–112. doi: 10.1093/jnen/64.2.104. [DOI] [PubMed] [Google Scholar]
- 26.Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novak M, Jakes R, Edwards PC, Milstein C, Wischik CM. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci U S A. 1991;88:5837–5841. doi: 10.1073/pnas.88.13.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, Spillantini MG, Crowther RA, Goedert M. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol. 2008;172:123–131. doi: 10.2353/ajpath.2008.070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alonso AD, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, Wisniewski HM, Alafuzoff I, Winblad B. Defective brain microtubule assembly in Alzheimer's disease. Lancet. 1986;ii:421–426. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- 31.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iqbal K, Zaidi T, Bancher C, Grundke-Iqbal I. Alzheimer paired helical filaments: restoration of the biological activity by dephosphorylation. FEBS Lett. 1994;349:104–108. doi: 10.1016/0014-5793(94)00650-4. [DOI] [PubMed] [Google Scholar]
- 33.Khatoon S, Grundke-Iqbal I, Iqbal K. Guanosine triphosphate binding to β-subunit of tubulin in Alzheimer's disease brain: role of microtubule-associated protein tau. J Neurochem. 1995;64:777–787. doi: 10.1046/j.1471-4159.1995.64020777.x. [DOI] [PubMed] [Google Scholar]
- 34.Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5305. [PubMed] [Google Scholar]
- 35.Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- 36.Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau: abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- 37.Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Mol Brain Res. 1996;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- 38.Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007;113:501–511. doi: 10.1007/s00401-007-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alonso AD, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 41.Liu SJ, Zhang JY, Li HL, Fang ZY, Wang Q, Deng HM, Gong CX, Grundke-Iqbal I, Iqbal K, Wang JZ. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J Biol Chem. 2004;279:50078–50088. doi: 10.1074/jbc.M406109200. [DOI] [PubMed] [Google Scholar]
- 42.Vandebroek T, Terwel D, Vanhelmont T, Gysemans M, Van Haesendonck C, Engelborghs Y, Winderickx J, Van Leuven F. Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3β or cdk5. J Biol Chem. 2006;281:25388–25397. doi: 10.1074/jbc.M602792200. [DOI] [PubMed] [Google Scholar]
- 43.Vandebroek T, Vanhelmont T, Terwel D, Borghgraef P, Lemaire K, Snauwaert J, Wera S, Van Leuven F, Winderickx J. Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry. 2005;44:11466–11475. doi: 10.1021/bi0506775. [DOI] [PubMed] [Google Scholar]
- 44.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 45.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alonso AD, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A. 2006;23:8864–8869. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- 48.Oyama F, Cairns NJ, Shimada H, Oyama R, Titani K, Ihara Y. Down's syndrome: up-regulation of β-amyloid protein precursor and tau mRNAs and their defective coordination. J Neurochem. 1994;62:1062–1066. doi: 10.1046/j.1471-4159.1994.62031062.x. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology. 2006;26:457–470. doi: 10.1111/j.1440-1789.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- 50.Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, Zhou J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem. 2008;283:28660–28669. doi: 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sengupta A, Grundke-Iqbal I, Iqbal K. Regulation of phosphorylation of tau by protein kinases in rat brain. Neurochem Res. 2006;31:1473–1480. doi: 10.1007/s11064-006-9205-9. [DOI] [PubMed] [Google Scholar]
- 52.Singh TJ, Grundke-Iqbal I, McDonald B, Iqbal K. Comparison of the phosphorylation of microtubule-associated protein tau by non-proline dependent protein kinases. Mol Cell Biochem. 1994;131:181–189. doi: 10.1007/BF00925955. [DOI] [PubMed] [Google Scholar]
- 53.Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer's disease brain: an update. J Alzheimer's Dis. 1999;1:329–351. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 54.Pei JJ, Sjogren M, Winblad B. Neurofibrillary degeneration in Alzheimer's disease: from molecular mechanisms to identification of drug targets. Curr Opin Psychiatry. 2008;21:555–561. doi: 10.1097/YCO.0b013e328314b78b. [DOI] [PubMed] [Google Scholar]
- 55.Iqbal K, Alonso A, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Gong CX, Wegiel J, Lidsky T, Zuck L, Avila J, Wisniewski HM, Grundke-Iqbal I, Iqbal K. Regulation of phosphorylation of neuronal microtubule-associated proteins MAP1b and MAP2 by protein phosphatase-2A and -2B in rat brain. Brain Res. 2000;853:299–309. doi: 10.1016/s0006-8993(99)02294-5. [DOI] [PubMed] [Google Scholar]
- 57.Bennecib M, Gong CX, Grundke-Iqbal I, Iqbal K. Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett. 2001;490:15–22. doi: 10.1016/s0014-5793(01)02127-5. [DOI] [PubMed] [Google Scholar]
- 58.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 59.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 60.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 61.Iqbal K, Grundke-Iqbal I. Tau phosphatase activity as a therapeutic target for Alzheimer's disease. Drug News Perspect. 1998;11:10–14. doi: 10.1358/dnp.1998.11.1.863668. [DOI] [PubMed] [Google Scholar]
- 62.Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry. 1995;34:1988–1996. doi: 10.1021/bi00006a020. [DOI] [PubMed] [Google Scholar]
- 65.Li M, Makkinje A, Damuni Z. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry. 1996;35:6998–7002. doi: 10.1021/bi960581y. [DOI] [PubMed] [Google Scholar]
- 66.Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- 67.Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 2005;579:363–372. doi: 10.1016/j.febslet.2004.11.097. [DOI] [PubMed] [Google Scholar]
- 68.Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. 2005;166:1761–1771. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iqbal K, Wang X, Blanchard J, Grundke-Iqbal I. Overexpression of inhibitor-1 or -2 of protein phosphatase-2A causes abnormal hyperphosphorylation of tau and cognitive impairment in rat. Alzheimer's Dementia. 2009;5:135. [Google Scholar]
- 70.Kentrup H, Becker W, Heukelbach J, Wilmes A, Schurmann A, Huppertz C, Kainulainen H, Joost HG. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J Biol Chem. 1996;271:3488–3495. doi: 10.1074/jbc.271.7.3488. [DOI] [PubMed] [Google Scholar]
- 71.Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, Ramakrishna N, Gong CX. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22:3224–3233. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem. 1996;271:28741–28744. doi: 10.1074/jbc.271.46.28741. [DOI] [PubMed] [Google Scholar]
- 73.Hart GW, Kreppel LK, Comer FI, Arnold CS, Snow DM, Ye Z, Cheng X, DellaManna D, Caine DS, Earles BJ, et al. O-GlcNAcylation of key nuclear and cytoskeletal proteins: reciprocity with O-phosphorylation and putative roles in protein multimerization. Glycobiology. 1996;6:711–716. doi: 10.1093/glycob/6.7.711. [DOI] [PubMed] [Google Scholar]
- 74.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain. 2009;132:1820–1832. doi: 10.1093/brain/awp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer's disease. J Neurochem. 2009;111:242–249. doi: 10.1111/j.1471-4159.2009.06320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]