

Abstract
Poly(amido amine) (PAMAM) dendrimers have shown promise in oral drug delivery. Conjugation of SN38 to PAMAM dendrimers has the potential to improve its oral absorption while minimizing gastrointestinal toxicity. In this work we evaluated G3.5 PAMAM dendrimer-SN38 conjugates with ester-linked glycine and β-alanine spacers for their suitability in oral therapy of hepatic colorectal cancer metastases. G3.5-βAlanine-SN38 was mostly stable while G3.5-Glycine-SN38 showed 10%, 20%, and 56% SN38 release in simulated gastric, intestinal and liver environments for up to 6, 24 and 48 hours, respectively. Short-term treatment of Caco-2 cells with G3.5-SN38 conjugates did not reduce cell viability, while comparable concentrations of SN38 caused significant cytotoxicity. G3.5-Glycine-SN38 and G3.5-βAlanine-SN38 showed IC50 values of 0.60 and 3.59 μM, respectively, in HT-29 cells treated for 48 hours, indicating the efficacy of the drug delivery system in colorectal cancer cells with longer incubation time. Both conjugates increased SN38 transepithelial transport compared to the free drug. Transport of G3.5-Glycine-SN38 was highly concentration-dependent whereas transport of G3.5-βAlanine-SN38 was concentration-independent, highlighting the influence of drug loading and spacer chemistry on transport mechanism. Together these results show that PAMAM dendrimers have the potential to improve the oral bioavailability of potent anti-cancer drugs.
Keywords: PAMAM dendrimers, oral drug delivery, 7-ethyl-10-hydroxy-camptothecin, transepithelial transport, Caco-2
Introduction
Polymer-based drug delivery systems have shown great promise due to their ability to improve the efficacy of traditional drugs [1]. Conjugation of small molecule therapeutics to a polymeric carrier can enhance the drug's solubility, increase accumulation at the target site and minimize non-specific toxicity. Because chemotherapy drugs are often plagued by poor water solubility and dose-limiting toxicities, they are promising candidates for polymeric drug delivery strategies. Attachment of chemotherapy drugs to water soluble polymers enhances solubility, allows for accumulation of the polymer-drug conjugate at the tumor site due to the enhanced permeability and retention effect, improves efficacy and reduces side effects [2]. Several polymer-drug conjugates using N-(2-hydroxypropyl)methacrylamide (HPMA) and poly(ethylene glycol) (PEG) as carriers are currently being evaluated in clinical trials [3, 4].
While conjugation of chemotherapy drugs to water-soluble polymers can improve their solubility and tumor uptake, the large size of these macromolecular constructs necessitates intravenous administration. Oral administration is typically limited to small, lipophilic drugs that can permeate the cell membrane, small, hydrophilic drugs that pass through the tight junctions or drugs that are substrates for intestinal transporters [5]. Research shows that compared to intravenous administration, oral chemotherapy is the preferred method of administration by cancer patients given similar efficacy for both treatments [6]. Several studies have compared the cost savings of oral versus intravenous treatments and have found significant reductions in patient costs with oral delivery due to reduction in inpatient procedures and outpatient visits, often associated with intravenous treatments [7, 8]. With cancer being one of the most significant contributors to health care costs in the United States, illustrated by a mean annual medical cost of $32,629 for cancer patients compared to $3,218 for control patients, controlling treatment costs is critical both for patients and the healthcare system [9]. Therefore, the combination of the distinct therapeutic advantages of polymer-drug conjugates with strong patient preference and lower costs of oral chemotherapy supports a significant need for orally bioavailable polymer therapeutics.
Poly(amido amine) (PAMAM) dendrimers have shown promise as polymer-based oral drug delivery carriers. They share many characteristics with traditional polymeric drug delivery systems including water solubility, a high capacity for drug loading and improved biodistribution [10-13]. In addition, dendrimers have the ability to translocate across the intestinal barrier due to their compact structure [14-18]. PAMAM dendrimers are known to cross the epithelial barrier by a combination of transcellular and paracellular routes and can transiently open tight junctions, thereby enhancing their own transport via the paracellular pathway [19-22]. Therefore, conjugation of anticancer drugs to PAMAM dendrimers can potentially render these drugs orally bioavailable.
Because of its low water solubility and poor bioavailability, SN38 (7-ethyl-10-hydroxy-campothecin), a potent topoisomerase-1 poison used to treat colorectal cancer and hepatic metastases, is an ideal candidate for polymeric delivery strategies. SN38 is the active metabolite of CPT-11 produced when CPT-11 is cleaved by carboxylesterase [23]. While SN38 shows 100-1000-fold higher activity than CPT-11 in vitro, its use is limited by low water solubility and significant intestinal toxicity including diarrhea [24, 25]. Conjugation of SN38 to PAMAM dendrimers has the potential to allow for oral administration while also improving water solubility and minimizing gastrointestinal toxicity. Previously we established the synthesis, characterization, and bioactivity of G3.5 PAMAM-SN38 conjugates against colorectal carcinoma cell lines [26]. To successfully advance these systems for oral administration, their stability in the gastrointestinal tract and transport across the epithelial barrier of the gut must be determined. In this work we have examined G3.5 PAMAM-SN38 conjugates for their in vitro release profiles, cytotoxicity against Caco-2 and HT-29 colorectal cancer cells, cellular uptake, and transepithelial transport across Caco-2 monolayers as models of the intestinal epithelial barrier.
Materials and Methods
Materials
PAMAM G3.5 dendrimers (reported molecular weight=12,931), lucifer yellow carbohydrazide (CH) dipotassium salt, pepsin from porcine gastric mucosa, pancreatin from porcine pancreas and carboxylesterase from rabbit liver were purchased from Sigma Aldrich (St. Louis, MO). Simulated Intestinal Fluid and Simulated Gastric Fluid were obtained from Ricca Chemical Company (Arlington, Texas). 7-ethyl-10-hydroxy camptothecin (SN38) was obtained from AK Scientific Company (Mountain View, CA). WST-1 cell proliferation reagent was purchased from Roche Applied Sciences (Indianapolis, IN). Caco-2 cells and HT-29 cells were obtained from American Type Cell Culture (Rockville, MD).
Synthesis and Characterization of G3.5-Gly-SN38 and G3.5-βAla-SN38 Conjugates
G3.5-Gly-SN38 and G3.5-βAla-SN38 were synthesized as previously described [26]. Briefly, SN38 was modified at the 20-OH position via an ester linker with glycine or β–alanine [27]. The modified SN38 molecules were then conjugated to carboxylic acid-terminated G3.5 dendrimers using EDC (1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide) / NHS (N-Hydroxysuccinimide) as a coupling agent. The products were dialyzed against distilled water using 3500 molecular weight cutoff (MWCO) membranes to remove low molecular weight impurities and further purified using preparative fast protein liquid chromatography (FPLC). 1H nuclear magnetic resonance (NMR) was used to quantify the number of SN38 molecules per dendrimer by comparing the area of the dendrimer protons between 2.1 and 3.8 ppm with the methyl protons of SN38 between 0.9-1.1 ppm. The drug loading (moles SN38 per mole dendrimer) was 2.9 and 4 for G3.5-Gly-SN38 and G3.5-βAla-SN38, respectively, corresponding to 8 and 10 wt %. The hydrodynamic radii of G3.5, G3.5-Gly-SN38 and G3.5-βAla-SN38 were determined previously to be 1.76 ± 0.11, 1.37 ± 0.06 and 1.33 ± 0.05 nm, respectively using a quasi-elastic light scattering detector (QELS) to measure the dendrimer peak on an FPLC system [26]. Scheme 1 illustrates the conjugation strategy used to synthesize G3.5-Gly-SN38 and G3.5-βAla-SN38.
Scheme 1.
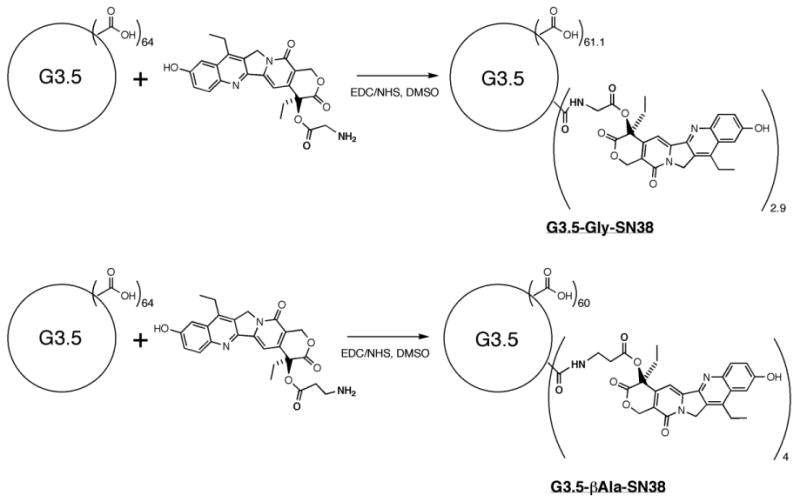
Conjugation of SN38 to G3.5 dendrimers via Glycine and β–Alanine linkers.
Drug Release Studies
G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates were prepared at 0.15 and 0.1 mg/ml respectively in the release buffer of interest and incubated at 37°C with rotation in glass vials. Release buffers included Simulated Gastric Fluid (SGF) with and without 0.32% w/v pepsin [28], Simulated Intestinal Fluid (SIF) with and without 1% w/v pancreatin [28] and phosphate buffered saline (PBS) with and without carboxylesterase (17.3 IU (international units)/ ml) [29]. Release in gastric conditions was monitored for up to 6 hours and in the intestinal conditions for up to 24 hours, mimicking the expected residence time in each of these compartments [30]. Enzyme activity was confirmed at each time point using bovine serum albumin [31], Z-Arg-AMC (benzyloxycarbonyl-L-arginine-4-methylcoumaryl-7-amide) [32] and p-nitrophenol acetate [33] as model substrates for pepsin, pancreatin and carboxylesterase, respectively.
During the release experiment, 100 μL of sample was taken at each time point and added to an additional 400 μL of release buffer. Released SN38 was separated from G3.5-SN38 conjugate by passing the sample through a 3000 MWCO Amicon Ultra 0.5 centrifugal concentrator (Millipore, Billerica, MA) by centrifuging at 14,000 × g for 30 minutes. Amicon concentrators were pre-treated with 5% v/v Triton X-100 to minimize non-specific binding as per the manufacturer's instructions. SN38 is known to exist in a pH-dependent equilibrium between a closed ring lactone form and an open ring carboxylate form, with the lactone form favored at low pH [34]. For SIF and PBS, 350 μL of the filtrate was taken and acidified to pH 2 with the addition of an appropriate volume of 1 N HCl and subsequently incubated at 37°C for 1 hour to convert SN38 to the lactone form. This acidification step was omitted for SGF since it is at a pH of 1-2. Next, the acidified SN38 was extracted by adding 200 μL of acetonitrile followed by 200 uL of chloroform. The solution was vortexed and centrifuged at 250 × g for 2 minutes to separate the layers. The organic layer was isolated, the chloroform addition was repeated two more times, and the organic extracts were pooled. The extracts were then dried under nitrogen gas, redissolved in 70 μL of 50/50 dimethyl sulfoxide (DMSO)/0.1N HCl and then measured by high pressure liquid chromatography (HPLC).
SN38 was quantified by HPLC using a system containing a Waters 1525 Binary Pump, Waters 717plus Autosampler and Waters 2487 dual wavelength ultraviolet (UV) detector (Waters Corporation, Milford, MA) set at 375 nm with a Phenomenex C18 column (250 × 4.6 mm, 5 μm) (Phenomenex, Torrance, CA). A gradient method with methanol and water with 0.1% trifluroacetic acid (TFA) was used with a total flow rate of 1 ml per minute and an injection volume of 20 μL. A calibration curve using the peak area versus concentration was generated for each release buffer by extracting known concentrations of SN38. Extraction efficiencies comparing extracted standards to direct injection of SN38 standards were found to be 86%, 91%, 75%, 80%, 70% and 83% in SGF, SGF/pepsin, SIF, SIF/pancreatin, PBS, and PBS/ carboxylesterase, respectively, and were time and concentration-independent.
Cell Culture
Caco-2 cells (passages 30–50) were grown at 37 °C in an atmosphere of 95% relative humidity and 5% CO2. Cells were maintained in T-75 flasks using Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 10,000 units/mL penicillin, 10,000 μg/mL streptomycin and 25 μg/mL amphotericin B. Media was changed every other day and cells were passaged at 80–90% confluence using a 0.25% trypsin/ethylenediamine tetraacetic acid (EDTA) solution. Incubation buffer used in the assays consisted of Hank's balanced salt solution (HBSS), supplemented with 10 mM N-(2-hydroxyethyl)piperazine-N′-179 (2 ethanesulfonic acid) hemisodium salt (HEPES) buffer (pH 7.4). HT-29 cells (passages 130-140) were cultured under the same incubation conditions as Caco-2 cells using McCoy's 5A media supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 10,000 units/mL penicillin, 10,000 μg/mL streptomycin and 25 μg/mL amphotericin B and were passaged at 80-90% confluence.
Potential Short Term Cytotoxicity of G3.5-SN38 Conjugates
Potential short-term cytotoxicity of G3.5-SN38 conjugates was assessed in Caco-2 cells to ensure cell viability during uptake and transport assays. Unmodified G3.5 dendrimers, G3.5-Gly-SN38 and G3.5-βAla-SN38 were prepared at 10 and 100 μM in HBSS transport buffer. SN38 was prepared as a concentrated stock in DMSO at 40,000 μM and used to make solutions in HBSS at 4, 40, and 400 μM. Cytotoxicity was assessed by the water soluble tetrazolium salt (WST-1) assay. Caco-2 cells were seeded at 50,000 cells per well in 96 well cell culture plates (Corning, Corning, NY) and maintained at 37 °C, 95% relative humidity and 5% CO2 for 48 hours. Cells were washed with warm HBSS buffer and incubated for 2 hours with 100 μL of varying concentrations of test compounds. After 2 hours, the inhibitor solutions were removed and the cells were washed with warm HBSS buffer. 10 μL WST-1 cell proliferation reagent in 100 μL of HBSS buffer was added to each well and incubated for 4 hours at 37 °C. Absorbance at 460 nm and background at 600 nm were measured using a SpectraMax 384 plate reader (Molecular Devices, Sunnyvale, CA). HBSS was used as a negative control for 100% cell viability and 0.01% Triton X-100 was used as a positive control. UV absorbance of G3.5-SN38 conjugates alone was also assessed to confirm that SN38 absorbance did not interfere with the cell viability dye (data not shown).
Potential Delayed Cytotoxicity of G3.5-SN38 Conjugates
Potential delayed cytotoxicity of G3.5-SN38 conjugates was assessed in Caco-2 cells by measuring cell viability 24 hours post-exposure. Cell viability 24 hours after short-term exposure mimics the potential long-term effects of the conjugates on the intestinal cells that may not be apparent when cell viability is measured immediately post-treatment. Caco-2 cells were prepared and treated the same as in the short-term toxicity assays. After the two-hour exposure, the cells were washed twice with HBSS and incubated with 100 μL of cell culture media for an additional 24 hours. The WST-1 assay was used to assess the cell viability 24 hours later, with HBSS used as a negative control for 100% cell viability and 0.01% Triton X-100 as a positive control.
Transepithelial Transport
Caco-2 cells were seeded at 80,000 cells/cm2 onto polycarbonate 12-well Transwell filters of 0.4 μm mean pore size with 1.0 cm2 surface area (Corning, Corning, NY). Caco-2 cells were maintained under standard incubation conditions where media was changed every other day and cells were used for transport experiments 21-25 days post-seeding. Prior to experiments, the transepithelial electrical resistance (TEER) of each monolayer was measured with an epithelial voltohmmeter (World Precision Instruments, Sarasota, FL). Monolayers with TEER > 600 Ω·cm2 were used for assays. Cell monolayers were washed with HBSS and then 0.5 ml of 10 or 100 μM G3.5-Gly-SN38, G3.5-βAla-SN38 or 4 μM SN38 was added to the apical compartment and 1.5 ml HBSS was added to the basolateral compartment. After incubating for 2 hours, samples were taken from the basolateral compartment. Transport was quantified by measuring fluorescence in the basolateral compartment using a SpectraMax Gemini XS spectrofluorometer (Molecular Devices, Sunnyvale, CA) with excitation and emission wavelengths of 375 and 550 nm, respectively, and compared to fluorescence standard curves for each conjugate and free SN38. Presence of free SN38 in the basolateral compartment of monolayers treated with G3.5-SN38 conjugates was determined by the extraction methods described in drug release. Equivalent SN38 flux was calculated by multiplying the measured molar flux of the conjugates with the number of SN38 molecules per dendrimer. Apical to basolateral flux is reported as the average of four replicates. Statistical significance was determined by analysis of variance followed by Tukey's multiple comparison test. Lucifer yellow CH (LY) permeability was also monitored in the presence of HBSS to ensure the integrity of the monolayers. LY apparent permeability was less than 1 × 10-6 cm/s, which is within the accepted range of LY permeability for differentiated monolayers (data not shown).
Cellular Uptake
Cellular uptake of G3.5-SN38 conjugates and free SN38 was assessed in differentiated Caco-2 monolayers. After the transport assay, the cells were washed twice with HBSS. 300 μl of 0.1% Triton X-100 was added to the apical side of each well and incubated for 2 hours at 37°C to solubilize the cells. The cells were then removed from the Transwell by pipette and transferred to a microcentrifuge tube. The cell debris was removed by centrifugation at 1000 rpm for 5 minutes. 100 μl of the clear supernatant was taken and the uptake of G3.5-SN38 conjugates and SN38 was quantified by fluorescence with excitation at 375 nm and emission at 550 nm as described in “Transepithelial Transport”. Presence of free SN38 in the cellular compartment was determined by the extraction methods described in drug release. Uptake is reported as an average of four replicates and normalized to total protein as determined by the Bradford Protein Assay (Bio-Rad, Hercules, CA). Statistical significance was determined by analysis of variance followed by Tukey's multiple comparison test.
IC50 in HT-29 Cells
The IC50 values (half maximal inhibitory concentrations) of G3.5-SN38 conjugates and free SN38 were determined in HT-29 cells to assess the efficacy of the conjugates in colorectal cancer cells compared to the free drug. HT-29 cells were seeded at 2,500 cells/ per well in 96 well cell culture plates and incubated at 37°C for 24 hours. After 24 hours, the cells were treated with different concentrations of SN38, G3.5-Gly-SN38 and G3.5-βAla-SN38 in media and incubated for an additional 48 hours. Cells were also treated with comparable concentrations of G3.5 dendrimer alone to ensure that the carrier did not cause any long-term cytotoxicity. After 48 hours, the media was aspirated by pipette, the cells were washed with HBSS buffer and the WST-1 assay was used to assess cell viability. Cell viability was determined by the % absorbance relative to the control cells, which were treated with media alone. GraphPad Prism software (La Jolla, CA) was used to generate the IC50 curves and values using the sigmoidal dose-response non-linear curve fitting routine.
Results
G3.5-SN38 Conjugate Stability
Stability of G3.5-SN38 conjugates in simulated gastric, intestinal and liver carboxylesterase conditions was assessed to determine the suitability of these conjugates for oral delivery of SN38 for the treatment of colorectal cancer hepatic metastases. In particular, stability of the conjugates was monitored in the presence of SGF with and without pepsin, SIF with and without pancreatin and PBS with and without carboxylesterase (Fig. 1).
Figure 1.
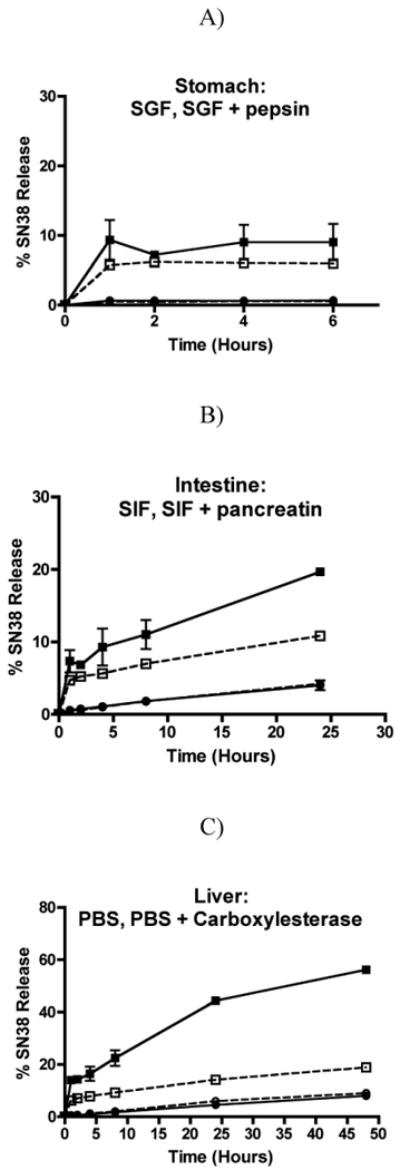
Stability of G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates. Release of SN38 was monitored in simulated conditions of the stomach for 2 hours (A), intestine for 24 hours (B) and liver for 48 hours (C). Each data point represents the average of two independent runs (mean ± standard deviation). G3.5-Gly-SN38 is represented by squares and G3.5-βAla-SN38 is represented by circles. Buffers without enzymes are depicted as dashed lines with open symbols and buffers with enzymes are depicted as solid lines with filled symbols.
Both G3.5-Gly-SN38 and G3.5-βAla-SN38 show little to no release of free SN38 in the gastric environment (Fig. 1A). G3.5-βAla-SN38 did not release more than 0.5% SN38 during 6 hours in SGF with or without pepsin. In contrast, G3.5-Gly-SN38 showed a burst release of SN38 after 1 hour in SGF with and without pepsin and ultimately released approximately 9% in SGF with pepsin and 6% in SGF without pepsin. This small amount of gastric release could be prevented by use of enteric coating. These studies illustrate that although G3.5-Gly-SN38 released more SN38 than G3.5-βAla-SN38 in the gastric environment, they were both relatively stable in acidic conditions and were not substrates for pepsin.
In comparison to SGF, G3.5-SN38 conjugates showed increased susceptibility for free drug release in the presence of SIF (Fig. 1B). G3.5-βAla-SN38 showed up to 4% SN38 release in 24 hours in the presence of SIF, but the addition of pancreatin did not increase this release. In contrast, G3.5-Gly-SN38 released up to 11% SN38 in the presence of SIF and up to 20% with the addition of pancreatin. These results show that both conjugates are inherently more susceptible to hydrolysis in the basic pH of SIF compared to the acidic pH of SGF, which is common for ester linkages. In addition, while G3.5-βAla-SN38 did not appear to be a substrate for pancreatin, G3.5-Gly-SN38 showed increased release in the presence of pancreatin compared to SIF alone. However, with a maximum of 20% release after 24 hours, both of these conjugates showed a low extent and slow rate of release in the intestinal environment.
Finally, we examined the stability profile of the conjugates in the presence of liver carboxylesterase and in PBS alone (Fig. 1C). Carboxylesterase is highly expressed in the liver environment and can be used to estimate the release in this milieu. Both conjugates showed similar release profiles in PBS and SIF up to 24 hours, and the linear release kinetics continued until 48 hours, suggesting similar rates of hydrolysis in SIF and PBS without enzymes. G3.5-βAla-SN38 did not show any additional release in the presence of carboxylesterase, suggesting that it is not a substrate for this enzyme. In contrast, G3.5-Gly-SN38 showed a significant increase in SN38 release in the presence of carboxylesterase, achieving 56% release after 48 hours, illustrating that the ester bond in G3.5-Gly-SN38 can be cleaved by carboxylesterase to release free SN38 in the liver environment. Taken together, these stability studies show that G3.5-βAla-SN38 is stable in all three environments and is not a substrate for pepsin, pancreatin or carboxylesterase, while G3.5-Gly-SN38 shows less stability in the gastric and intestinal environments and the greatest release in the presence of carboxylesterase, making it a potential candidate for oral delivery of SN38 to colorectal hepatic metastases.
Short-Term Cytotoxicity
Short-term cytotoxicity of G3.5 dendrimers, G3.5-SN38 conjugates and SN38 was assessed in Caco-2 cells by the WST-1 cell viability assay. This assay interrogates the short-term effects of the conjugates on the intestinal barrier and also serves to ensure that cell viability is not compromised during the 2-hour time needed for transport and uptake assays. Figure 2 shows the cell viability of Caco-2 cells treated for 2 hours with unmodified G3.5 dendrimers and G3.5-SN38 conjugates. SN38 is also tested at 4, 40 and 400 μM for comparison, corresponding to 1, 10 or 100% of the drug loading on the G3.5-βAla-SN38 conjugate.
Figure 2.
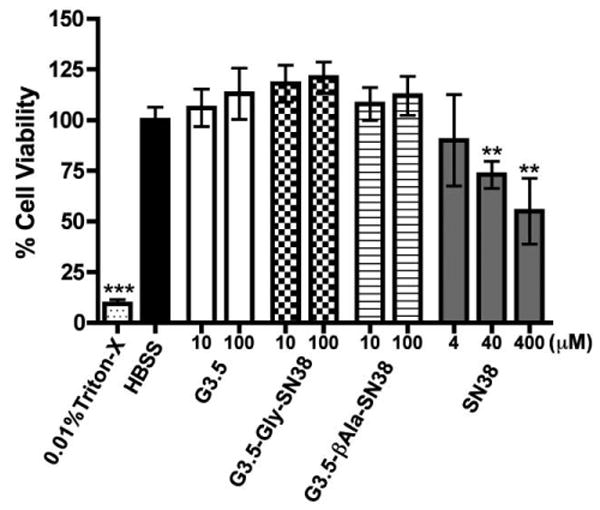
Caco-2 Cell viability after treatment for 2 hours with G3.5 dendrimers, G3.5-SN38 conjugates and SN38. Data is shown as mean ± standard deviation (n=6). (**) and (***) indicate a statistically significant decrease in cell viability compared to HBSS control with p<0.01 and p<0.001, respectively. G3.5 dendrimers and G3.5-SN38 conjugates do not show a reduction in cell viability up to 100 μM while SN38 shows a significant cytotoxic effect at 40 and 400 μM.
As shown in Figure 2, G3.5 dendrimers and G3.5-SN38 conjugates did not cause a significant reduction in cell viability up to 100 μM. In contrast, despite the short treatment time, SN38 shows a significant reduction in cell viability at 40 and 400 μM, corresponding to approximately 10% and 100% of the drug loading on the conjugates. This suggests that conjugation of SN38 to G3.5 dendrimers is able to significantly reduce intestinal toxicity of SN38. In addition, it is observed that G3.5-Gly-SN38 and G3.5-βAla-SN38 can be used in transport and uptake assays up to 100 μM without compromising cell viability.
Delayed Cytotoxicity
In order to assess the potential long-term effects of G3.5 dendrimers and G3.5-SN38 conjugates on the intestinal barrier, we performed a delayed cytotoxicity assay. In this assay, Caco-2 cells were treated for 2 hours, the treatment was removed and the cells were incubated for an additional 24 hours in cell culture media. This allows for the assessment of any potential delayed-onset responses of the cells to dendrimer treatment (e.g. apoptosis), which can be detected after 24 hours (Fig. 3).
Figure 3.
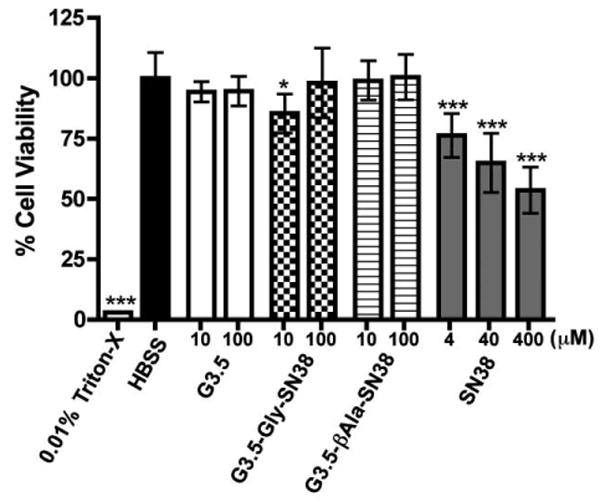
Caco-2 cell viability 24 hours after 2-hour treatment with G3.5, G3.5-SN38 conjugates and SN38. Data is shown as mean ± standard deviation (n=6). (*) and (***) indicate a statistically significant decrease in cell viability relative to HBSS control with p<0.05 and p<0.001, respectively.
Figure 3 illustrates that even after 24 hours, G3.5 dendrimers and G3.5-SN38 conjugates did not cause a statistically significant decrease in cell viability, with the exception of G3.5-Gly-SN38 at a 10 μM concentration, which displayed 85.4 % +/- 8.1% viability. While this is a statistically significant decrease from the HBSS control, 85% viability is still considered to be acceptable in such viability assays and 10 μM treatment with G3.5-Gly-SN38 does not show a significant difference from 100 μM treatment with the same conjugate. In contrast, SN38, which is known to cause apoptosis by inhibition of topoisomerase-1 [23], had a significant impact on cell viability 24 hours post treatment at 4, 40 and 400 μM concentrations. This illustrates that by conjugating SN38 to dendrimers, intestinal toxicity is minimized compared to the free drug and that G3.5-SN38 conjugates should be safe for oral administration.
Transepithelial Transport
Transepithelial transport of G3.5-SN38 conjugates and free SN38 was measured across differentiated Caco-2 monolayers in the apical to basolateral direction and expressed as the equivalent SN38 flux calculated at 2 hours (Fig. 4).
Figure 4.
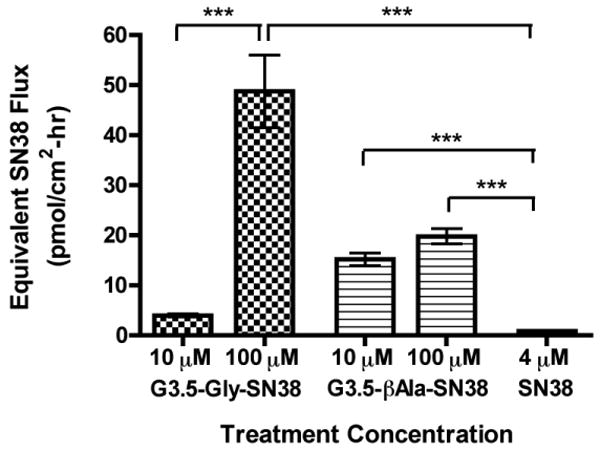
Equivalent SN38 flux across differentiated Caco-2 monolayers treated with G3.5-SN38 conjugates and SN38. Treatment concentrations of conjugates were 10 and 100 μM, corresponding to 29, 290 and 40, 400 μM equivalents of SN38 for G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates, respectively. Equivalent SN38 flux was calculated by multiplying the measured molar flux of the conjugates with the number of SN38 molecules per dendrimer. Data is shown as mean ± standard deviation (n=4). (***) indicates a significant difference with p<0.001.
In this study SN38 was tested at 4 μM concentration since significant cytotoxicity was observed in Caco-2 cells treated with SN38 at 40 and 400 μM. It has been shown that SN38 is transported across Caco-2 cell monolayers by an active transport pathway, and that increasing the concentration from 2.5 μM to 25 μM does not increase transepithelial flux [35]. This suggests that 4 μM SN38 should be beyond the saturation limit for transport and uptake, thus permitting direct comparison of SN38 flux to that of G3.5-SN38 conjugates despite the difference in total drug concentration.
Presence of free SN38 in the basolateral compartment was found to be less than 5% of the amount transported for G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates (data not shown). Hence the measured flux is due almost entirely to transport of intact conjugate. G3.5-Gly-SN38 at 100 μM and G3.5-βAla-SN38 at 10 μM and 100 μM showed a statistically significant increase in apical to basolateral SN38 flux relative to free drug (p<0.001). Taking into account the drug loading on each conjugate (2.9 SN38 per dendrimer for G3.5-Gly-SN38 and 4.0 SN38 per dendrimer for G3.5-βAla-SN38), the overall SN38 flux increase compared to free drug is 13, 159, 69 and 89-fold for G3.5-Gly-SN38 at 10 μM and 100 μM and G3.5-βAla-SN38 at 10 μM and 100 μM, respectively. These significant increases in the amount of SN38 transported across the monolayer indicate that G3.5 dendrimers are effective oral drug delivery carriers and can increase the permeability of SN38.
Interestingly, the impact of concentration on transport is different for G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates. G3.5-Gly-SN38 shows approximately a 10-fold increase in transport with a 10-fold increase in concentration, indicating that diffusion-driven processes (i.e. paracellular transport) are predominantly involved. In contrast, G3.5-βAla-SN38, shows minimal increase in transport with a 10-fold increase in concentration, suggesting that its epithelial flux may be controlled by an energy-dependent mechanism with minimal paracellular transport. Dendrimers have been shown to cross epithelia both by transcellular and by paracellular pathways and the relative importance of each has been shown to be charge and surface chemistry dependent [18]. Factors such as the number of conjugated surface groups as well as the ability of these groups to shield the dendrimer surface charge have been found to impact the ability of dendrimers to open tight junction and overall mechanism of transport. Therefore, both the number of SN38 molecules conjugated and the identity of the linker could potentially impact the ultimate transport pathway. These studies further confirm the importance of dendrimer surface chemistry and drug linker chemistry in the degree and mechanism of transport. More detailed mechanistic studies, however, are required to explain the differences in transepithelial transport profiles of the two conjugates.
Cellular Uptake
Cellular uptake studies shed light on the contribution of the transcellular pathway to overall transepithelial transport as well as the ability of the carrier to promote drug uptake. Cellular uptake of G3.5-SN38 conjugates and SN38 was measured in differentiated Caco-2 monolayers after a 2-hour incubation time (Fig. 5).
Figure 5.
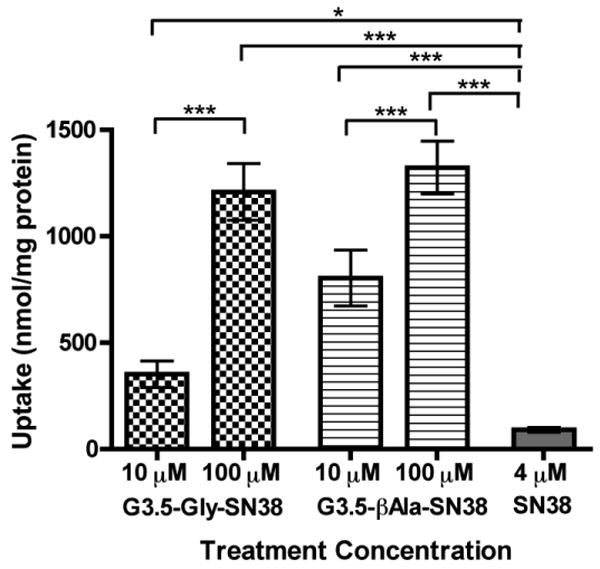
Cellular uptake of G3.5-SN38 conjugates and free SN38 in differentiated Caco-2 monolayers after treatment for 2 hours on the apical side. Data is shown as mean ± standard deviation (n=4). (*), (**), and (***) show statistical differences in uptake between groups with p < 0.05, 0.01 and 0.001, respectively. All conjugates show a statistically significant increase in uptake relative to SN38.
Similar to the transport studies, free SN38 in the cells after a 2-hour treatment was found to be less than 5% of conjugate uptake (data not shown). All conjugates tested showed a significant increase in uptake relative to free SN38. This illustrates that G3.5 dendrimers can enter cells more efficiently than SN38, and thus are suitable for cellular delivery. Both G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates show a significant increase in uptake (p<0.001) with increase in concentration. However, for both conjugates the increase in uptake is less than the corresponding increase in concentration, suggesting the involvement of a saturable uptake mechanism, such as receptor-mediated endocytosis. Interestingly, both conjugates show similar uptake for the100 μM treatment, suggesting that differences in overall transport may be due to differences in paracellular transport rather than transcellular transport at this concentration. These uptake results confirm that cellular uptake of G3.5-SN38 conjugates in Caco-2 cells plays a significant role in G3.5-SN38 conjugate transport.
IC50 in HT-29 Cells
Toxicity of G3.5-SN38 conjugates and free SN38 was determined in HT-29 cells for 48 hours in order to compare the activity of the free drug and dendrimer-drug conjugates (Fig. 6). HT-29 cells are derived from human colon adenocarcinoma, are well suited for IC50 studies of anti-cancer activity due to their uniform cell growth and morphology.
Figure 6.
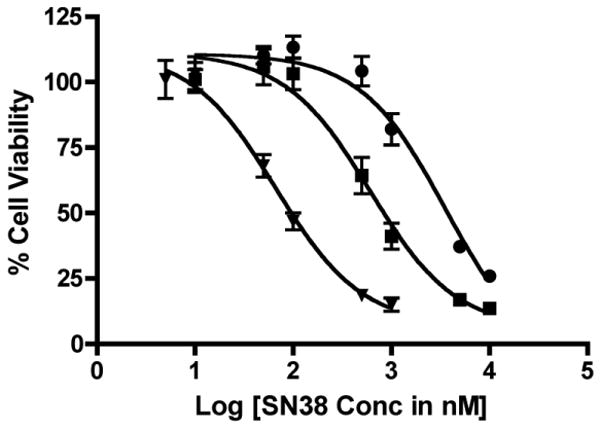
IC50 Curves of SN38 (triangle), G3.5-Gly-SN38 (square) and G3.5-bAla-SN38 (circle) in HT-29 cells. Data is shown as mean ± standard deviation (n=6). IC50 values were determined from nonlinear sigmoidal dose response curve fitting by GraphPad Prism software and are 66.3 nM, 0.60 μM and 3.59 μM for SN38, G3.5-Gly-SN38 and G3.5-βAla-SN38, respectively
SN38 shows the highest activity against HT-29 cells with an IC50 value of 66.3 nM. In contrast, SN38 conjugated to G3.5 dendrimers shows much lower activity with IC50 values of 0.60 μM and 3.59 μM for G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates, respectively, corresponding to approximately 10 and 60 times less potency than the free drug. Treatment with comparable G3.5 dendrimer concentrations did not cause any reduction in cell viability (data not shown), confirming that the carrier does not cause any toxicity. Since cytotoxicity is dependent on free drug release from the dendrimer backbone, the lower IC50 value for G3.5-Gly-SN38 compared to G3.5-βAla-SN38 is consistent with the higher release rate of SN38 from the G3.5-Gly-SN38 conjugate. Because the conjugates show some release under acidic conditions and greater release at neutral pH, it is likely that the activity in HT-29 cells is due to a combination of extracellular release of SN38 in cell culture media and intracellular release. Further detailed studies are required to examine the relative contribution of extra- and intracellular drug release to cytotoxcity. Importantly, despite the loss in efficacy compared to the free drug, the IC50 values of the G3.5-SN38 conjugates are still in the nanomolar to micromolar range, which is acceptable for therapy.
Discussion
Dendrimers have shown promise as oral drug delivery carriers due to their ability to translocate across the epithelial layer of the gut. The impact of dendrimer properties such as generation, charge and concentration on transepithelial transport have been described in detail, but comparatively few reports have been published using dendrimers to translocate drugs across the intestinal barrier. Previously Kolhatkar and co-workers [17] reported the use of G4 dendrimers complexed with SN38 through non-covalent interactions as a potential oral drug delivery system. While the conjugates increased the transepithelial transport and cellular uptake of SN38, they released 40% of the drug within 24 hours in PBS and 90% of the drug within 30 minutes at pH 5. Because of the instability in acidic conditions, these dendrimer-SN38 complexes would have significant limitations for oral administration. In addition, because of the high intrinsic toxicity of the G4 dendrimer carrier [36], these complexes could only be used up to a 10 μM concentration, limiting the amount of drug transported across the intestinal barrier and into the bloodstream.
In our previous work Vijayalakshmi and colleagues [26] reported the preliminary synthesis and characterization of G3.5-SN38 conjugates with glycine and β–alanine linkers. In our current work, we investigated the potential of these G3.5-SN38 conjugates for oral therapy of colorectal hepatic metastases. The G3.5-SN38 conjugates have several advantages compared to the previously reported G4-SN38 complexes. Because the drug is covalently conjugated to the dendrimer rather than complexed, the conjugates are relatively stable in the gastric and intestinal environments, minimizing premature drug release upon oral administration. In addition, the G3.5-Gly-SN38 conjugates release the drug in the presence of carboxylesterase, allowing for the targeted treatment of colorectal hepatic metastasis. These systems can be used at higher concentrations (100 μM vs. 10 μM) due to the low intrinsic toxicity of G3.5 dendrimers, allowing for greater drug transport across the gastrointestinal barrier and a higher dose at the site of action. Therefore, these conjugates show distinct advantages over previously published dendrimer-SN38 complexes and are a significant step towards a functional dendrimer-based oral drug delivery system.
In order to determine their suitability for oral delivery, the stability of the conjugates in the gastrointestinal milieu as well as in the presence of liver carboxylesterase was investigated. G3.5-βAla-SN38 conjugates were significantly more stable than G3.5-Gly-SN38 under the conditions studied, releasing a maximum of 0.7% drug in SGF after 6 hours, 4% in SIF after 24 hours and 8% in carboxylesterase after 48 hours. In addition, in each of the release media, the addition of an enzyme did not increase the release of SN38, illustrating that G3.5-βAla-SN38 is not susceptible to enzymatic cleavage. In contrast, G3.5-Gly-SN38 showed much higher release in all of the conditions and showed increased rate and extent of release in the presence of pancreatin and carboxylesterase compared to buffer alone. Previous studies conjugating CPT to poly(l-lysine) dendrimers using glycine and β-alanine linkages at the 20-OH position showed similar results with the glycine linker indicating higher rates of hydrolysis than the stable β–alanine linker [37]. Although the β–alanine linker provides an extra methyl group as a spacer, this group appears to stabilize the bond against hydrolysis and enzymatic degradation.
The impact of linker chemistry on SN38 release shows a direct correlation with the IC50 values in which the G3.5-Gly-SN38 conjugates had six-fold greater efficacy than G3.5-βAla-SN38 conjugates, illustrating the importance of SN38 release for anti-cancer activity. Since the conjugates were relatively stable at lower pH it is likely that only a small percentage of the drug was released in the intracellular acidic environment with the majority released in the extracellular media. Stability studies in PBS for 48 hours showed a two-fold increase in SN38 release from G3.5-Gly-SN38 compared to G3.5-βAla-SN38, suggesting additional enzymatic mechanisms may play a role in enhancing G3.5-Gly-SN38 efficacy by 6-fold. Comparing the release profiles with and without enzymes, an increased release of SN38 from G3.5-Gly-SN38 in the presence of pancreatin and carboxylesterase was observed, but no such increase for G3.5-βAla-SN38 was seen, indicating the increased susceptibility of the glycine linker to enzymatic degradation.
In addition to differing release and toxicity profiles, G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates showed significant differences in transepithelial transport. Both conjugates showed increased flux of SN38 relative to free drug, which is critical for improving the oral bioavailability of SN38. In addition, neither conjugate shows short- or long-term effects on Caco-2 cells after a 2-hour treatment, illustrating that conjugation of SN38 to G3.5 dendrimers can minimize intestinal toxicity while maximizing transport. Interestingly, transport of G3.5-Gly-SN38 was highly concentration-dependent while G3.5-βAla-SN38 flux was unchanged between treatment with 10 and 100 μM concentrations. This suggests that G3.5-Gly-SN38 may be transported primarily by a concentration gradient-driven process, such as paracellular diffusion, whereas a saturable process, such as transcellular transport, governs G3.5-βAla-SN38 transport. This phenomenon, however, needs further examination. While the uptake data suggests comparable uptake of the conjugates at 100 μM this measurement could include surface bound dendrimer or dendrimer that would be degraded in the cell and not transcytosed. Therefore, reduction in G3.5-βAla-SN38 transport at 100 μm compared to G3.5-Gly-SN38 transport could be due to decreases in both transcellular and paracellular pathways. In previous reports we have shown the impact of surface chemistry on the mechanism of transport and uptake of PAMAM dendrimers. Specifically, reduction of surface charge on G3.5 dendrimers by the addition of low molecular weight polyethylene glycol was found to reduce tight junction opening, transepithelial transport and uptake in Caco-2 cells [18]. Thus, it is possible that addition of a methyl group in the alanine linker as well as the increase of drug loading from 2.9 to 4.0 molecules of SN38 increased the hydrophobicity of the conjugates, hence reducing the degree to which G3.5-βAla-SN38 conjugates opened the tight junctions, resulting in an overall transport mechanism dominated by the transcellular route. Future studies will address this hypothesis in more detail. Importantly, these studies illustrate the choice of drug linker and the degree of drug loading not only impact drug release and ultimate efficacy but can also impact transport. Therefore, dendrimer-drug conjugates for oral delivery must be carefully designed to meet transport, release and efficacy demands.
Conclusion
In this work we investigated G3.5-SN38 conjugates for oral delivery of SN38 by determining their in vitro release profiles in simulated gastric and intestinal conditions and in the presence of carboxylesterase, their toxicity against intestinal cells and target colorectal cancer cells, and their transepithelial transport and cellular uptake in Caco-2 monolayers. We demonstrated that conjugation of SN38 to G3.5 dendrimers increased the transepithelial transport while simultaneously reducing intestinal toxicity compared to free SN38, illustrating the potential for these conjugates in oral drug delivery. A significant impact of linker chemistry on drug release and efficacy in HT-29 cells was shown with G3.5-Gly-SN38 showing lower in vitro stability and higher efficacy than G3.5-βAla-SN38 conjugates. The drug linker chemistry and drug loading also impacted the transport pathway with G3.5-Gly-SN38 having a concentration-dependent transport profile and G3.5-βAla-SN38 conjugates having a saturable transport profile. G3.5-Gly-SN38 shows promise for oral delivery of SN38 for the treatment of colorectal hepatic metastases. Treatment with 100 μM G3.5-Gly-SN38 caused a 159-fold increase in SN38 transepithelial transport compared to free SN38 illustrating its potential to enhance SN38 bioavailability. In addition, G3.5-Gly-SN38 was relatively stable in gastric and intestinal milieu with increased release in the presence of liver carboxylesterase. Finally, G3.5-Gly-SN38 shows an IC50 of 0.60 μM in HT-29 cells, which is acceptable for cancer therapy. Together these results show that PAMAM dendrimers have the potential to improve the oral bioavailability of potent anti-cancer therapeutics and that appropriate selection of drug linker is a critical step in designing dendrimers for oral drug delivery applications.
Acknowledgments
Financial Support was provided by the Fischell Fellowship in Bioengineering to D. Goldberg and NIH R01EB07470.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2(5):347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 2.Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006;24(1):39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Kopecek J, Kopeckova P. HPMA copolymers: origins, early developments, present, and future. Adv Drug Delivery Rev. 2010;62(2):122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasut G, Veronese FM. PEG conjugates in clinical development or use as anticancer agents: an overview. Adv Drug Delivery Rev. 2009;61(13):1177–1188. doi: 10.1016/j.addr.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 6.Findlay M, von Minckwitz G, Wardley A. Effective oral chemotherapy for breast cancer: pillars of strength. Ann Oncol. 2008;19(2):212–222. doi: 10.1093/annonc/mdm285. [DOI] [PubMed] [Google Scholar]
- 7.Le Lay K, Myon E, Hill S, Riou-Franca L, Scott D, Sidhu M, Dunlop D, Launois R. Comparative cost-minimisation of oral and intravenous chemotherapy for first-line treatment of non-small cell lung cancer in the UK NHS system. Eur J Health Econ. 2007;8(2):145–151. doi: 10.1007/s10198-006-0034-1. [DOI] [PubMed] [Google Scholar]
- 8.Pashko S, Johnson DH. Potential cost savings of oral versus intravenous etoposide in the treatment of small cell lung cancer. Pharmacoeconomics. 1992;1(4):293–297. doi: 10.2165/00019053-199201040-00006. [DOI] [PubMed] [Google Scholar]
- 9.Chang S, Long SR, Kutikova L, Bowman L, Finley D, Crown WH, Bennett CL. Estimating the cost of cancer: results on the basis of claims data analyses for cancer patients diagnosed with seven types of cancer during 1999 to 2000. J Clin Oncol. 2004;22(17):3524–3530. doi: 10.1200/JCO.2004.10.170. [DOI] [PubMed] [Google Scholar]
- 10.Milhem OM, Myles C, McKeown NB, Attwood D, D'Emanuele A. Polyamidoamine Starburst dendrimers as solubility enhancers. Int J Pharm. 2000;197(1-2):239–241. doi: 10.1016/s0378-5173(99)00463-9. [DOI] [PubMed] [Google Scholar]
- 11.Tomalia DA, Reyna LA, Svenson S. Dendrimers as multi-purpose nanodevices for oncology drug delivery and diagnostic imaging. Biochem Soc Trans. 2007;35(Pt 1):61–67. doi: 10.1042/BST0350061. [DOI] [PubMed] [Google Scholar]
- 12.Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Adv Drug Delivery Rev. 2005;57(15):2215–2237. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 13.Lee CC, MacKay JA, Fréchet JM, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23(12):1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 14.Wiwattanapatapee R, Carreño-Gómez B, Malik N, Duncan R. Anionic PAMAM dendrimers rapidly cross adult rat intestine in vitro: a potential oral delivery system? Pharm Res. 2000;17(8):991–998. doi: 10.1023/a:1007587523543. [DOI] [PubMed] [Google Scholar]
- 15.Kitchens KM, Kolhatkar RB, Swaan PW, Eddington ND, Ghandehari H. Transport of poly (amido amine) dendrimers across Caco-2 cell monolayers: Influence of size, charge and fluorescent labeling. Pharm Res. 2006;23(12):2818–2826. doi: 10.1007/s11095-006-9122-2. [DOI] [PubMed] [Google Scholar]
- 16.D'Emanuele A, Jevprasesphant R, Penny J, Attwood D. The use of a dendrimer-propranolol prodrug to bypass efflux transporters and enhance oral bioavailability. J Control Release. 2004;95(3):447–453. doi: 10.1016/j.jconrel.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Kolhatkar RB, Swaan PW, Ghandehari H. Potential oral delivery of 7-ethyl-10-hydroxy-camptothecin (SN-38) using poly (amido amine) dendrimers. Pharm Res. 2008;25(7):1723–1729. doi: 10.1007/s11095-008-9572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweet D, Kolhatkar R, Ray A, Swaan P, Ghandehari H. Transepithelial transport of PEGylated anionic poly (amido amine) dendrimers: implications for oral drug delivery. J Control Release. 2009;138(1):78–85. doi: 10.1016/j.jconrel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Sayed M, Rhodes CA, Ginski M, Ghandehari H. Transport mechanism(s) of poly (amido amine) dendrimers across Caco-2 cell monolayers. Int J Pharm. 2003;265(1-2):151–157. doi: 10.1016/s0378-5173(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 20.Kitchens KM, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis inhibitors prevent poly (amido amine) dendrimer internalization and permeability across Caco-2 cells. Mol Pharm. 2008;5(2):364–369. doi: 10.1021/mp700089s. [DOI] [PubMed] [Google Scholar]
- 21.Goldberg DS, Ghandehari H, Swaan PW. Cellular entry of G3.5 poly (amido amine) dendrimers by clathrin- and dynamin-dependent endocytosis promotes tight junctional opening in intestinal epithelia. Pharm Res. 2010;27(8):1547–1557. doi: 10.1007/s11095-010-0153-3. [DOI] [PubMed] [Google Scholar]
- 22.Jevprasesphant R, Penny J, Attwood D, D'Emanuele A. Transport of dendrimer nanocarriers through epithelial cells via the transcellular route. J Control Release. 2004;97:259–267. doi: 10.1016/j.jconrel.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 23.Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51(16):4187–4191. [PubMed] [Google Scholar]
- 24.Xu G, Zhang W, Ma MK, McLeod HL. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin Cancer Res. 2002;8(8):2605–2611. [PubMed] [Google Scholar]
- 25.Rothenberg ML. Irinotecan (CPT-11): recent developments and future directions-colorectal cancer and beyond. Oncologist. 2001;6(1):66–80. doi: 10.1634/theoncologist.6-1-66. [DOI] [PubMed] [Google Scholar]
- 26.Vijayalakshmi N, Ray A, Malugin A, Ghandehari H. Carboxyl terminated PAMAM-SN38 conjugates: synthesis, characterization, and in vitro evaluation. Bioconjug Chem. 2010;21(10):1804–1810. doi: 10.1021/bc100094z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, Horak ID. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem. 2008;19(4):849–859. doi: 10.1021/bc700333s. [DOI] [PubMed] [Google Scholar]
- 28.Roger E, Lagarce F, Benoit JP. The gastrointestinal stability of lipid nanocapsules. Int J Pharm. 2009;379(2):260–265. doi: 10.1016/j.ijpharm.2009.05.069. [DOI] [PubMed] [Google Scholar]
- 29.Ahlmark M, Vepsäläinen J, Taipale H, Niemi R, Järvinen T. Bisphosphonate prodrugs: synthesis and in vitro evaluation of novel clodronic acid dianhydrides as bioreversible prodrugs of clodronate. J Med Chem. 1999;42(8):1473–1476. doi: 10.1021/jm9810809. [DOI] [PubMed] [Google Scholar]
- 30.Gabor F, Fillafer C, Neutsch L, Ratzinger G, Wirth M. Improving oral delivery. Handb Exp Pharmacol. 2010;(197):345–398. doi: 10.1007/978-3-642-00477-3_12. [DOI] [PubMed] [Google Scholar]
- 31.Kimsey R, Harding E. A spectrophotometric assay optimizing conditions for pepsin activity. Amer Biol Teach. 1998;60(3):200–201. [Google Scholar]
- 32.Mullally M, OCallaghan D, FitzGerald R, Donnelly W, Dalton J. Proteolytic and peptidolytic activities in commercial pancreatic protease preparations and their relationship to some ehey protein hydrolysate characteristics. J Agric Food Chem. 1994;42:2973–2961. [Google Scholar]
- 33.Wheelock CE, Severson TF, Hammock BD. Synthesis of new carboxylesterase inhibitors and evaluation of potency and water solubility. Chem Res Toxicol. 2001;14(12):1563–1572. doi: 10.1021/tx015508+. [DOI] [PubMed] [Google Scholar]
- 34.Stewart CF, Zamboni WC, Crom WR, Houghton PJ. Disposition of irinotecan and SN-38 following oral and intravenous irinotecan dosing in mice. Cancer Chemother Pharmacol. 1997;40(3):259–265. doi: 10.1007/s002800050656. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto W, Verweij J, de Bruijn P, de Jonge MJ, Takano H, Nishiyama M, Kurihara M, Sparreboom A. Active transepithelial transport of irinotecan (CPT-11) and its metabolites by human intestinal Caco-2 cells. Anticancer Drugs. 2001;12(5):419–432. doi: 10.1097/00001813-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Kitchens KM, El-Sayed ME, Ghandehari H. Transepithelial and endothelial transport of poly (amido amine) dendrimers. Adv Drug Delivery Rev. 2005;57(15):2163–2176. doi: 10.1016/j.addr.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 37.Fox ME, Guillaudeu S, Frechet JM, Jerger K, Macaraeg N, Szoka FC. Synthesis and in vivo antitumor efficacy of PEGylated poly(l-lysine) dendrimer-camptothecin conjugates. Mol Pharm. 2009;6(5):1562–1572. doi: 10.1021/mp9001206. [DOI] [PMC free article] [PubMed] [Google Scholar]