

Abstract
Alveolar development comprises the transition of lung architecture from saccules to gas-exchange units during late gestation and early postnatal development. Exposure to hyperoxia disrupts developmental signaling pathways and causes alveolar hypoplasia as seen in bronchopulmonary dysplasia affecting preterm human newborns. Expanding literature suggests that epigenetic changes due to environmental triggers during development may lead to genetically heritable changes in gene expression. Given recent data on altered histone deacetylase (HDAC) activity in lungs of humans and animal models with airspace enlargement/emphysema, we hypothesized that alveolar hypoplasia from hyperoxia exposure in neonatal mice is a consequence of cell cycle arrest and reduced HDAC activity and up-regulation of the cyclin-dependent kinase inhibitor, p21. We exposed newborn mice to hyperoxia and compared lung morphologic and epigenetic changes to room air controls. Further, we pretreated a subgroup of animals with the macrolide antibiotic azithromycin (AZM), known to possess anti-inflammatory properties. Our results showed that hyperoxia exposure resulted in alveolar hypoplasia and was associated with decreased HDAC1 and HDAC2 and increased p53 and p21 expression. Further, AZM did not confer protection against hyperoxia-induced alveolar changes. These findings suggest that alveolar hypoplasia due to hyperoxia is mediated by epigenetic changes affecting cell cycle regulation/senescence during lung development.
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is a chronic lung disease associated with significant mortality and morbidity in premature infants. Despite the advent of antenatal steroids, surfactant replacement therapy, and use of gentle ventilation, the incidence of BPD remains unchanged (1). The pathogenesis of BPD is complex with interplay of genetic and environmental factors that include sepsis (antenatal and/or post-natal), ventilator-induced lung injury, and hyperoxia (1). Characteristic histopathologic changes in lungs of premature infants with BPD include enlarged distal airspaces, decreased alveolar secondary septation, and dysmorphic capillary configuration resulting in alveolar simplification (2).
Alveolar formation is a highly coordinated process between development of airways and pulmonary vasculature (3). During this critical stage of lung development, multiple etiological factors can lead to disruption of normal vascular growth and alveolar formation. Pulmonary inflammation arrests lung development and contributes to the pathogenesis of BPD as demonstrated by increased pro-inflammatory mediators and cells in tracheal aspirates from premature infants with respiratory distress who later developed BPD (4). Hyperoxia exposure is known to disrupt alveolar formation and growth by upregulating inflammatory mediators (5), impairing signaling of vascular endothelial growth factor (VEGF) (6), and accelerating apoptosis (5, 7).
Epigenetic changes, such as acetylation of histone, result in unwinding of the DNA to facilitate gene transcription. Histone deacetylase enzymes (HDACs) remove acetyl moieties from lysine residues of histones, causing rewinding of the DNA and silencing of gene transcription. p21CIP1/WAF1/SDI1 (p21) is a potent inhibitor of cyclin-dependent kinases (cdks) that arrests the cell cycle at the G1/S interface (8). Decreased HDAC activity triggers p21 transcription and leads to an arrest of cell proliferation (8). HDACs are known to contribute to the pathogenesis of chronic inflammatory lung diseases in adults (9, 10) but their potential role during the pathogenesis of BPD in premature infants remains unknown. Furthermore, the effect of hyperoxia on HDAC activity in an animal model of BPD has not been studied. We thus hypothesize that alveolar hypoplasia due to hyperoxia exposure in neonatal mice is due, in part, to cell cycle arrest as a consequence of reduced HDAC activity and up-regulation of p21. Azithromycin (AZM) is a macrolide antibiotic that has been shown previously to improve lung development in hyperoxic neonatal rats (11). We further hypothesize that AZM reverses hyperoxia-induced alveolar hypoplasia in neonatal mice by restoring HDAC activity and decreasing transcription of p21. To test this hypothesis, neonatal mice were exposed to 14 days of hyperoxia with and without AZM treatment to assess inflammatory responses as well as HDAC and p21 expression associated with alveolar hypoplasia.
METHODS
Reagents
Unless otherwise stated, biochemical reagents were purchased from Sigma (St. Louis, MO). Antibodies for Western blotting, immunohistochemistry, immunofluorescence including HDAC1 (sc-6298), HDAC2 (sc-7899), and p21 (sc-397) were from Santa Cruz Biotechnology (Santa Cruz, CA), proliferating cell nuclear antigen (PCNA) (ab2426) from Abcam Inc. (Cambridge, MA), and p53 (#2524) from Cell Signaling Technology (Danvers, MA). Azithromycin was from Abraxis Pharmaceutical Products (Schaumburg, IL).
Animal hyperoxia
Timed-pregnant FVB/n mice (Charles River Laboratories, Inc.) delivered newborn litters that were culled to equal sizes (n = 9) and separated into 4 groups: 1) saline-room air (Sal-RA), 2) saline-hyperoxia (Sal-O2), 3) azithromycin-room air (AZM-RA), and 4) azithromycin-hyperoxia (AZM-O2). Starting day 0, pups were weighed and injected daily with either AZM (40 mg/kg i.p.) or saline until day of sacrifice. Hyperoxia was delivered via oxygen concentrator (AirSep, VisionAire Corp., Buffalo, NY) into a 20-gallon chamber at 5 Lpm constant flow and fraction of inspired oxygen (FiO2) levels were recorded daily by oxygen sensor (Hudson RCI, TeleFlex Medical, Inc., Research Triangle Park, NC). Animals in hyperoxia were maintained in FiO2 80% starting day 1 and lactating dams were switched daily between hyperoxia and RA litters to minimize prolonged oxygen toxicity. Mice were fed food and water ad libitum and maintained under approved IACUC protocols at UCLA until the time of sacrifice.
Lung bronchoalveolar lavage and tissue collection
Mice were euthanized using pentobarbital (100 mg/kg i.p.) and a 27 gauge angiocatheter was used to cannulate the trachea and collect bronchoalveolar lavage (BAL) by instilling 0.5 ml PBS + 5mM EDTA as previously described (12). Retrieved solutions were analyzed for total cell counts and cytospin differentials. Lung tissue was processed for histological analysis or snap-frozen for further analysis.
Lung histology and morphometric analysis
A 27 gauge angiocatheter was used to cannulate the trachea and inflation-fix the lungs in 4% paraformaldehyde at 25 cm H2O for 1 min. Tissue was immersion-fixed overnight, dehydrated through graded alcohols, washed in 100% ethanol and CitriSolvR (Decon Labs, King of Prussia, PA), and embedded in paraffin. Serial 5-μm-thick sections were stained with hematoxylin and eosin (H&E) for histopathological analysis and photographed using a SPOT Insight QE camera and software to measure alveolar morphometric data. Radial alveolar counts were measured as previously described (13).
Protein extraction from lung tissues
Lung tissue was homogenized (Pro 200 homogenizer) in 0.5 ml of ice-cold RIPA (radioimmunoprecipitation assay) buffer containing complete protease inhibitor cocktail (14). Protein levels were measured by bicinchoninic acid (BCA) colorimetric assay (Thermo Scientific, Rockford, IL) using BSA as a standard, and samples were stored at -80 °C for Western blotting and total HDAC activity.
Western blot analysis
Proteins from lung tissue homogenates were separated on a SDS-polyacrylamide gel and transferred onto nitrocellulose membranes (Amersham, Arlington Heights, IL). Membranes were probed with a specific primary antibody (1:1000 dilution) at 4°C overnight. After washing, the membrane was probed with secondary anti-rabbit, anti-mouse, or anti-goat antibody (1:10,000 dilution) linked to horseradish peroxidase, and detected using enhanced chemiluminescence (Perkin Elmer, Waltham, MA). Equal loading of the gel was determined by quantitation of protein as well as reprobing membranes for β-actin.
Total HDAC activity assay
Whole lung homogenates were incubated with assay buffer and Color de Lys substrate (Biomol) and read on a standard microplate reader at 405 nm. Total HDAC activity was expressed relative to standard curve generated from 0 – 500 μM Color de Lys deacetylated standard.
Immunohistochemical staining for HDAC1 and HDAC2
For localization and assessment of HDAC1 and HDAC2 abundance,tissues sections were incubated with anti-HDAC1 or anti-HDAC2 antibody overnight at 4°C at a titer of 1:100. Tissues were then washed and incubated with secondary antibody at a titer of 1:1000 for 30 min. 3,3'-diaminobenzidine (Vector laboratories, Burlingame, CA) was used as peroxidase substrate. Tissue slides were counterstained with H&E and analyzed under light microscope for image capture at x200 magnification using Advance SPOT software. Immunohistochemical staining for HDAC1 and HDAC2 was scored semi-quantitatively in a blinded manner.
Cytokine analysis
The level of pro-inflammatory cytokine monocyte chemotatic protein (MCP)-1 in lung homogenates was measured by enzyme-linked immunosorbent assay (ELISA) using duo-antibody kit (R&D Systems, Minneapolis, MN).
Proliferation/Apoptosis assay
Tissue sections were incubated overnight with rabbit anti-PCNA antibody at a titer of 1:100 before incubating with donkey anti-rabbit Cy3 at a titer of 1:50 (Millipore, Billerica, MA). Images were captured using a Photometrix digital camera mounted on a Nikon laser microscope with MetaMorph imaging software (MDS Analytical Technologies, Downingtown, PA). Apoptosis was assessed using a terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) detection kit (Roche Applied Science, Indianapolis, IN). Slides were analyzed by fluorescence microscopy and quantified by performing manual cell count of TUNEL-positive cells for each section.
Statistical analysis
Data are presented as mean ± SEM. Statistical significance was calculated using one-way Analysis of Variance (ANOVA) followed by Tukey’s post-hoc test for multigroup comparisons using StatView software. P < 0.05 was considered significant.
RESULTS
Hyperoxia-induced alveolar hypoplasia is not reversed by azithromycin
Animal models of alveolar hypoplasia resembling human bronchopulmonary dysplasia include exposure to hyperoxia during the critical period of alveolar development (postnatal days 5–21 in rodents) (8). Azithromycin (AZM) is a macrolide antibiotic with anti-inflammatory properties (15) that was shown previously to improve lung development in neonatal rats receiving >95% oxygen from postnatal days 4-14 (11). To determine whether AZM confers similar protection in mice, we exposed newborn mice treated with AZM (40 mg/kg i.p. daily, starting day 0) or saline to 80% oxygen for 14 d. Similarly-treated room-air littermate controls were used for comparisons between the 4 groups (Sal-RA, Sal-O2, AZM-RA, and AZM-O2). Results from H&E stained histology sections showed fewer alveolar airspaces in saline-treated animals exposed to hyperoxia compared to saline-treated room air controls (Figure 1A). AZM treatment showed no significant improvement in alveolar formation as quantified by radial alveolar counts (Figure 1A & B). Animals in all 4 groups maintained normal weight gain and lung/body weight ratios with no significant differences between hyperoxia or room-air groups (Figure 1C & D). Interestingly, AZM treatment alone resulted in slightly increased lung/body weight ratios as compared to saline-treated room air controls. These data demonstrate that hyperoxia-induced alveolar hypoplasia is not reversed by AZM treatment.
Figure 1.

Lung morphology and weights in neonatal mice exposed to hyperoxia. (A) H&E stained lungs from saline and AZM-treated groups exposed to room air (RA) or hyperoxia (O2) (10X magnification; Scale bar = 100 μm). Thin lines used to calculate radial alveolar counts shown in (B); data shown as mean±SEM (n=4 mice per group; *P<0.05 compared to respective saline or AZM group). (C) Daily body weights (triangle dotted line = AZM-O2; triangle solid line = AZM-RA; circle solid line = Sal-RA; circle dotted line = Sal-O2). (D) Lung/body weight ratios (n=5 mice per group; *P<0.05 compared to Sal-RA group; †P<0.05 compared to AZM-RA group).
Hyperoxia and lung inflammation
Since AZM did not reverse hyperoxia-induced alveolar hypoplasia, we investigated whether exposure to 80% oxygen elicited an inflammatory response and whether AZM had any anti-inflammatory effect. Results showed no significant change in total cells or inflammatory cell subsets (lymphocytes, macrophages, and neutrophils) detected in BAL fluid at 15 days between hyperoxia and room-air controls (Figure 2A & B). Consistent with the lack of inflammatory cells, there was no significant change in levels of chemokine MCP-1 (Monocyte chemotactic protein-1) in lung tissue at 3 days and 15 days (Figure 2C). Treatment with AZM thus had no detectable effect on BAL inflammatory cells or chemokine levels. These data demonstrate that 80% oxygen did not elicit a persistent inflammatory response and that AZM treatment had no significant impact on baseline inflammatory status.
Figure 2.

Lung inflammation in neonatal mice exposed to hyperoxia. (A) Total cell counts from bronchoalveolar lavage (BAL) in 15 days saline or AZM-treated neonatal mice exposed to hyperoxia. (B) Differential cell counts for lymphocytes (Lymph), macrophages (Mac), and neutrophils (Neut). (C) MCP-1 measured by ELISA. Data are shown as mean±SEM (n=4 mice per group; □ = 3 days; ■ = 15 days).
Hyperoxia decreases total HDAC activity
Decreased HDAC activity has been shown in conditions of uncontrolled inflammation such as asthma and chronic obstructive pulmonary disease (COPD) (16). To determine whether hyperoxia and/or AZM affect HDAC activity during alveolar development, we measured total HDAC activity at day 3 and day 15 in our hyperoxia model. Results showed that although no change in total HDAC activity was noted at day 3, hyperoxia significantly reduced total HDAC activity in saline and AZM-treated hyperoxia groups at day 15 (Figure 3A & B). These findings demonstrate that oxidative stress imposed by hyperoxia decreases HDAC activity and that AZM is unable to reverse this process.
Figure 3.
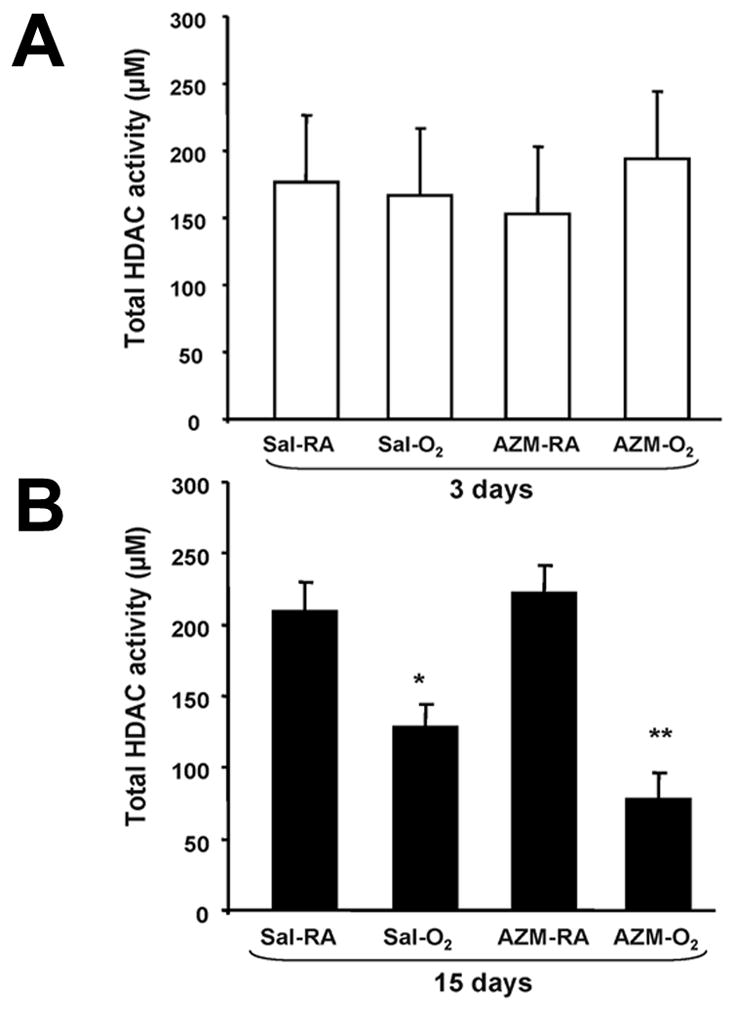
Total HDAC activity in neonatal mice exposed to hyperoxia. (A) Total HDAC activity from lung homogenates of neonatal mice exposed to room air or hyperoxia with saline or AZM treatment at 3 days. (B) Total HDAC activity at 15 days. Data are shown as mean±SEM (n = 4 mice per group; *P<0.01 compared with respective Sal-RA group; **P<0.001 compared with respective AZM-RA group).
Hyperoxia decreases HDAC1 and HDAC2 expression in alveolar and airway epithelium
Since total HDAC activity was decreased following hyperoxia, we investigated which HDAC proteins were altered in lung airway and alveolar compartments. HDAC1 and HDAC2 are isoforms of the Class I HDAC family that are ubiquitous nuclear proteins involved in transcriptional repression (17). HDAC1 is a critical repressor of genes involved in cell proliferation including p21 (18), and HDAC2 is a redox-sensitive protein prone to alteration by oxidants and/or free radicals (16). Results showed that HDAC1 and HDAC2 abundance was decreased at day 15 in both alveolar and airway epithelium of saline and AZM-treated mice exposed to hyperoxia compared to respective room air controls (Figure 4A-D). These results establish that reduction of total HDAC activity following hyperoxia is largely due to decreased HDAC1 and HDAC2 protein expression throughout the lung epithelium, and that AZM treatment does not inhibit this process. Levels of HDAC1 and HDAC2 proteins were slightly reduced in response to hyperoxia and AZM treatments in crude lung homogenates of heterogeneous mixture of lung cells (data not shown).
Figure 4.

HDAC1 and HDAC2 lung localization in neonatal mice exposed to hyperoxia. (A) HDAC1 and (C) HDAC2 abundance by immunohistochemistry in lung tissue of neonatal mice exposed to hyperoxia (15 days) compared to room air; Scale bar = 100 μm; 200X magnification. (B) Immunostaining scores for HDAC1 and (D) HDAC2 in alveolar and airway epithelium of neonatal mouse lung. Dark arrows indicate intense staining of HDAC1 or HDAC2. Black solid bars, intense staining; grey shaded bars, moderate/weak staining; open bars, no staining. (*P<0.05, **P<0.01, and ‡P<0.001 compared with Sal-RA group).
Hyperoxia and azithromycin induce p21 and p53 expression in lungs
p21 is a potent inhibitor of cyclin-dependent kinases (cdks) that arrests the cell cycle at the G1/S interface (8). p21 expression increases during hyperoxia (8, 19, 20) and has been shown to inhibit cell proliferation as a mechanism of inducing cell senescence to allow cells to repair damaged DNA (21, 22). Since reduction in HDAC activity is known to up-regulate transcription of p21 (22), we investigated whether decreased HDAC activity alters p21 expression following hyperoxia in our model. Results showed that hyperoxia significantly increased p21 expression at day 3 and day 15 following hyperoxia exposure (Figure 5A & B). Interestingly, AZM treatment did not decrease p21 levels during hyperoxia, and moreover, AZM alone induced p21 expression. These data confirm previous findings that p21 is induced by hyperoxia, but shows that it is also induced by AZM.
Figure 5.

p21 levels in neonatal mice exposed to hyperoxia. (A) p21 abundance assessed by immunoblotting using lung homogenates of neonatal mice exposed to room air or hyperoxia with saline or AZM treatment at 3 days and 15 days. β-actin was used as a loading control. After densitometric analysis, values were normalized against loading controls (B). Gel pictures are representative of at least three separate experiments. Open bars = 3 days; Solid bars = 15 days. Data are shown as mean±SEM (n=4 mice per group; **P<0.01, ‡P<0.001 compared with Sal-RA group).
p53 is a transcription factor that regulates the expression of several target genes related to cell cycle arrest, cell death, and DNA repair (23). Hyperoxia exposure caused a significant increase in the levels of p53 protein abundance after 3 days in Sal-O2 and AZM-O2 treatment groups (Figure 6A & B). Interestingly, hyperoxia exposure for 15 days showed a significant increase in Sal-O2 treatment group only, whereas AZM-O2 treatment group showed a significant reduction in p53 levels. These findings suggest that p21 at both early and late phase may be induced through a HDAC-independent mechanism, such as via p53, where cell cycle inhibition occurs in response to hyperoxia.
Figure 6.

p53 levels in neonatal mice exposed to hyperoxia. (A) p53 abundance assessed by immunoblotting using lung homogenates of neonatal mice exposed to room air or hyperoxia with saline or AZM treatment at 3 days and 15 days. β-actin was used as a loading control. After densitometric analysis, values were normalized against loading controls (B). Gel pictures are representative of at least three separate experiments. Open bars = 3 days; Solid bars = 15 days. Data are shown as mean±SEM (n=4 mice per group; *P < 0.05 compared with Sal-RA group; † P <0.05 compared with AZM-RA group).
Hyperoxia decreases alveolar proliferation
Given increased p21 expression and its implications on altering cell cycle activity, we studied changes in cell proliferation and/or apoptosis following hyperoxia exposure. Immunofluorescence to detect proliferating cell nuclear antigen (PCNA) revealed a significant decrease in proliferating cells in hyperoxia-treated lungs as compared to room air controls (Figure 7A & B). No significant difference in cell apoptosis was noted by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (data not shown). AZM treatment had no effect on proliferation or apoptosis in hyperoxia or control groups. These changes in cell proliferation imply that hyperoxia arrests cell cycle activity and renders the alveolar epithelium in a state of cell senescence and growth inhibition.
Figure 7.
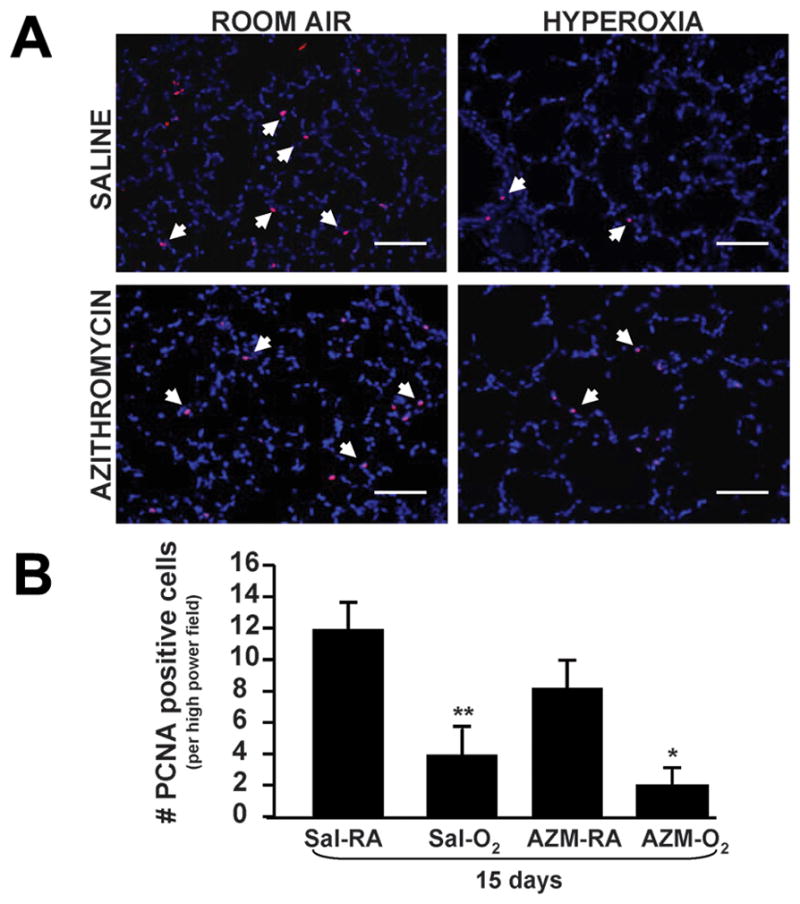
Proliferating cells in neonatal mice exposed to hyperoxia. (A) Immunofluorescence to detect proliferating cell nuclear antigen (PCNA) signal. Arrowheads indicate PCNA-positive cells (20X magnification; Scale bar = 100 μm). (B) Quantification of PCNA-positive cells. Data are shown as mean±SEM (n=4 measurements per group; *P<0.05 compared with AZM-RA group; **P<0.01 compared with Sal-RA group).
DISCUSSION
Alveolar hypoplasia is the phenotypic hallmark of bronchopulmonary dysplasia. Although several animal models recapitulate arrested alveolar development through hyperoxia exposure, the exact mechanisms driving this process remain poorly understood. Based on mounting evidence that environmental triggers exert changes in lung architecture through altered epigenetic mechanisms (9, 10), we hypothesized that hyperoxia exposure disrupts alveolar development through imbalance of histone deacetylases (HDACs) and their signaling mediators. Our results demonstrate that hyperoxia decreases expression of HDAC1 and HDAC2 and increases expression of the cyclin-dependent kinase p21, resulting in cell cycle arrest. To our knowledge, this is the first report detailing the effects of hyperoxia in arresting alveolar development through altered HDAC activity in neonatal mice.
To date, studies describing the effects of hyperoxia on alveolar formation have focused on inflammation and oxidative mechanisms that cause lung injury and disrupt alveolar architecture (5). Elevated oxygen levels induce superoxide radicals and pro-inflammatory cytokines responsible for neutrophil recruitment into the lung. Minimal data exist, however, on whether alveolar changes are a direct result of lung inflammation and injury or due to permanent disruption of normal developmental signaling pathways. We found no evidence of persistent inflammation or increased cytokine expression following 15 days of exposure to 80% oxygen consistent with previous reports (24). These observations raise the possibility that alveolar changes due to hyperoxia may be due to epigenetic regulation of developmental genes rather than ongoing inflammation or injury.
HDACs have the ability to deacetylate non-histone proteins such as nuclear factor κB (NF-κB) that are central to inflammatory signaling pathways. Decreased HDAC1 expression has been shown in bronchial biopsies of patients with asthma, thus favoring increased expression of inflammatory genes. Similarly, markedly decreased HDAC2 activity has been shown in lungs of patients with chronic obstructive pulmonary disease (COPD) and in a mouse model of emphysema due to oxidative and nitrative stress from exposure to cigarette smoke (16, 25). We determined the localization and abundance of HDAC1 and HDAC2 in lung sections exhibiting hypoplasia in response to hyperoxia. Our results showed that total HDAC activity and HDAC1 and HDAC2 abundance in alveolar and airway epithelium significantly decreased following hyperoxia. In addition to transcription factor repression, class I HDACs, and especially HDAC1, also repress genes involved in cell differentiation and proliferation including the cyclin-dependent kinase inhibitor p21WAF1/CIP1 (17, 26). Decreased HDAC activity with inhibition of cell proliferation may thus promote a state of cell cycle arrest and inhibit alveolar growth.
Our results demonstrated that hyperoxia in newborn pups increased p21 and p53 levels at 3 days and 15 days, consistent with data in adult mice showing increased p53 gene transcription, protein levels, and activity following hyperoxia (27, 28). Though recent reports indicate that the p53 pathway can elicit hyperoxia-induced senescence independent of p21 activation (29), the exact role of p53 in modulating cellular response to hyperoxia remains to be investigated. p21 may thus act as a molecular switch involved in triggering a ‘danger’ signal that attempts to protect the cell from further damage by inducing irreversible cell cycle growth arrest.
Azithromycin (AZM) is a macrolide antibiotic previously shown to suppress activation of NF-κB by tumor necrosis factor-alpha (TNF-α) in cultured tracheal aspirate cells from premature infants (30). Studies in neonatal rats showed that daily treatment with AZM during exposure to > 95% oxygen from day of life 4 to 14 improved survival and decreased lung damage and inflammation when compared to saline controls (11). Finally, a prospective double-blind, randomized, placebo-controlled pilot study in preterm humans investigated whether AZM treatment conferred protection from developing BPD. Although no difference was seen in incidence of BPD or mortality, a significant reduction in need for postnatal steroids and mechanical ventilation was noted in the treatment group (31). Results from our study showed no difference in hyperoxia-induced alveolar hypoplasia between animals treated with AZM versus saline controls. Moreover, treatment with AZM may confer a potential negative impact on lung growth homeostasis as uncovered by the finding that p21, and therefore cell senescence/mitosis, is induced by AZM (32).
Several possibilities may account for the difference between our study and the previous study in neonatal rats. First, it is possible that species-specific differences related to AZM efficacy may render mice less protected than rats. Second, the level of oxygen concentration differed between the two studies. We chose 80% oxygen based on minimal mortality for pups and lactating dams as compared to significant toxicity seen with 95% oxygen. Lower oxygen concentration is also associated with a lower degree of lung inflammation (24). Since immunomodulatory properties of AZM confer protection from inflammation, it is possible that any protective effect of AZM was less at 80% oxygen compared to 95% oxygen. A limitation of our study is that we have focused on a limited panel of molecules that participate in epigenetic regulation. Several additional classes of HDAC including sirtuins as well as HAT (histone acetyltransferase) family members and co-activators may also influence these developmental signaling pathways. Furthermore, our experimental animal design using hyperoxia serves only as a surrogate model for human BPD and represents one component of a complex and multifactorial pathologic process. In-depth studies on how varying oxygen levels and exposure times affect various signaling pathways may lead to additional interventions to prevent hyperoxic injury (33).
In summary, we have utilized an established newborn mouse model to provide novel evidence that the mechanism, in part, by which hyperoxia induces alveolar hypoplasia is through decreased HDACs and increased p21. Alveolar hypoplasia thus results from arrested lung growth and cell cycle arrest induced by hyperoxia. Further understanding of these epigenetic mechanisms in alveolar senescence may help to identify new molecular targets to ultimately prevent and treat human BPD. Future studies with these goals using HDAC inducers such as caffeine/theophylline (methylxanthine derivates) in protecting against hyperoxia-induced alveolar hypoplasia are currently underway.
Acknowledgments
We thank Dr. Bo-Chul Shin and the Devaskar Lab, Neonatal Research Center, UCLA, for use and assistance with the immunofluorescence microscope. We also thank Dr. John Belperio in the Division of Pulmonary and Critical Care at UCLA for use and assistance with the hyperoxia apparatus.
This study was funded by grants from NIH/NHLBI, no. KO8 HL076538, and UCLA Faculty Research Grant [to V.L.] and a grant from NIH/NHLBI, no R01 HL085613, [to I.R.]
ABBREVIATIONS
- AZM
azithromycin
- AZM
RA azithromycin-room air
- BAL
bronchoalveolar lavage
- BPD
bronchopulmonary dysplasia
- HDAC
histone deacetylase
- PCNA
proliferating cell nuclear antigen
- Sal-RA
saline-room air
Footnotes
Publisher's Disclaimer: Pediatric Research Articles Ahead of Print contains articles in unedited manuscript form that have been peer-reviewed and accepted for publication. As a service to our readers, we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting and review of the resulting proof before it is published in its final definitive form. Please note that during the production process errors may be discovered, which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bhandari A, Bhandari V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics. 2009;123:1562–1573. doi: 10.1542/peds.2008-1962. [DOI] [PubMed] [Google Scholar]
- 2.Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol. 2003;8:73–81. doi: 10.1016/s1084-2756(02)00193-8. [DOI] [PubMed] [Google Scholar]
- 3.Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol. 2005;67:623–661. doi: 10.1146/annurev.physiol.67.040403.102229. [DOI] [PubMed] [Google Scholar]
- 4.Aghai ZH, Saslow JG, Meniru C, Porter C, Eydelman R, Bhat V, Stahl G, Sannoh S, Pyon K, Hewitt C, Bhandari V. High-mobility group box-1 protein in tracheal aspirates from premature infants: relationship with bronchopulmonary dysplasia and steroid therapy. J Perinatol. 2010;30:610–615. doi: 10.1038/jp.2010.16. [DOI] [PubMed] [Google Scholar]
- 5.Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275:L110–L117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 6.Remesal A, Pedraz C, San Feliciano L, Ludena D. Pulmonary expression of vascular endothelial growth factor (VEGF) and alveolar septation in a newborn rat model exposed to acute hypoxia and recovered under conditions of air or hyperoxia. Histol Histopathol. 2009;24:325–330. doi: 10.14670/HH-24.325. [DOI] [PubMed] [Google Scholar]
- 7.McGrath-Morrow SA, Stahl J. Apoptosis in neonatal murine lung exposed to hyperoxia. Am J Respir Cell Mol Biol. 2001;25:150–155. doi: 10.1165/ajrcmb.25.2.4362. [DOI] [PubMed] [Google Scholar]
- 8.McGrath SA. Induction of p21WAF/CIP1 during hyperoxia. Am J Respir Cell Mol Biol. 1998;18:179–187. doi: 10.1165/ajrcmb.18.2.2964m. [DOI] [PubMed] [Google Scholar]
- 9.Ito K, Caramori G, Lim S, Oates T, Chung KF, Barnes PJ, Adcock IM. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med. 2002;166:392–396. doi: 10.1164/rccm.2110060. [DOI] [PubMed] [Google Scholar]
- 10.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 11.Ballard HO, Bernard P, Qualls J, Everson W, Shook LA. Azithromycin protects against hyperoxic lung injury in neonatal rats. J Investig Med. 2007;55:299–305. doi: 10.2310/6650.2007.00011. [DOI] [PubMed] [Google Scholar]
- 12.Londhe VA, Belperio JA, Keane MP, Burdick MD, Xue YY, Strieter RM. CXCR2/CXCR2 ligand biological axis impairs alveologenesis during dsRNA-induced lung inflammation in mice. Pediatr Res. 2005;58:919–926. doi: 10.1203/01.PDR.0000181377.78061.3E. [DOI] [PubMed] [Google Scholar]
- 13.Emery JL, Mithal A. The number of alveoli in the terminal respiratory unit of man during late intrauterine life and childhood. Arch Dis Child. 1960;35:544–547. doi: 10.1136/adc.35.184.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao H, Edirisinghe I, Rajendrasozhan S, Yang SR, Caito S, Adenuga D, Rahman I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1174–L1186. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]
- 15.Ivetic Tkalcevic V, Bosnjak B, Hrvacic B, Bosnar M, Marjanovic N, Ferencic Z, Situm K, Culic O, Parnham MJ, Erakovic V. Anti-inflammatory activity of azithromycin attenuates the effects of lipopolysaccharide administration in mice. Eur J Pharmacol. 2006;539:131–138. doi: 10.1016/j.ejphar.2006.03.074. [DOI] [PubMed] [Google Scholar]
- 16.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 17.Gopal YN, Van Dyke MW. Depletion of histone deacetylase protein: a common consequence of inflammatory cytokine signaling? Cell Cycle. 2006;5:2738–2743. doi: 10.4161/cc.5.23.3522. [DOI] [PubMed] [Google Scholar]
- 18.Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Reilly MA, Staversky RJ, Watkins RH, Maniscalco WM. Accumulation of p21(Cip1/WAF1) during hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 1998;19:777–785. doi: 10.1165/ajrcmb.19.5.3200. [DOI] [PubMed] [Google Scholar]
- 20.McGrath-Morrow SA, Cho C, Soutiere S, Mitzner W, Tuder R. The effect of neonatal hyperoxia on the lung of p21Waf1/Cip1/Sdi1-deficient mice. Am J Respir Cell Mol Biol. 2004;30:635–640. doi: 10.1165/rcmb.2003-0049OC. [DOI] [PubMed] [Google Scholar]
- 21.Yao H, Yang SR, Edirisinghe I, Rajendrasozhan S, Caito S, Adenuga D, O'Reilly MA, Rahman I. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol. 2008;39:7–18. doi: 10.1165/rcmb.2007-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajendrasozhan S, Yao H, Rahman I. Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD. 2009;6:291–297. doi: 10.1080/15412550903049132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 24.Rogers LK, Tipple TE, Nelin LD, Welty SE. Differential responses in the lungs of newborn mouse pups exposed to 85% or >95% oxygen. Pediatr Res. 2009;65:33–38. doi: 10.1203/PDR.0b013e31818a1d0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol. 2009;40:464–473. doi: 10.1165/rcmb.2008-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol. 2009;53:275–289. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- 27.O'Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol. 1998;18:43–50. doi: 10.1165/ajrcmb.18.1.2950m. [DOI] [PubMed] [Google Scholar]
- 28.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF. Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol. 1998;19:573–581. doi: 10.1165/ajrcmb.19.4.3173. [DOI] [PubMed] [Google Scholar]
- 29.Klimova TA, Bell EL, Shroff EH, Weinberg FD, Snyder CM, Dimri GP, Schumacker PT, Budinger GR, Chandel NS. Hyperoxia-induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. FASEB J. 2009;23:783–794. doi: 10.1096/fj.08-114256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aghai ZH, Kode A, Saslow JG, Nakhla T, Farhath S, Stahl GE, Eydelman R, Strande L, Leone P, Rahman I. Azithromycin suppresses activation of nuclear factor-kappa B and synthesis of pro-inflammatory cytokines in tracheal aspirate cells from premature infants. Pediatr Res. 2007;62:483–488. doi: 10.1203/PDR.0b013e318142582d. [DOI] [PubMed] [Google Scholar]
- 31.Ballard HO, Anstead MI, Shook LA. Azithromycin in the extremely low birth weight infant for the prevention of bronchopulmonary dysplasia: a pilot study. Respir Res. 2007;8:41. doi: 10.1186/1465-9921-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribeiro CM, Hurd H, Wu Y, Martino ME, Jones L, Brighton B, Boucher RC, O'Neal WK. Azithromycin treatment alters gene expression in inflammatory, lipid metabolism, and cell cycle pathways in well-differentiated human airway epithelia. PLoS ONE. 2009;4:e5806. doi: 10.1371/journal.pone.0005806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright CJ, Dennery PA. Manipulation of gene expression by oxygen: a primer from bedside to bench. Pediatr Res. 2009;66:3–10. doi: 10.1203/PDR.0b013e3181a2c184. [DOI] [PMC free article] [PubMed] [Google Scholar]