

Abstract
Impaired neurological development in premature infants frequently arises from periventricular white matter injury (PWMI), a condition associated with myelination abnormalities. Recently, exposure to hyperoxia was reported to disrupt myelin formation in neonatal rats. To identify the causes of hyperoxia-induced PWMI, we characterized cellular changes in the white matter (WM) using neonatal wild-type, 2–3-cyclic nucleotide 3-phosphodiesterase–enhanced green fluorescent protein (EGFP) and glial fibrillary acidic protein (GFAP)–EGFP transgenic mice exposed to 48 h of 80% oxygen from postnatal day 6 (P6) to P8. Myelin basic protein expression and CC1+ oligodendroglia decreased after hyperoxia at P8, but returned to control levels during recovery between P12 and P15. At P8, hyperoxia caused apoptosis of NG2+O4− progenitor cells and reduced NG2+ cell proliferation. This was followed by restoration of the NG2+ cell population and increased oligodendrogenesis in the WM after recovery. Despite apparent cellular recovery, diffusion tensor imaging revealed WM deficiencies at P30 and P60. Hyperoxia did not affect survival or proliferation of astrocytes in vivo, but modified GFAP and glutamate-aspartate transporter expression. The rate of [3H]-d-aspartic acid uptake in WM tissue was also decreased at P8 and P12. Furthermore, cultured astrocytes exposed to hyperoxia showed a reduced capacity to protect oligodendrocyte progenitor cells against the toxic effects of exogenous glutamate. This effect was prevented by 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide treatment. Our analysis reveals a role for altered glutamate homeostasis in hyperoxia-induced WM damage. Understanding the cellular dynamics and underlying mechanisms involved in hyperoxia-induced PWMI will allow for future targeted therapeutic intervention.
Introduction
The most common pathology observed in preterm infants is periventricular white matter injury (PWMI), a disorder that has been associated with myelination disturbances (Cheong et al., 2009; Tzarouchi et al., 2009), impaired neurological development (Back, 2006; Deng et al., 2008; Khwaja and Volpe, 2008), and neuronal/axonal disease (Pierson et al., 2007; Volpe, 2009). Therefore, children born preterm are more likely to develop future motor and cognitive deficits (Aylward, 2002; Bhutta et al., 2002; Wocadlo and Rieger, 2008).
Causes associated with PWMI include perinatal infection and hypoxia/ischemia (Volpe, 2008). Clinical evidence suggests that the use of high oxygen in preterm infants can also lead to poor neurological outcome (Collins et al., 2001; Deulofeut et al., 2007), however the role of oxygen in the etiology of PWMI has not been extensively characterized. In utero, paO2 is maintained at ∼24–28 mmHg. After birth, preterm infants experience a several-fold increase in arterial oxygen tension to 65–80 mmHg (Hoffmann, 2002), even without supplemental oxygen (Castillo et al., 2008).
In an experimental rat model, exposure to 80% oxygen at postnatal day 7 (P7) resulted in cell death in the gray matter and subcortical white matter (WM) that is not observed with exposure at later postnatal ages (Felderhoff-Mueser et al., 2004). Although cellular changes were not characterized in these studies, cell death in the brain was associated with increased intracranial expression of proinflammatory cytokines (Felderhoff-Mueser et al., 2005). Subsequent in vivo studies revealed that hyperoxia produced a maturation-dependent reduction in myelin basic protein (MBP) expression in neonatal rats (Gerstner et al., 2008). In vitro, 80% oxygen caused caspase-dependent cell death in cultured O4+O1− preoligodendrocytes (pre-OLs), but not in mature O4+O1+MBP+ oligodendrocytes (Gerstner et al., 2008). These findings indicate that high levels of oxygen cause oligodendroglial and WM damage; however, the overall effects of hyperoxia on oligodendroglia and astrocytes in the immature WM remain unclear.
Glutamate-mediated excitotoxicity has been shown to cause extensive damage to the developing brain in animal models of hypoxia/ischemia (Silverstein et al., 1986) and trauma (Bittigau et al., 1999). In addition, the overactivation of non-NMDA glutamate receptors (GluRs) leads to oligodendroglial cell death (Follett et al., 2000; Deng et al., 2004), decreased oligodendrocyte progenitor cell (OPC) proliferation, and attenuated oligodendroglia lineage progression (Gallo et al., 1996; Yuan et al., 1998). Vesicular release of glutamate within the developing WM occurs from both myelinated (Bezzi et al., 2001; Ziskin et al., 2007) and unmyelinated axons (Montana et al., 2004). Furthermore, the uptake of glutamate through membrane transporters is critical for its extracellular clearance (Bergles et al., 1999; Danbolt, 2001). Glial transporters, particularly in astrocytes, are believed to perform the majority of glutamate uptake in the brain (Schousboe and Waagepetersen, 2005), and altered transport has been demonstrated in experimental models of neural dysfunction (Zugno et al., 2007) and Down syndrome (Begni et al., 2003).
In this study, we sought to characterize hyperoxia-induced cellular changes within the developing WM and investigate the role of astrocytes and astrocyte-mediated glutamate uptake as potential contributors to oligodendroglial damage and altered WM development.
Materials and Methods
Transgenic animals.
Transgenic mice expressing enhanced green fluorescent protein (EGFP) under the control of the human glial fibrillary acidic protein (GFAP) promoter were generated on an FVB/N background (from Dr. Frank Kirchhoff, Max Planck Institute of Experimental Medicine, Göttingen, Germany) (Nolte et al., 2001). The FVB/NTgN (GFAP-EGFP) mice were previously characterized to express EGFP in 50 to 80% of GFAP-positive astrocytes, depending on age and brain region (Wehner et al., 2003). In our studies on external capsule (EC) and cingulum (CG), we found an overlap of glutamine synthetase (GS) immunostaining and EGFP expression in 50% of astrocytes at P8 and 80% at P12. Transgenic mice expressing EGFP under the control of the 2–3-cyclic nucleotide 3-phosphodiesterase (CNP) promoter have been described previously (Yuan et al., 2002). CNP-EGFP expression is detected in cells of the oligodendroglial lineage at early embryonic stages of development, and this expression is maintained throughout brain maturation. In accordance, we detected GFP+ oligodendroglia within the subcortical white matter and cortex of CNP-EGFP transgenic mice throughout development. No obvious differences were observed in brain or white matter size within either type of transgenic mouse. All procedures were performed according to the Institutional Animal Care and Use Committee, Children's National Medical Center, and National Institutes of Health guidelines.
Hyperoxia exposure, blood gas levels, and metabolic panel measurements.
Six-day-old (P6) C57BL/6 wild-type (WT) mice, GFAP-EGFP transgenic mice, and CNP-EGFP transgenic mice were subjected to hyperoxia and subsequently used for immunohistology, with only C57BL/6 wild-type mice being used for Western blot analysis. Litters composed of both male and female neonatal mice were divided into hyperoxia and control groups. Pups exposed to hyperoxia were placed along with their mothers in a chamber containing 80% O2 for 6 or 48 h. The control pups of each litter were kept in room air with a second lactating mother. The mothers of the two groups were replaced after 24 h to prevent oxygen-induced acute lung injury (Taglialatela et al., 1998). During recovery in room air, all pups exposed to hyperoxia were reunited with their biological mother until being killed. The pups appeared normal and did not suffer weight loss (control, P7, 4.14 ± 0.24 g; P8, 5.31 ± 0.26 g; P12, 6.83 ± 0.29 g; hyperoxia, P7, 4.26 ± 0.21 g; P8, 5.22 ± 0.11 g; P12, 6.76 ± 0.31 g) or changes in body temperature (control, P7, 30.65 ± 1.11°C; P8, 30.07 ± 0.77°C; P12, 31.37 ± 0.57°C; hyperoxia, P7, 30.75 ± 0.90°C; P8, 30.10 ± 0.37°C; P12, 31.10 ± 0.49°C; n = 6 for each time point) during or after the hyperoxia exposure. Body temperature was monitored by using a Micro Therma 2T device with a neonatal mouse (RET-4) rectal probe (Braintree Scientific).
To analyze oxygenation and metabolic status, blood gas levels and a metabolic panel were obtained in both experimental groups at P8. Animals exposed to hyperoxia were decapitated within the oxygen chamber. The blood sample was taken immediately with a capillary tube from the upper cervical region, and caution was taken to prevent the formation of air bubbles in the sample. pH, pO2, pCO2, Na+, K+, Ca2+, Cl−, glucose, and lactate were measured with a blood analyzer (Rapidlab 1200; Siemens).
Bromodeoxyuridine injections.
Acute bromodeoxyuridine (BrdU) incorporation within cells of the WM was determined by administering an intraperitoneal injection of 10 mg/kg BrdU in both hyperoxia-exposed and litter-matched control mice 2 h before they were killed. For BrdU analysis at P8, pups undergoing hyperoxia exposure where administered the BrdU injection within the oxygen chamber to prevent room-air exposure. During further analysis of BrdU incorporation after recovery in room air at P10 and P12, pups again received an intraperitoneal injection of BrdU 2 h before they were anaesthetized and subjected to transcardial perfusion of fixative. To evaluate cumulative BrdU incorporation during recovery in room air from P8 to P12, both experimental groups were subjected to a cumulative (pulse-chase) BrdU protocol where the animals were administered repeat intraperitoneal injections of 10 mg/kg BrdU every 24 h, starting immediately after hyperoxia at P8 and continuing for 4 d until P12. At P12, all mice were killed and transcardially perfused, and tissue was then fixed and analyzed using immunohistological techniques.
Immunofluorescence.
Mice at P8, P10, P12, P15, and P30 were anesthetized following National Institutes of Health guidelines and transcardially perfused with PBS and then 4% paraformaldehyde (PFA). Brains were dissected out and postfixed with 4% PFA overnight at 4°C. Fixed brains were preserved in 10% glycerol in PBS. For tissue sections, brains were rinsed in 1× PBS and then frozen in tissue freezing medium (Triangle Biomedical Sciences) and mounted on a Microm HM400 microtome (Microm International). Sections were cut (30–40 μm) and stored in a 1× PBS, 0.05% sodium azide solution. For immunohistochemistry, sections were blocked at room temperature for at least 1 h in blocking solution [1% bovine serum albumin (BSA), 0.3% Triton X-100, and 20% normal goat serum (NGS) in 1× PBS]. Primary antibodies were diluted using carrier solution (1% BSA, 0.3% Triton X-100, and 1% NGS in 1× PBS). Polyclonal rabbit antibody to NG2 chondroitin sulfate proteoglycan (Millipore Bioscience Research Reagents) was diluted 1:400. Monoclonal mouse antibody to MBP (Covance) was diluted 1:500. Polyclonal mouse antibody to GFAP (Sigma-Aldrich) and polyclonal chicken antibody to GFAP (Abcam) were diluted 1:500. The monoclonal rabbit Ki67 antibody (Vector Laboratories) was diluted 1:250. Monoclonal mouse BrdU antibody was diluted 1:500 (Axyll). Tissue analyzed for BrdU incorporation was pretreated with 2N HCl for 30 min followed by 0.1 m boric acid for 15 min at room temperature. Monoclonal rabbit Olig2 antibody and the monoclonal mouse CC1 antibody were diluted 1:500 (Abcam; Calbiochem). Polyclonal guinea pig antibodies for glutamate-aspartate transporter (GLAST)/excitatory amino acid transporter 1 (EAAT1) (Millipore Life Science) and glutamate transporter-1 (GLT-1)/EAAT2 (Millipore Life Science) were diluted to 1:500. Polyclonal rabbit antibody for GS (Abcam) was diluted to 1:500. Brain sections were incubated in primary antibodies at 4°C overnight. Rinses were performed in carrier solution at room temperature, with three changes of solution every 10 min. All secondary antibodies used were from Jackson Immunoresearch Laboratories in carrier solution: FITC-conjugated goat anti-mouse IgG (1:200), FITC-conjugated goat anti-rabbit IgG (1:200), FITC-conjugated donkey anti-chicken IgG (1:500); CY5-conjugated goat anti-mouse IgG (1:500), CY5-conjugated goat anti-rabbit IgG (1:500), CY5-conjugated goat anti-rat (1:500), and CY5-conjugated goat anti-guinea pig; CY3/Rhodamine-conjugated goat anti-mouse IgG (1:200), CY3/Rhodamine-conjugated goat anti-rabbit IgG (1:200), and CY3/Rhodamine-conjugated goat anti-guinea pig (1:200). Incubation was performed at room temperature for 1 h followed by three washes as described above. Sections were then stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min and, after three washings with PBS, mounted with Mowiol.
Cell cultures.
Primary mixed glial cultures were prepared from embryonic day 19 pregnant Sprague Dawley rats by mechanical dissociation according to the method of McCarthy and de Vellis (1980) as described previously (Gallo and Armstrong, 1995; Gallo et al., 1996). Mixed cultures (7–10 d old) were shaken overnight to detach OPCs from the astrocyte monolayer. To minimize contamination by microglial cells, the detached cell suspension was incubated in succession for 45 min each in 60 mm dishes. OPCs enriched by this method contained >95% GD31 cells labeled by the LB1 monoclonal antibody (Levi et al., 1986; Curtis et al., 1988), with <0.05% GFAP+ astrocytes and <0.05% Ox42+ microglia. Attached astrocytes were passaged after overnight shaking and transferred into T75 culture flasks (BD Falcon) at a density of 2 × 106 cells per flask with 10 ml of DMEM (Invitrogen) containing 10% FCS, which was changed after overnight incubation to remove nonattached OPCs. Once cells became confluent, flasks were shaken overnight and media containing nonattached OPCs and microglia was removed. For immunocytochemistry, astrocyte layers were trypsinized and plated on poly-lysine-coated (0.1 mg/ml) 25 mm coverslips in 35 mm dishes using 2 × 105 cells in 2 ml of DMEM with 10% FCS until confluent after several days. For Western blot protein analysis, astrocytes, after trypsinization, were plated on six-well plates in a density of 2 × 105 cells per well in 2 ml of DMEM with 10% FCS until confluent. For subconfluent astrocyte cultures, 1 × 105 cells in 2 ml of DMEM containing 10% FCS were transferred to six-well plates and cultured for 48 h, and media were changed the day after trypsinization. The six-well plates were then used for experiments at either 80 or 21% O2.
Immunocytochemistry.
Live staining for cell surface antigens with A2B5, O4, and O1 antibodies (Bansal et al., 1989) was performed as described previously (Yuan et al., 1998). Briefly, live cells were incubated at room temperature for 1 h with primary antibodies diluted 1:10 in DMEM followed by fluorescein-conjugated goat anti-mouse IgM for 45 min. After three washes in PBS, cells were fixed in 4% paraformaldehyde (pH 7.3 in PBS) for 10 min at room temperature and washed in PBS. Coverslips were then mounted in DAPI-containing Vectashield. For double staining with GFAP, cells, after live staining, fixation, and washing, were blocked again in 10% NGS in DMEM containing 0.1% Triton X-100 for permeabilization for 20 min at room temperature. Incubation with GFAP mouse antibody (1:500; Sigma-Aldrich) followed for 1 h at room temperature. After washing, the cells were incubated with Rhodamine anti-mouse IgG antibodies (1:200; Jackson Immunoresearch Laboratories). The cells were then washed and mounted in Vectashield with DAPI.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assays.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays were performed according to the manufacturer's directions (In Situ Cell Death Detection Kit, Rhodamine; Roche Applied Science). TUNEL immunostaining was performed on tissue obtained after hyperoxia or from animals kept in room air (control). Tissue was then processed for NG2 immunolabeling as described above (Immunofluorescence). Sections were then permeabilized using 0.2% Triton X-100 in 1× PBS for 1 h, rinsed with 1× PBS, and incubated with TUNEL solution for 1 h at 37°C. After three washes in 1× PBS, tissue was mounted on slides, allowed to dry, and placed in Vectashield with DAPI. Cells grown on 25 mm coverslips were fixed in paraformaldehyde for 10 min at room temperature and then rinsed in PBS. Cells were then permeabilized for 2 min on ice before labeling with 50 μl of the TUNEL reaction mixture and incubation at 37°C for 1 h in a humidified chamber under Parafilm on coverslips. After washing with PBS, paraformaldehyde fixation was followed by treatment of cells with 0.07 NaOH in PBS for 10 min at room temperature. After washing, cells were fixed again for 10 min and permeabilized in 0.1% Triton X-100 in PBS for 1 min. After washing, cells were incubated with 10% goat serum for 15 min, followed by monoclonal anti-GFAP antibodies (1:500; Sigma-Aldrich) for 1 h at room temperature, followed by fluorescein-conjugated goat anti-mouse IgG (1:200; Jackson Immunoresearch Laboratories) for for 30 min at room temperature. Coverslips were then mounted in DAPI-containing Vectashield.
3-(4,5-Dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay.
After exposure of astrocytes in 12-well plates to hyperoxia or normoxia in DMEM containing 10% FCS, measurements of reduction of 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were performed according to the manufacturer's directions (TACS MTT Assays; R & D Systems). MTT reagent (100 μl) was added to each well of a 12-well plate containing 1 ml of growth media during the final 4 h of incubation. Detergent reagent (300 μl) was then added to each well to solubilize the dark blue crystals overnight. Supernatants (250 μl) were finally transferred to 96-well plates and read on a Molecular Devices ThermoMax 96-well plate reader, using a test wavelength of 570 nm.
Western blotting.
Protein analysis was only conducted in WT mice. Microdissected tissue of the WM, including corpus callosum (CC), cingulum, and external capsule, was homogenized in 4°C radioimmunoprecipitation assay (RIPA) buffer solution for protein extraction. For analysis of cultured astrocytes, cells were harvested and lysed in 4°C RIPA buffer for 30 min on ice. After cell lysis, lysates were centrifuged at 4°C for 30 min. Aliquots were then assayed for protein concentration using the Pierce BCA kit with a 30 min incubation at 60°C before spectrophotometry at 590 nm. Total proteins were equally loaded (10–40 μg per lane) on 4–20% mini precast Tris-glycine gels. The gels were transferred onto polyvinylidene fluoride membranes at 4°C overnight and blocked in 4% nonfat dry milk in Tris-buffered saline–Tween 20 (TBST). Primary antibodies were diluted 1:500 to 1:2000 in 4% milk. Horseradish-peroxidase-conjugated secondary antibodies (anti-rabbit and anti-mouse, BD Biosciences PharMingen; anti-goat, Santa Cruz Biotechnology; anti-guinea pig, Millipore Bioscience Research Reagents) were diluted 1:2000 in 4% milk in TBST. Chemiluminescent detection was performed using ECL Plus (GE Healthcare) or Supersignal West Pico (Pierce) kits according to manufacturers' directions. The antibodies used were as follows: monoclonal mouse MBP, 1:1000 (Covance); polyclonal mouse GFAP, 1:1000 (Sigma-Aldrich); polyclonal rabbit GLAST/EAAT1, 1:250 (Abcam); polyclonal guinea pig GLT-1/EAAT2, 1:250 (Millipore Bioscience Research Reagents); monoclonal mouse β-actin, 1:1250 (Millipore Bioscience Research Reagents).
Microscopy and cell density measurements.
A Zeiss LSM 510 confocal laser scanning microscopic system was used for the analysis of fluorescence after immunohistochemical staining in wild-type, CNP-EGFP, and GFAP-EGFP mice. Optical sections were acquired with field depth of 5–10 μm using a 20×, 40×, or 60× objective and the LSM 510 software. Four different laser lines were used to image localization of FITC (488 nm excitation; 522/35 emission filter), CY3 (560 nm excitation; 605/32 emission filter), Cy5 (647 nm excitation; 680/32 emission filter), and DAPI (400 nm excitation). Data acquisition and processing were controlled by LSM software. Analysis of immunofluoresence was performed on confocal z-stacks. Cells were counted in 225 × 225 × 10 μm (X, Y, Z planes) images for cells/volume quantifications. An average of three images were taken from the CG and EC regions depicted in supplemental Figure 1 (available at www.jneurosci.org as supplemental material) within two to three sections for each animal analyzed. Confocal Assistant 4.02 and Image J (NIH) software were used to merge images for analysis. Merged images were processed in Photoshop 7.0 with minimal manipulation of contrast. Cells were counted in a blinded fashion and double or triple labeled by analyzing the merged image for each confocal z-stack and identifying positive immunofluorescence for each individual channel.
Exposure of primary astrocytes to hyperoxia.
At the beginning of the experiment, cultures were placed in six-well plates using 2 ml of DMEM growth media per well as mentioned and transferred to a humidified oxygen chamber (Billups-Rothenberg). The chamber was filled with 80% O2 using a gas tank containing 80% O2, 5% CO2, and 15% N2 (Roberts Oxygen) with a flow regulator (Biospherix) and maintaining a flow of 20 L/min for 10 min, in accordance with the manufacturer's instructions. Exposures were performed for 24 and 48 h, respectively. Normoxia plates for control were kept under 21% O2, 5% CO2, and 15% N2 at 37°C.
Astrocyte–OPC cocultures.
Monolayers of confluent astrocytes were obtained on coverslips, as described previously. Media were then renewed with DMEM without FCS containing 100 μm dibutyryl (db)-cAMP (Sigma-Aldrich) and exposed to a mixture of 80% O2, 5% CO2, and 15% N2 for 48 h. The dishes were removed from the oxygen chamber briefly before the OPC suspensions were prepared for plating. For control, dishes containing astrocytes on coverslips were kept at 21% O2, 5% CO2, and 75% N2 in an incubator. To reduce the number of microglia in the mixed glia cell cultures, media were changed after shaking for 2 h. After an overnight shake for 12–16 h, OPCs were harvested and transferred to 15 ml tubes for centrifugation at 1500 rpm for 5 min. Resuspended cells were transferred to a 60 mm culture dish and incubated for 45 min at 37°C for attachment of microglia. OPCs were removed by gentle flushing of the media. The OPC suspension was collected into a 15 ml tube and centrifuged for 5 min at 1500 rpm.
To label OPCs Qtracker-Orange CMRA (Invitrogen) was used according to manufacturer's instructions in a final concentration of 10 μm in 10 ml of DMEM containing the OPCs to be plated. After incubation for 45 min at 37°C, the tube was centrifuged for 5 min at 1500 rpm and media was aspirated. Pellets were resuspended in 2 ml of DMEM containing 10% FCS per 300,000 OPCs. Two milliliters of this OPC suspension were placed on top of the astrocytes exposed to either 80% or 21% O2. After 1 h incubation to allow OPC attachment, media was carefully aspirated and replaced by 2 ml of DMEM without FCS for each dish/coverslip. For glutamate challenge, 40 μl of glutamate 50 mm was added to the dish to obtain a final glutamate concentration of 1 mm/2 ml culture media. For protection experiments, the non-NMDA-receptor antagonist 2,3-dioxo-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX; 100 μm; Sigma-Aldrich) was added to the culture media 1 h before glutamate challenge. The cocultures were maintained for 24 h for analysis of A2B5+/Dye+ cells, and for 48 h for O4+/Dye+ cells.
Measurement of [3H]-d-aspartic acid uptake by cultured astrocytes.
Primary astrocytes cultures were grown on 24-well plates until confluent. Media was changed to DMEM without FCS containing db-cAMP, and hyperoxia experiments were performed as described previously. Plates, after 48 h exposure to either 80 or 21% O2, were placed at the surface of a 37°C water bath, and rinsed twice with 1 ml of preheated Krebs' buffer [containing (in mm) 25 HEPES, pH 7.4, 4.8 KCl, 1.2 KH2PO4, 1.3 CaCl2, 1.2 MgSO4, 6 glucose, and 140 NaCl]. Instead of l-glutamate, we used its transportable analog d-aspartic acid, which is not metabolized and does not interact with any form of glutamate receptors. [3H]-d-aspartic acid (50 nm; PerkinElmer) was diluted with unlabeled d-aspartic acid to achieve the various final concentrations of 1, 5, 10, 100, and 500 μm. The uptake was stopped after 10 min by three rinses with ice-cold sodium-free Krebs' buffer in which NaCl was replaced by chloride of the same osmolarity (120 mm). The cells were then lysed with 500 μl of 1N NaOH, and the radioactivity of 200 μl of the lysate was determined by liquid scintillation counting. A fraction of the lysate was also used for protein determination. To correct for nonspecific [3H]-d-aspartic acid uptake, we subtracted data obtained under conditions using Na-free Krebs' buffer. All data are expressed as the rate of Na-dependent uptake per milligram of protein per minute. For conversion of scintillation counts into femtomoles of aspartate, we used online software, GraphPad Radioactivity Calculator.
[3H]-d-aspartic acid uptake assay with WM tissue.
[3H]-d-aspartic acid uptake assays at all time points (P8, P12, P15, and P30) were performed using WM tissue isolated by microdissection and a modified assay protocol (Weller et al., 2008). Brains were removed and placed in D1 solution (1× HBSS, containing 6% glucose, 15% sucrose, and 1% PenStrep) on ice until being sliced into coronal sections. The WM was then carefully removed and placed in 1 ml of homogenizing buffer (50 mm Tris, 0.3 m sucrose, pH 7.4) on ice. After homogenization, suspension was centrifuged at 13,500 rpm for 10 min at 4°C. Pellet was then resuspended in KRH (Krebs' Ringer's HEPES solution). Duplicate 200 μl assays were performed for each experimental condition. Assays containing 100 μm dihydrokainic acid (DHK; Sigma-Aldrich) and 75 μm dl-threo-b-benzyloxyaspartic acid (TBOA); Tocris Bioscience) were incubated at 37°C for 10 min. After each respective agent was added to the sample, 50 nm [3H]-d-aspartic acid (PerkinElmer) was placed in each sample and allowed to incubate for 5 min at 37°C. Preparations were then centrifuged at 13,500 rpm for 5 min. Samples were then washed by resuspending the pellet in sodium-free KRH minus Na (sodium-free Krebs' solution containing the same molar concentration of “normal” Krebs' solution but with choline chloride in place of sodium chloride) twice to stop additional sodium-dependent [3H]-d-aspartic acid uptake. Cells in each sample were then lysed with 1N NaOH, and 150 μl of the lysate was added to Fischer Scientific ScintiVerse scintillation fluid and allowed to extract overnight. Protein concentration of each sample was measured by Bradford assay using the remaining 50 μl of lysate. [3H]-d-aspartic acid uptake was determined using a Beckman LS 6500 scintillation counter and calculated as picomoles per milligram of protein per minute. Values obtained from samples containing TBOA were subtracted from the total and non-GLT values to show sodium-dependent [3H]-d-aspartic acid uptake for each respective sample in all experiments.
Magnetic resonance imaging and diffusion tensor imaging.
Magnetic resonance imaging (MRI) was performed using a 7 tesla rodent scanner (Pharmascan 70/16; Bruker BioSpin) with a 16 cm horizontal bore magnet and a 9 cm (inner diameter) shielded gradient with an H-resonance frequency of 300 MHz and a maximum gradient strength of 300 mT/m. For imaging, a 1H-phased-array surface coil for mouse head and a 1H-radio frequency-volume resonator (72 mm) for transmission were used. Data acquisition and image processing were performed with the Bruker software Paravision 4.0. During the examinations, mice were anesthetized with 2.0–1.0% isoflurane (Forene; Abbot) delivered in a O2/N2O mixture under constant ventilation monitoring (Small Animal Monitoring and Gating System; SA Instruments). To ensure physiological body temperature, animals were placed on a heated circulating water blanket. To localize the CC, we first used a T2-weighted two-dimensional turbo spin-echo sequence [repetition time (TR), 4200 ms; echo time (TE), 36 ms; rare factor, 8; four averages] with a field of view (FOV) of 2.56 × 2.56 cm and a matrix size of 256 × 256 with 20 slices at 0.5 mm to cover the whole brain. For subsequent diffusion tensor imaging (DTI), a 1 mm slice with an FOV of 2.60 × 2.60 cm and a matrix size of 128 × 128 was placed over the CC. Imaging parameters are as follows: DTI echo-planar imaging with spin echo preparation and 16 diffusion directions; b = 1000 s/mm2; diffusion duration, δ = 4 ms; diffusion separation, Δ = 20 ms; TR, 3000 ms, TE, 34 ms; one average. If there were movement artifacts, the whole data set of the animal was excluded from analysis. On a pixel-by-pixel basis, fractional anisotropy (FA) as well as radial and axial apparent diffusion coefficients (ADCs; determined perpendicularly and parallel to the length of CC fibers respectively) were calculated for the CC using Bruker software Paravision 4.0. One value per animal was obtained by averaging the measurements of nine separate pixels along the length of CC fibers. In addition, directionally encoded color (DEC) maps were used to represent anisotropy in three directional manners in coronal images: red for lateral–medial, blue for anterior–posterior, green for in–out (Pajevic and Pierpaoli, 1999). Given the axonal directory in the CC, the dominant direction for fractional anisotropy in this region is medial–lateral (red). Mean, SD, and statistical significance (Mann–Whitney rank test) of FA and ADC values for control versus hyperoxia were calculated using GraphPad 5.0 software.
Results
Hyperoxia delays white matter maturation in newborn mice
To induce hyperoxia in the neonatal mouse, WT mice were exposed to 80% oxygen for 48 h from P6 to P8. Analysis of blood gas levels was performed immediately after the full exposure at P8. As shown in Table 1, animals exposed to hyperoxia exhibited a 2.32-fold increase (n = 5; p < 0.001) in average oxygen tension (oxygen partial pressure) compared to control animals kept in room air. No change in any metabolic panel value was observed in the hyperoxia group when compared to control (Table 1).
Table 1.
Blood gas levels and metabolic pane
| pH | pO2 (mmHg) | pCO2 (mmHg) | Na+ (mmol/L) | K+ (mmol/L) | Ca2+ (mmol/L) | Cl− (mmol/L) | Glucose (mg/dl) | Lactate (mmol/L) | |
|---|---|---|---|---|---|---|---|---|---|
| Control (n = 5–8) | 7.40 ± 0.06 | 61.82 ± 9.17 | 22.24 ± 5.84 | 134.10 ± 6.17 | 6.82 ± 1.46 | 1.27 ± 0.15 | 107.50 ± 7.90 | 36.33 ± 11.93 | 0.76 ± 0.21 |
| Hyperoxia (n = 5–8) | 7.38 ± 0.33 | 141.48 ± 9.23* | 18.96 ± 6.29 | 132.82 ± 8.59 | 8.10 ± 1.28 | 1.28 ± 0.34 | 103.60 ± 13.05 | 40.00 ± 12.69 | 0.85 ± 0.17 |
pO2 levels in mice exposed to hyperoxia (48 h, from P6 to P8) were significantly higher than those of mice kept in room air over the same time period. No change was observed for any metabolic panel parameter between the hyperoxia and control groups. Data are shown as mean ± SD, using an unpaired t test comparing control versus hyperoxia, where *p < 0.001.
MBP is part of the major family of myelin proteins (Baumann and Pham-Dinh, 2001; Back, 2006), and MBP expression marks the onset of developmental myelination. We therefore sought to investigate the effect of hyperoxia on MBP expression through immunofluorescence and Western blot analyses in the developing WM. When compared to controls, animals exposed to hyperoxia showed a marked decrease in MBP immunofluorescence at P8, P10, and P12 (Fig. 1A). A reduction in MBP protein expression immediately after exposure at P8 (56.4% decrease; n = 4; p < 0.025), after 2 d of recovery at P10 (95.3% decrease; n = 3; p < 0.005), and after 4 d of recovery at P12 (66.0% decrease; n = 4; p < 0.025) confirmed these observations (Fig. 1B,C). To determine whether hyperoxia caused a chronic disturbance in MBP expression, levels were measured after more prolonged periods of recovery. Both immunostaining and Western blots revealed a compensation in MBP expression to control levels at P15 and P30 (Fig. 1A–C). These results indicate two distinct phases of myelin regulation after hyperoxia exposure: an acute phase from P8 to P12, where MBP is reduced, followed by a secondary phase during continued recovery that involves compensatory MBP formation and the return of MBP to control levels at P15 and P30.
Figure 1.
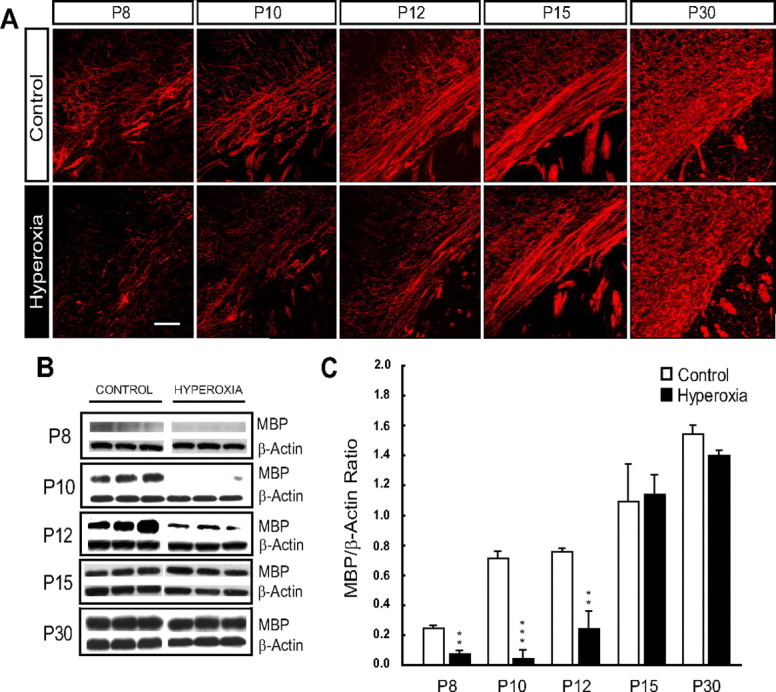
Hyperoxia delays MBP expression in the developing white matter. Six-day-old (P6) mice were exposed to normoxia (21% oxygen, room air) or hyperoxia (80% oxygen) for 48 h. Tissue was microdissected from the white matter at each of the time points indicated. A, Time course of white matter MBP immunohistochemical staining from P8 (immediately after hyperoxia) to P30 (after 3 weeks of recovery) in control versus hyperoxia animals. All images are of the EC. Scale bar, 50 μm. B, Western blots for MBP and β-actin at all represented time points (P8 to P30). C, Densitometric analysis of MBP Western blots showing decreased MBP expression after hyperoxia at P8, P10 and P12. MBP expression returned to control levels at P15 and P30. Values represent standardized mean ratios of MBP and β-actin. For each group and time point, n = 3–6 brains. An unpaired t test comparing control versus hyperoxia, where **p < 0.025, and ***p < 0.005, was used for statistical analysis.
Hyperoxia alters oligodendroglia number in the immature white matter
Cells of the oligodendroglial lineage progress through a series of maturational steps to become myelinating oligodendrocytes: (1) OPCs, (2) pre-OLs, (3) immature/premyelinating oligodendrocytes, and (4) mature oligodendrocytes (Baumann and Pham-Dinh, 2001; Back, 2006).
We characterized the oligodendroglia population at P4, P6, and P8 in neonatal mice kept in room air (control) to determine the cellular distribution of the various stages of oligodendroglial development at these ages. We found that the number of total oligodendroglia (Olig2+ cells) and mature oligodendrocytes (CC1+ cells) increased throughout early postnatal development in the WM (Table 2). The OPC (NG2+O4− cells) population appeared to be the predominant cell type within the OL lineage in the first postnatal week of WM development. However, as the WM matures, the relative contribution of NG2+O4− cells (percentage of Olig2+ cells) to the overall oligodendroglia population decreases over time. From P4 to P8, the percentage of Olig2+ cells displaying a pre-OL phenotype (NG2+O4+ and O4+NG2− cells) remained consistent throughout this time period, even though the actual number of pre-OLs increased throughout WM development (Table 2). Last, in agreement with our observation that the developing WM contains a low percentage of CC1+ cells between P4 and P8, MBP expression was almost undetectable in the WM at P4 and P6 (data not shown), with levels increasing slightly at P8 (Fig. 1). Similar to the findings of Gerstner et al. (2008), we observed changes in WM MBP expression after hyperoxia exposure during the first postnatal week of development. Therefore, during this critical developmental time window, the WM of the rodent is of an immature state and an insult during this period is more likely to result in injury.
Table 2.
Oligodendroglia lineage analysis during early WM postnatal development
| P4 |
P6 |
P8 |
||||
|---|---|---|---|---|---|---|
| Cells/106 μm3 | % Olig2 + cells/106μm3 | Cells/106μm3 | % Olig2 + cells/106μm3 | Cells/106μm3 | % Olig2 + cells/106μm3 | |
| Olig2+ | 157.66 ± 8.10 | 100.00 ± 5.16 | 215.48 ± 14.45 | 100.00 ± 6.71 | 303.91 ± 13.37 | 100.00 ± 4.39 |
| NG2 + O4− | 84.23 ± 13.22 | 53.65 ± 8.42 | 91.32 ± 12.12 | 42.47 ± 5.64 | 118.07 ± 18.63 | 38.85 ± 6.13 |
| NG2 + O4+ | 19.90 ± 0.62 | 12.8 ± 0.40 | 20.84 ± 2.34 | 9.69 ± 1.09 | 32.17 ± 1.33 | 10.58 ± 0.89 |
| O4 + NG2− | 43.19 ± 4.41 | 27.51 ± 2.81 | 63.99 ± 3.89 | 29.76 ± 1.81 | 102.38 ± 7.69 | 33.69 ± 2.53 |
| CC1+ | 8.50 ± 0.52 | 5.55 ± 0.63 | 18.55 ± 1.73 | 8.63 ± 0.98 | 48.84 ± 1.97 | 12.78 ± 2.02 |
Numbers of cells at various stages of oligodendroglia development in the postnatal WM. The Olig2+ (total oligodendroglia) population expands throughout early postnatal development. NG2+O4− cells (OPCs) appear to be the predominant phenotype during this time period, with the percentages of pre-OLs, NG2+O4+, and O4+NG2− cells remaining relatively constant over this time period. The CC1+ (mature oligodendrocytes) cell population increased over time to a represent larger percentage of the overall oligodendroglia population. Note that NG2+O4+ cells were counted as a separate group and were not included in either the NG2+O4− or the O4+NG2− groups. Data are shown as the mean ± SD for the number of cells in 106 μm3, where n = 3–6.
Our next goal was to determine the cellular changes underlying the observed hyperoxia-induced delay in MBP expression in the developing WM. Consistent with the changes in MBP, newborn mice exposed to 48 h of hyperoxia displayed a reduction in the number of Olig2+ cells (total oligodendroglia) and CC1+ cells (mature oligodendrocytes) in both the EC and CG regions of the WM at P8 (Fig. 2A–E). At P10, the hyperoxia group only showed a reduction in the number of CC1+ cells, and no difference in the Olig2+ cell population was observed between the two groups in either WM region of interest (Fig. 2F). Interestingly, the number of Olig2+ cells in the CG was elevated by 27% (n = 6; p < 0.050) in the hyperoxia-exposed animals, when compared to controls after 4 d of recovery in room air (P12) (Fig. 2G,H–K). In addition, despite the decrease in MBP expression in pups exposed to hyperoxia, the number of CC1+ oligodendroglia returned to control levels in both WM regions at P12 (Fig. 2I–K). This analysis points to a hyperoxia-induced reduction in the oligodendroglial population within the developing WM, followed by compensation in both total oligodendroglia (Olig2+) and CC1+ oligodendrocytes after recovery in room air. Changes in the CC1+ cell population are likely responsible for the observed biphasic response in MBP expression after hyperoxia.
Figure 2.
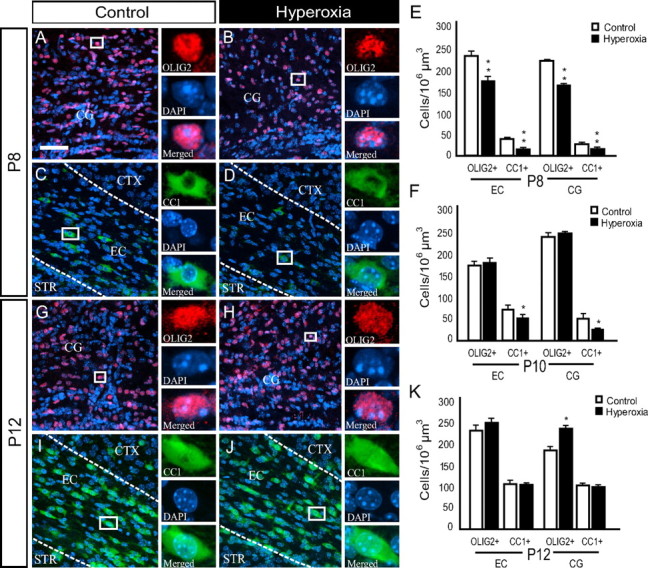
Changes in the Olig2+ and CC1+ oligodendroglia population of the immature white matter are associated with hyperoxia exposure. P6 mice underwent exposure to normoxia (21% oxygen, room air) or hyperoxia (80% oxygen) for 48 h. CTX, Cortex; STR, striatum. A–D, Confocal images (40×) showing a reduction in the number of Olig2+ and CC1+ oligodendroglia in hyperoxia versus control mice at P8. The areas of interest included the EC and CG. E, F, Total number of Olig2+ and CC1+ within the EC and CG at P8 and P10. G, H, Images showing an increase in the number of Olig2+ oligodendroglia within the CG of hyperoxia-exposed pups versus control mice at P12. I, J, CC1+ oligodendroglia in hyperoxia versus control mice at P12 in the EC. No significant difference was found between the two groups in the EC or CG. K, Total number of Olig2+ and CC1+ within the EC and CG at P12. Scale bar, 50 μm. Data shown are expressed as mean ± SD (n = 3–6 brains for each group, using an unpaired t test comparing control vs hyperoxia; *p < 0.05; **p < 0.025) for the number of cells in 106 μm3.
Changes in NG2+ oligodendrocyte progenitor cells after hyperoxia: damage and repair
It is well established that OPCs and pre-OLs differentiate into mature oligodendrocytes within the developing brain (Baumann and Pham-Dinh, 2001; Nishiyama et al., 2009), and OPCs play a crucial role in remyelination after a demyelinating lesion (Zhao et al., 2005; Franklin and Ffrench-Constant, 2008). We analyzed changes in WM NG2+ cells, which may underlie the hyperoxia-induced decrease in MBP expression and the subsequent recovery of MBP in the WM at P15. We also analyzed total NG2+ cell proliferation in newborn WT mice using BrdU immunofluorescence. Immediately after 48 h of hyperoxia at P8, there was a significant decrease in the number of NG2+ cells found within the EC and CG when compared to control values (Fig. 3A,B,I). In addition, hyperoxia diminished the number of proliferating NG2+ cells (NG2+BrdU+) in both WM regions of interest (Fig. 3C,D,I). After 2 d of recovery in room air (P10), we found no significant difference between hyperoxia and controls in the total number of NG2+ cells or NG2+BrdU+ cells in the WM (Fig. 3J). After 4 d of recovery at P12, we observed a 43% increase in the number of NG2+ cells and a 62% increase in NG2+BrdU+ cells within the CG of the hyperoxia-exposed animals compared with control animals (Fig. 3E,H–K). No difference in the number of NG2+ or NG2+BrdU+ cells was detected in the EC at this time point (Fig. 3K).
Figure 3.
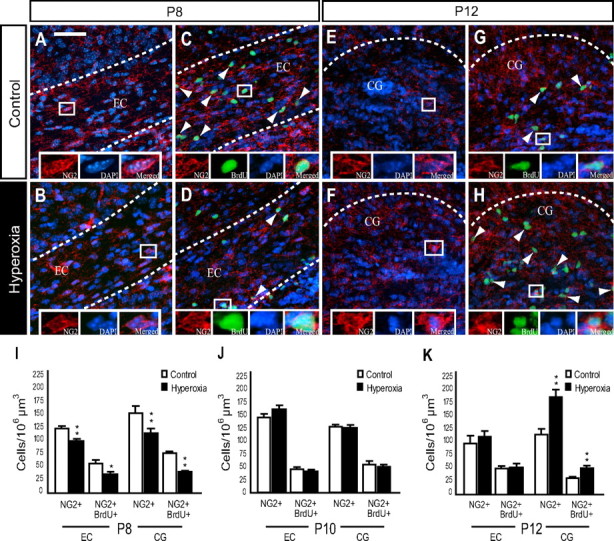
Time-dependent changes in white matter NG2+ oligodendrocyte progenitor cells after hyperoxia. A, B, Confocal images of NG2+ progenitors under control conditions or after hyperoxia in WT mice at P8. C, D, Confocal images of NG2+BrdU+ progenitors in control versus hyperoxia WT mice at P8. E, F, Images of NG2+ progenitors in the control versus hyperoxia group in WT mice at P12. G, H, NG2+BrdU+ progenitors control versus hyperoxia in WT mice at P12. Arrowheads indicate respective triple immunolabeling. I, Decrease in the total number of NG2+ and NG2+BrdU+ cells in the EC and in the CG at P8. J, At P10, the total number of NG2+ and NG2+BrdU+ cells in the hyperoxia group returned to control levels in both WM areas of interest. K, Increase in NG2+ and NG2+BrdU+ cells within the CG of the hyperoxia group at P12. Total numbers of NG2+ and NG2+BrdU+ returned to control levels within the EC. Scale bar, 50 μm. Data are shown as mean ± SD (n = 3–5 brains for each group, using an unpaired t test comparing control vs hyperoxia; *p < 0.05; **p < 0.025).
It has been shown that NG2 expression is not always restricted to oligodendroglia (Nishiyama et al., 2009). Therefore, to verify that changes in the NG2+ cell population observed in WT mice were cells of the oligodendroglial lineage, the number of NG2+ cells in CNP-EGFP transgenic mice exposed to hyperoxia and room air was evaluated. In the CNP-EGFP mouse, cells of the oligodendroglial lineage are labeled with green fluorescent protein (GFP), which is under the control of the CNP promoter (Yuan et al., 2002). Notably, virtually all NG2+ cells in the WM regions analyzed were GFP+ (data not shown). A region-specific and temporal cellular response similar to that observed in WT mice was found in NG2+GFP+ progenitors of the CNP-EGFP transgenic mice. At P8, the number of NG2+GFP+ cells was significantly decreased (n = 3; p < 0.05) after hyperoxia (supplemental Fig. 2A,B,E, available at www.jneurosci.org as supplemental material). After 4 d of recovery at P12, NG2+GFP+ cells were markedly increased (n = 3; p < 0.05) selectively in the CG, but not in the EC (supplemental Fig. 2C,E, available at www.jneurosci.org as supplemental material).
These results indicate that hyperoxia alters the growth of the NG2+ oligodendroglia population. NG2+ cells are also likely involved in the repair of the developing WM after hyperoxia. This is supported by our observation that hyperoxia causes an acute reduction in the number of NG2+ and NG2+BrdU+ cells in the WM immediately after exposure at P8. Furthermore, we found a greater number of NG2+ and NG2+BrdU+ cells when compared to controls at P12. This was associated with a restoration of the CC1+ cell population after recovery at the same time point (Fig. 2).
Hyperoxia-induced oligodendroglial lineage apoptosis in the developing white matter
The immature rat brain, especially the WM, is susceptible to hyperoxia-induced apoptosis (Felderhoff-Mueser et al., 2004) with cells of the oligodendroglial lineage exhibiting several maturation-dependent factors that enhance susceptibility to oxidative stress (Back et al., 2007; Khwaja and Volpe, 2008; Bradl and Lassmann, 2009). Recent in vitro studies revealed immature oligodendroglia to be most vulnerable to caspase-dependent apoptosis, whereas mature oligodendrocytes remain resistant (Gerstner et al., 2008). To assess overall hyperoxia-induced apoptosis in the developing WM in vivo, we first analyzed the EC and CG regions together for the presence of cleaved caspase-3α immunopositive (Casp3+) cells and TUNEL positive (TUNEL+) cells. In wild-type mice, hyperoxia caused a significant increase in the number of Casp3+ cells at P6 (after 6 h of hyperoxia), P8, and P12, with the peak of cell death occurring at P8 (Fig. 4A). Hyperoxia-induced apoptosis was virtually undetectable at P15 and P30 (Fig. 4A).
Figure 4.
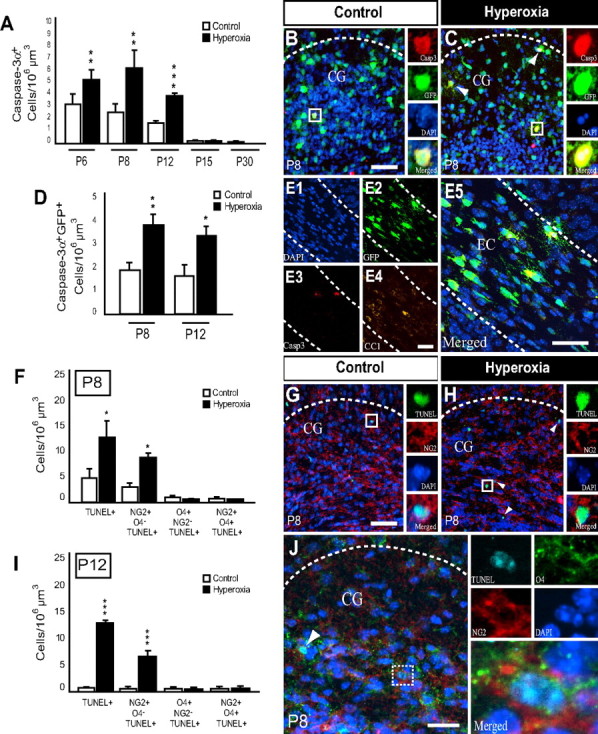
Hyperoxia causes cell death of NG2+ oligodendroglia in the developing white matter. A, Quantification of Casp3+ cells in WT mice within the EC and CG regions combined during hyperoxia (P6, 6 h of exposure) and at various times after hyperoxia (P8, P12, P15, and P30). Counts revealed an increase in the number of Casp3+ cells at P6, P8, and P12 with no difference observed at P15 and P30. B, C, Images of activated Casp3+ cells in P8 CNP-EGFP mice under control conditions or after hyperoxia show more apoptosis in pups exposed to hyperoxia. Arrowheads indicate Casp3+GFP+ positive cells. D, Numbers of Casp3+GFP+ cells in CNP-EGFP mice within both regions combined at P8 and P12 showed increased cell death at both time points. E, Confocal images from a P8 animal exposed to hyperoxia, showing Casp3+GFP+ cells to be negative for CC1, indicating the cells undergoing apoptosis were immature, not mature oligodendroglia. F, Histogram showing the number of TUNEL+, NG2+O4−TUNEL+, O4+NG2−TUNEL+, and NG2+O4+TUNEL+ cells at P8. G, H, Confocal images of TUNEL+ and NG2+O4−TUNEL+ cells in control and hyperoxia exposed animals. Arrowheads indicate double immunopositive cells (NG2+TUNEL+). I, Quantification of TUNEL+, NG2+O4−TUNEL+, O4+NG2−TUNEL+, and NG2+O4+TUNEL+ cells at P12. J, Representative image from a P8 hyperoxia-exposed animal showing several NG2+O4+TUNEL+ cells. Scale bars, 50 μm. Data are shown as mean ± SD (n = 3–6 brains for each group using an unpaired t test comparing control vs hyperoxia; *p < 0.05; **p < 0.025; ***p < 0.005).
To verify our results obtained in WT mice, we also analyzed the CNP-EGFP transgenic mouse and found that these mice when exposed to hyperoxia exhibited a similar pattern of apoptotic cell death in the WM. We observed a significant elevation in the number of Casp3+ GFP+ double-positive cells at P8 and after 4 d recovery at P12, when compared to controls at these time points (Fig. 4B–D).
To identify the developmental stage of oligodendroglia susceptible to hyperoxia-induced toxicity in vivo, CNP-EGFP transgenic mice were first immunostained for Casp3 and CC1. No Casp3+GFP+CC1+ cells were found at any time point after hyperoxia exposure, indicating oligodendroglia undergoing cell death to be of immature state (Fig. 4E). Based on these findings, further immunohistochemical analysis was performed using TUNEL and antibodies against NG2 and O4. There was a significant increase in the number of TUNEL+ and NG2+O4−TUNEL+ cells in the hyperoxia group at P8 (Fig. 4F–H). No difference in O4+NG2−TUNEL+ or NG2+O4+TUNEL+ was found between the two experimental groups (Fig. 4F). At P12, we also observed a significant increase in TUNEL+ and NG2+O4−TUNEL+ cells in the hyperoxia group with very little cell death in control animals (Fig. 4I). Again, no difference was found when comparing the numbers of O4+ NG2−TUNEL+ and NG2+O4+TUNEL+ cells in each group at P12 (Fig. 4I).
In summary, hyperoxia causes apoptosis in the OPC population (NG2+ cells) of the developing WM. This increased OPC cell death, both during and immediately after hyperoxia exposure, may in part explain the changes in (1) Olig2+/CC1+ cell numbers and (2) MBP expression.
Regeneration of mature oligodendroglia after hyperoxia exposure
The generation of mature, myelinating oligodendrocytes occurs throughout development and is also crucial in WM repair after demyelination (Franklin and Ffrench-Constant, 2008). To assess oligodendrogenesis during the recovery period from P8 to P12, the generation of newly formed oligodendrocytes was determined through analysis of BrdU and CC1 double immunopositive cells after cumulative BrdU labeling as described in Materials and Methods. At P12, the number of newly generated BrdU+CC1+ oligodendrocytes was increased in the hyperoxia-exposed group within both the EC and CG (Fig. 5A,B).
Figure 5.

Increased oligodendrogenesis after hyperoxia exposure throughout the immature white matter. Six-day-old (P6) mouse pups were exposed to normoxia (room air, 21% oxygen) or hyperoxia (80% oxygen) for 48 h. Animals in both experimental groups were kept in room air from P8 to P12. BrdU injections were started immediately at the end of exposure and were given every 24 h, and newly generated oligodendrocytes were labeled by immunofluorescence for CC1 and BrdU at P12. A, Confocal images of CC1+BrdU+ oligodendrocytes in control versus hyperoxia-exposed mice at P12. Arrowheads indicate triple immunolabeling for CC1, BrdU, and DAPI. Scale bar, 50 μm. B, Total numbers of newly formed CC1+BrdU+ oligodendrocytes in the EC and CG after cummulative BrdU injections from P8 to P12. Data are shown as mean ± SD (n = 3–5 brains for each group, using an unpaired t test comparing control vs hyperoxia; *p < 0.05; **p < 0.025).
In addition, a comparison of the CC1+ cell populations between P8 and P12 revealed that hyperoxia caused an 8.41-fold average increase in the total number of CC1+ cells in the two WM regions of interest compared to a 4.01-fold increase in controls (p < 0.05; one-way ANOVA; Holm-Sidak). This indicates that more CC1+ oligodendroglia were generated in the experimental group exposed to hyperoxia over this time period.
Together with our OPC observations described previously, these findings indicate that during the recovery phase, restoration of the NG2+ population contributes to newly formed CC1+ oligodendroglia in the WM at later time points. The generation of new CC1+ cells is then responsible for the restoration of MBP expression observed at P15 and P30.
Neonatal exposure to hyperoxia reduces WM diffusivity in the adult mouse
We hypothesized that the delay in oligodendroglia development could produce long-term effects on WM integrity. To determine whether chronic disturbances could be detected in the WM of mice exposed to hyperoxia, FA was measured by DTI. The measurements of diffusivity by DTI using MRI represents a technique of high sensitivity for changes of myelination and WM organization both during development and after injury (Basser and Pierpaoli, 1996; Inder et al., 1999). In addition, FA is more sensitive than conventional T2-weighted MRI in detecting WM injury (Counsell et al., 2003; Huppi and Dubois, 2006). FA in the white matter increases during brain development and has been associated with myelination in the WM (Moseley, 2002), fiber density (Horsfield and Jones, 2002), and with alignment and packing of axons (Huppi and Dubois, 2006). In a hypoxia model of WM damage, where neonatal mice were exposed from P3 to P11, the CC was established as a reliable region to determine FA decreases at age P45 to P51, and FA in the CC was markedly decreased at ages P45 and P51 (Chahboune et al., 2009). Moreover, low FA in the CC significantly correlated to poor neurological outcome assessed by neurodevelopmental tests (Chahboune et al., 2009). Therefore, we performed T2-weighted MRI sequences to select the coronal slice for DTI measurement in the CC and generated DEC maps to represent anisotropy in the lateral–medial directional manners of the CC. In the anisotropy measurements, FA was significantly decreased in P30 animals that had been exposed to hyperoxia from P6 to P8 (Fig. 6B,D1,F1,F2,I) (mean, 0.450; 25th–75th percentile, 0.433–0.471) compared to litter-matched control animals always kept in room air (Fig. 6A,C1, E1,E2,I) (mean, 0.580; 25th–75th percentile, 0.524–0.601; n = 6; Mann–Whitney rank test; p < 0.005). This significant decrease of FA in the CC was maintained even at P60 (Fig. 6G1–I) (mean hyperoxia, 0.525; 25th–75th percentile, 0.495–545; mean control, 0.623; 25th–75th percentile, 0.580–0.680; n = 6; p < 0.005). To distinguish between diffusivity that is parallel or perpendicular to the directory of CC fibers, we determined radial (perpendicular) and axial (parallel) ADCs. The radial ADC was increased after hyperoxia both at P30 (median, 0.615 × 10−3 mm2/s; range, 0.441–0.809) and at P60 (median, 0.715 × 10−3 mm2/s; range, 0.528–0.734) compared to controls (P30 median, 0.420 × 10−3 mm2/s; range, 0.420–0.612; P60 median, 0.465 × 10−3 mm2/s; range, 0.411–0.491; t test, p < 0.01 and p < 0.05, respectively; n = 6–7). The axial ADC values were similar in both groups, with 1.21 × 10−3 mm2/s in controls and 1.154 × 10−3 mm2/s after hyperoxia at P30, and 1.24 × 10−3 mm2/s and 1.14 × 10−3 mm2/s, respectively, at P60. Both the FA and ADC data indicate that, despite the restoration of the WM oligodendroglia population and subsequent recovery of MBP expression by P15, neonatal exposure to hyperoxia produces long-term damage to the WM.
Figure 6.

Changes in FA detected in P30 and P60 mice after exposure to hyperoxia from P6 to P8. A, B, T2-weighted coronal MRI images of P30 mice kept in room air (control) and age-matched mice after exposure to hyperoxia from P6 to P8. Red box shows region of CC that was selected for DTI measurements. C1, D1, FA obtained by DTI in control and hyperoxia-exposed animals, respectively. E1, F1, Using DEC maps, FA of medial–lateral directory representing fibers in the CC is shown in red. G1, H1, FA in the CC is significantly decreased at P30 and P60 in mice after neonatal exposure to hyperoxia as compared to controls. C2, D2, Enlarged from the boxes in C1 and D1, respectively. E2–H2, Enlargements of boxes from E1–H1. Data are shown as mean ± SD (n = 6–7 animals per group, using a Mann–Whitney rank test; *p < 0.005).
Hyperoxia alters astrocyte phenotype in the immature white matter
In the course of characterizing hyperoxia-induced changes within the oligodendroglial lineage, we considered that changes in the functional properties of other WM cell types could interfere with the survival or growth of oligodendroglia. Astrocytes have been found to influence CNS damage (D'Amelio et al., 1990; Kuroda and Shimamoto, 1991; Levine et al., 1999) and repair (Davies et al., 1997; Li et al., 2008). We hypothesized that astrocytes may contribute to hyperoxia-induced WM damage, and therefore analyzed the properties of WM astrocytes to determine their reactivity in response to hyperoxia.
Hyperoxia exposure caused a biphasic astrocytic response in the developing WM. Immunofluorescence and Western blots were first used to assess GFAP expression. At P8, immediately after 48 h of hyperoxia, GFAP immunoreactivity was reduced in the hyperoxia experimental group when compared with controls in both the CG and EC (Fig. 7A–D,I). Western blots of microdissected WM tissue collected from P8 pups also revealed a decrease in GFAP protein expression after hyperoxia (Fig. 7J,K). In contrast, at P12, the intensity of GFAP immunofluorescence was elevated in the hyperoxia treated group compared to controls (Fig. 7E–I). The increase of GFAP expression was confirmed by Western blot analysis (Fig. 7J,K).
Figure 7.
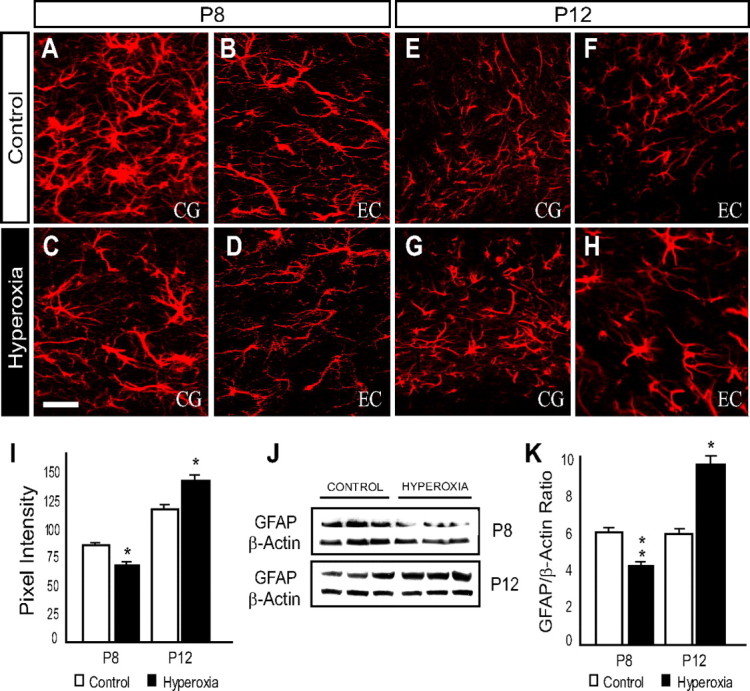
GFAP expression in the developing white matter is altered by hyperoxia exposure. Expression for GFAP was measured immediately after 48 h of hyperoxia exposure at P8 and after 4 d of room air recovery at P12. A–H, Confocal images of GFAP immunohistochemistry within the CG and EC at P8 and P12. Scale bar, 50 μm. I, At P8, analysis (pixel intensity) of the CG and EC regions combined revealed a decrease in GFAP immunoreactivity within the hyperoxia group when compared to controls. After 4 d of recovery in room air, increased GFAP expression was observed at P12 within the white matter. J, K, Western blot analysis was performed using microdissected tissue of the white matter at each time point indicated. The hyperoxic group exhibited a decrease in GFAP protein expression at P8 and an increase at P12. Data are shown as mean ± SD (n = 5–8 brains for each group, using an unpaired t test comparing control vs hyperoxia). *p < 0.01; **p < 0.001).
This initial decrease followed by an increase in GFAP expression has also been observed in astrocytes of the hippocampus in a model of transient global ischemia (Ouyang et al., 2007). The similarity between the pattern of GFAP expression and that of changes in the oligodendroglia lineage after hyperoxia suggests a role for astrocyte dysfunction in WM damage.
To quantify the number of astrocytes in the WM under both experimental conditions we performed coimmunolabeling for GFAP and GS.
GFAP+ processes of fibrous astrocytes are often at a distance from the cell soma or nucleus, making identification and quantification of individual GFAP+ cells difficult. In contrast, expression of GS, the enzyme responsible for the conversion of glutamate and ammonia to glutamine, is highly confined to the soma of all astroglia in the WM (Miyake and Kitamura, 1992). We observed no change in the number of GFAP+GS+ astrocytes at P8 or P12 (Fig. 8D,H). We also performed GS immunostaining in GFAP-EGFP mice, a transgenic mouse where astrocytes are labeled with GFP expressed under the control of the human GFAP promoter (Nolte et al., 2001) to asses astrocyte proliferation (Ki67) and apoptosis (Casp3) after hyperoxia. At all time points evaluated, no Ki67+GFP+GS+ astrocytes were observed in either the hyperoxia or control group (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and no cell death was observed in GFP+GS+ WM astrocytes (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), even at time points at which immature oligodendroglia displayed susceptibility to hyperoxia-induced apoptosis (Fig. 3).
Figure 8.

Hyperoxia does not affect astrocyte number, but decreases astrocytic GLAST expression in the immature WM. A1–B5, Immunofluorescence labeling with antibodies for GFAP, GS, GLAST, and the nuclear fluorescent stain DAPI to determine GLAST expression in GFAP+GS+ astrocytes (GFAP+GS+GLAST+ cells) in WT mice at P8. Arrowheads indicate cells positive for each respective marker and all four in merged image. C1–C5, Enlarged image of boxed region in A showing three GFAP+GS+GLAST+DAPI+ cells from a control mouse. D, Histrogram showing no difference in the number of astrocytes or glutamate transporter GLT-1 positive astrocytes (GFAP+GS+GLT-1+ cells) between the control and hyperoxia experimental groups at P8. However, hyperoxia reduced the number of astrocytes expressing GLAST. E, Data indicating that hyperoxia decreases the percentage of GLAST-expressing astrocytes without changing the number of astrocytes expressing GLT-1 at P8. F, G, Immunolabeling with antibodies for GFAP, GS, GLT-1, and DAPI to analyze astrocyte numbers and the amount of astrocytes expressing GLT-1 in WT mice at P12. Arrowheads indicate cells positive for each respective marker and all four in merged image. H, I, No change in the total number of GFAP+GS+GLAST+ cells or GFAP+GS+GLT-1+ cells was observed at P12. J, K, Densitometric analysis of GLAST and GLT-1 Western blots at P8 and P12. Hyperoxia caused a decrease in GLAST protein levels immediately after exposure (J). After 4 d of room air recovery (K), WM GLAST protein expression returned to the level of controls. No changes in GLT-1 levels were observed in hyperoxia-exposed animals. Values represent standardized mean ratios for the protein of interest (POI) and β-actin. L, Representative Western blots of GLAST, GLT-1, and β-actin protein expression using microdissected WM tissue from P8 and P12 animals in each experimental group. For each group and time point, n = 3–5 brains. An unpaired t test comparing control versus hyperoxia, where **p < 0.025 and ***p < 0.005, were used for statistical analysis.
GFAP+GS+ astrocytes express high-affinity glutamate uptake transporters; GLAST (EAAT-1), GLT-1 (EAAT-2), and EAAC1 (EAAT-3) (Danbolt, 2001; Fukamachi et al., 2001; Miralles et al., 2001; Liu et al., 2006; Arranz et al., 2010) and the expression of these transporters undergoes region specificity and is regulated during postnatal development throughout the rodent brain (Furuta et al., 1997; Regan et al., 2007). GLAST and GLT-1 are expressed on the astrocytic cell membrane, function in vivo, and are responsible for the majority of astrocyte-mediated glutamate uptake (Anderson and Swanson, 2000; Danbolt, 2001). EAAC1 is thought to be primarily neuronal (Danbolt, 2001), and the functional significance of EAAC1 in glutamate uptake performed by astrocytes has yet to be fully established. Even though cultured astrocytes do express EAAC1, it was not found to be expressed at the cell surface (Dallas et al., 2007; Arranz et al., 2010).
Intriguingly, recent evidence points to a functional interaction between GFAP and GLAST (Sullivan et al., 2007), and both increased GFAP expression in reactive astrocytes (Cavaliere et al., 2007) as well as ablation of GFAP have been associated with reduced glutamate clearance (Hughes et al., 2004). Modifications in glutamate uptake have been shown to cause glutamate-mediated excitotoxic cell death of immature oligodendroglia (Matute et al., 2002, 2007; Deng et al., 2006), and activation of oligodendroglia non-NMDA GluRs can lead to decreased OPC proliferation and the inhibition of lineage progression (Gallo et al., 1996; Yuan et al., 1998). Therefore, we reasoned that the hyperoxia-induced modulation in GFAP expression might be associated with altered astrocyte-mediated glutamate uptake, which may ultimately have detrimental effects on the oligodendroglia population of the immature WM.
To investigate a possible link between hyperoxia and altered glutamate homeostasis in the developing WM, we analyzed the number of astrocytes expressing GLAST or GLT-1. At P8 and P12, a decrease in the number of GFAP+GS+GLAST+ cells and a 14.08% reduction in the astrocyte population expressing GLAST was found at P8 in the hyperoxia group, with no difference in either parameter detected at P12 (Fig. 8A1–E). When quantifying the number of astrocytes expressing GLT-1, there was no statistically significant difference in total GFAP+ GS+GLT-1+ cells or the percentage of GFAP+GS+ cells positive for GLT-1 between the hyperoxia and control groups at either P8 or P12 (Fig. 8D–I). In addition, the majority (∼95% at P8 and 85% at P12) of cells expressing either GLAST or GLT-1 were GFAP+GS+ astroglia. GLAST+ astrocytes of the WM, in both experimental groups, also displayed a differential expression pattern, where GLAST immunoreactivity could be observed in the cell soma or astrocytic processes or in both locations (Fig. 8C). In contrast, GFAP+GS+GLT-1+ cells exhibited a more uniform distribution of GLT-1 immunofluorescence.
Protein expression in microdissected WM tissue was also measured at both P8 and P12. GLAST was reduced by 34% in the WM of mice exposed previously to hyperoxia when compared to age-matched controls (n = 8; p < 0.005), and GLAST expression returned to control levels at P12 (Fig. 8J–L). No change in the amount of GLT-1 was found at either P8 or P12 (Fig. 8J–L). These results suggest that hyperoxia may affect glutamate uptake by altering GLAST expression on GFAP+GS+ astrocytes in vivo.
Astrocyte glutamate uptake transporter function is attenuated by hyperoxia exposure
To determine whether hyperoxia directly affects astrocyte-mediated sodium-dependent glutamate uptake during early postnatal development, [3H]-d-aspartic acid uptake analysis was performed in tissue extracts of the WM and in culture. The amount of total and non-GLT-1 mediated (100 μm DHK was used to selectively block GLT-1) sodium-dependent [3H]-d-aspartic acid uptake was determined by analyzing WM taken at P8, P12, P15, and P30. TBOA, a nontransportable agent that selectively blocks all EAATs was also administered to a subset of samples, at a concentration of 75 μm. At P8, the rate of total [3H]-d-aspartic acid uptake was 1.11 ± 0.13 pmol/mg protein · min−1 in the control group decreased to 0.75 ± 0.24 pmol/mg protein · min−1 in the hyperoxia group (Fig. 9A). Non-GLT-1-mediated [3H]-d-aspartic acid uptake was also decreased from 0.83 ± 0.07 pmol/mg protein · min−1 in controls to 0.47 ± 0.16 pmol/mg protein · min−1 in the hyperoxia group (Fig. 9A) (n = 7; p < 0.001). A similar pattern was observed at P12, where total [3H]-d-aspartic acid uptake and non-GLT-1 uptake were reduced in hyperoxia when compared to control (n = 4; p < 0.001; control, total, 1.28 ± 0.11 pmol/mg protein · min−1; non-GLT, 0.73 ± 0.04 pmol/mg protein · min−1; hyperoxia, total, 0.68 ± 0.09 pmol/mg protein · min−1; non-GLT, 0.23 ± 0.04 pmol/mg protein · min−1) (Fig. 9B). No change in total or non-GLT-1 [3H]-d-aspartic acid uptake was found between control and hyperoxia groups at either P15 (Fig. 9C) or P30 (data not shown). There was a significant difference between total and non-GLT-mediated (+DHK) [3H]-d-aspartic acid uptake in both control and hyperoxia groups at P8 and P12 (Fig. 9A,B). Last, no difference was observed at any time point in samples containing TBOA when comparing hyperoxia and control groups (Fig. 9A–C).
Figure 9.

Astrocyte-mediated sodium-dependent [3H]-d-aspartic acid uptake is decreased after hyperoxia in tissue and in culture. [3H]-d-aspartic acid uptake assays at P8, P12, and P15. Microdissected WM was used to perform experiments and sodium-dependent [3H]-d-aspartic acid uptake was measured in pmol/mg protein · min−1. A, The hyperoxia group exhibited a decrease in total and non-GLT-1-mediated (+DHK) [3H]-d-aspartic acid uptake when compared to control (n = 7). B, Lower total and non-GLT-1-mediated [3H]-d-aspartic acid uptake was also observed at P12 (n = 4). C, No difference in total or non-GLT-1-mediated [3H]-d-aspartic acid uptake was found at P15 (n = 5). Note that the y-axis scale in C is different from those in A and B. Addition of TBOA nearly abolished all sodium-dependent [3H]-d-aspartic acid uptake at all time points analyzed. Data are shown as mean ± SD; statistical significance between groups was determined using an ANOVA followed by a Holm–Sidak test, where *p < 0.001 was considered to be significant. D, Representative measurement of the maximum velocity of sodium-dependent [3H]-d-aspartic acid uptake in purified astrocytes after 48 h of hyperoxia (80% oxygen) and normoxia (21% oxygen). Hyperoxia decreased the rate of [3H]-d-aspartic acid uptake in confluent cultured astrocytes. E, Representative Western blot of GLAST protein expression in cultured astrocytes stimulated with db-cAMP (all GLAST bands 65, 150, and 180 kDa). Analysis revealed a decrease in total GLAST levels.
Confluent cultures (treated with db-cAMP) exposed to 80% oxygen for 48 h demonstrated a significantly reduced rate of sodium-dependent [3H]-d-aspartic acid uptake, with a Vmax value of 9.3 ± 0.52 fmol/mg protein · min−1 compared with 12.9 ± 0.82 fmol/mg protein · min−1 in control astrocytes (Fig. 9D). Statistical significance was confirmed by a two-sided t test for uptake values at 100 μm [3H]-d-aspartic acid (95% confidence interval; p < 0.01). The KD in astrocytes exposed to hyperoxia was also decreased compared with controls, but this trend did not reach statistical significance (hyperoxia, 6.5 ± 1.6 fmol/mg protein · min−1; controls, 10.6 ± 2.8 fmol/mg protein · min−1). Levels of GLAST protein expression were also determined in the cultured astrocytes exposed to hyperoxia. Western blot analysis revealed a 40% decrease in total GLAST (Fig. 9E) (summation of monomeric 65 kDa, and multimeric 150 and 180 kDa bands) (Haugeto et al., 1996) expression in cultured astrocytes exposed to 80% oxygen for 48 h compared with cultured astrocytes kept at 21% oxygen [control, 2.26 ± 0.23 (GLAST/β-actin ratio); hyperoxia, 1.87 ± 0.18 (GLAST/β-actin ratio); data are mean ± SD; n = 4; p < 0.05, using an unpaired t test comparing control vs hyperoxia].
Together, these data suggest that hyperoxia exposure decreases sodium-dependent glutamate uptake transporter activity in astrocytes through a non-GLT-mediated mechanism. This is supported by our previous data showing GLAST to be the primary glutamate transporter present on astrocytes in the WM of neonatal mice around the time of hyperoxia exposure. The ability of DHK to reduce [3H]-d-aspartic acid uptake by the same extent in both experimental groups at P8 and P12 (Fig. 9A,B) indicates that the GLT-1-mediated component of sodium-dependent glutamate uptake is functionally intact after hyperoxia exposure, and the observed decrease in [3H]-d-aspartic acid uptake may be contributed largely by GLAST. This is in agreement with our observation that there was no change in the number or percentage of astrocytes expressing GLT-1 (Fig. 8D–I) and no change in overall WM GLT-1 protein expression (Fig. 8J–L). A reduction in astrocyte-mediated glutamate uptake/clearance has been implicated in animal models in which oxidative stress is prominent (Chen et al., 2005; Yeh et al., 2005). However, unlike our study, GLT-1 appears to be the vulnerable glutamate uptake transporter in this model of brain injury in the adult rodent.
Cell growth of cultured astrocytes is differentially affected by hyperoxia
To analyze the role of astrocyte dysfunction in hyperoxia-induced WM damage, we studied the effects of 80% oxygen on purified rat astrocytes under various culture conditions. In subconfluent astrocyte cultures, the numbers of GFAP+ astrocytes were decreased after 48 h of hyperoxia (Fig. 10A). Immature vimentin+ astrocytes were also reduced in the hyperoxia-exposed cultured astrocytes (Fig. 10A). In addition, the number of proliferating GFAP+ and vimentin+ astrocytes, as measured by Ki67 coimmunolabeling, were decreased in hyperoxic cultures (Fig. 10A–E).
Figure 10.
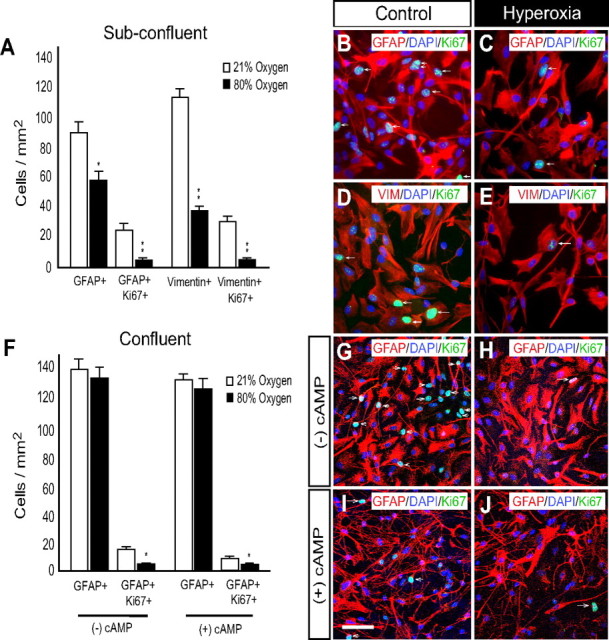
Hyperoxia decreases cell proliferation in primary cultured astrocytes. A–C, In subconfluent cultures of GFAP+ astrocytes, 48 h of hyperoxia decreased GFAP+ cell number and astrocyte proliferation (GFAP+Ki67+ cells) when compared to normoxia. A, D, E, Hyperoxia reduced the total number of subconfluent vimentin+ astrocytes along with the number of proliferating vimentin+GFAP+ immature astrocytes. F–H, Confluent primary astrocyte cultures were exposed to 48 h of hyperoxia in the absence of db-cAMP stimulation. These conditions yielded no change in the number of GFAP+ astrocytes but did decrease the low number of proliferating GFAP+Ki67+ astrocytes. F, I, J, In confluent astrocytes differentiated with db-cAMP, cell proliferation is significantly reduced by hyperoxia with no change in GFAP+ astrocyte number. Arrows indicate respective triple labeling. Scale bar, 50 μm. Data are shown as mean ± SD using an unpaired t test comparing control versus hyperoxia. *p < 0.01; **p < 0.001.
It has been well described that cAMP stimulation of cultured astrocytes can convert flat polygonal shaped astroglia into process-bearing stellate astrocytes (Favero and Mandell, 2007), which more closely resemble astrocytes in vivo with regard to morphology and differentiation (Shapiro, 1973; Narumi et al., 1978). In our confluent cultures not treated with db-cAMP, 48 h of hyperoxia (80% oxygen) decreased levels of proliferating GFAP+ astrocytes when compared to cultures kept at 21% oxygen (Fig. 10F–H). Furthermore, in agreement with the expectation that astrocyte proliferation declines during differentiation, the percentage of Ki67+GFAP+ cells was reduced by db-cAMP in both experimental groups. However, hyperoxia and db-cAMP stimulation in combination produced a further decrease in the percentage of Ki67+GFAP+ cells in culture (Fig. 10F,I,J). No cell death, measured by TUNEL immunofluorescence, was observed in cultured astrocytes (confluent or subconfluent) exposed to 48 h of hyperoxia or 21% oxygen (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). In confluent cultures, hyperoxia did not alter the numbers of GFAP+ cells despite a modest, but significant, reduction in Ki67 expression. The reason for this is unknown, but may reflect the induction of cellular arrest in G0/early G1 of the cell cycle. The cultures treated with db-cAMP exhibited stellation and very low rates of cell proliferation, which are properties closely resembling astrocytes in vivo. In addition, hyperoxia caused an upregulation of GFAP levels (supplemental Fig. 5B,C, available at www.jneurosci.org as supplemental material). It should not be surprising that some differences between the properties of astrocytes in vivo and in culture are observed. Both hyperoxia paradigms were designed to result in oxygen levels twofold to fourfold above normal conditions. The oxygen levels in culture are therefore not close to the oxygen tensions observed in vivo, and these extreme conditions are likely to underlie some of the observed differences in biological response of the astrocytes to hyperoxia. Nevertheless, the changes in glutamate transport after hyperoxia are in agreement in both systems; therefore, this mechanism of damage is being further investigated in culture.
Hyperoxia increases glutamate-induced OPC toxicity by reducing astrocyte-mediated protection
Reduced glutamate transport or uptake in the presence of a pathological glutamate challenge can inflict excitotoxic damage to surrounding cells (Matute et al., 2007). The overactivation of non-NMDA GluRs can cause decreased OPC proliferation and inhibit oligodendroglia lineage progression (Gallo et al., 1996; Yuan et al., 1998). We wanted to determine whether the reduction in astrocyte-mediated glutamate uptake induced by hyperoxia altered the OPC (A2B5+ cells) and/or O4+ oligodendroglia populations in the presence of high extracellular glutamate levels. To test this, we established an astrocyte–OPC coculture system in which dye-labeled OPCs were added to primary astrocytes exposed previously to hyperoxia or normoxia. After the presentation of an exogenous glutamate challenge (1 mm), OPCs were immunostained and analyzed for changes in cell number. A2B5+ cells were analyzed after 24 h, and O4+ cells after 48 h, in culture. Glutamate did not significantly affect the number of OPCs in cocultures prepared with normoxic (control) astrocytes. (Fig. 11A–D,I). However, glutamate challenge significantly decreased the numbers of A2B5+Dye+ and O4+Dye+ cells in cocultures containing astrocytes that were exposed previously to hyperoxia (Fig. 11A,B,E,F,I). Among the experimental groups that contained astrocytes that were exposed to 80% oxygen, but did not receive a glutamate challenge, hyperoxia produced a significant decrease in only the numbers of O4+Dye+ and did not affect the numbers of total dye-labeled or A2B5+Dye+ cells (Fig. 11I). The toxic effect of glutamate was prevented by exposure to the non-NMDA-receptor antagonist NBQX (100 μm) 1 h before glutamate challenge (Fig. 11, compare E, G for A2B5; compare F, H for O4).
Figure 11.
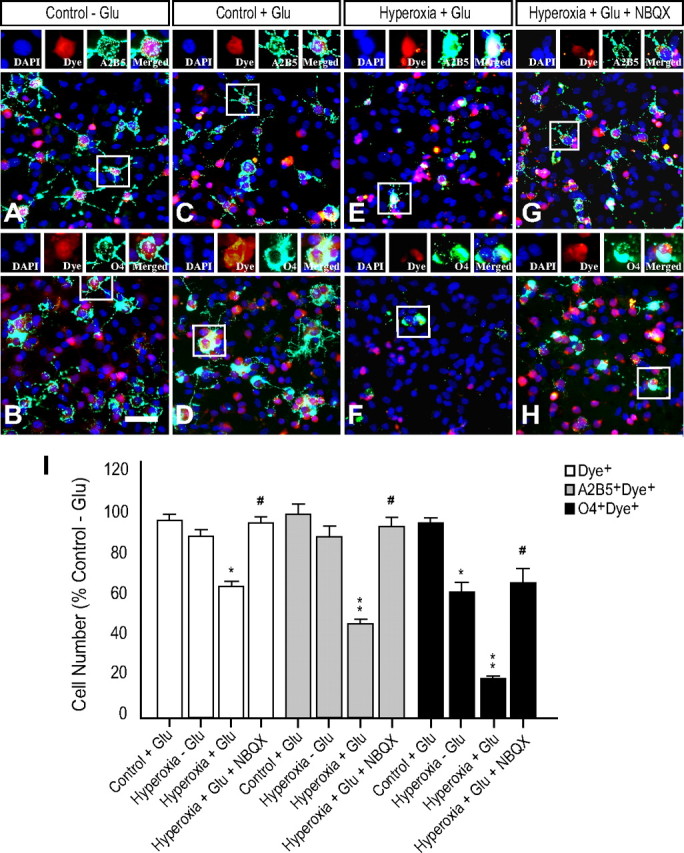
Reduced astrocyte-mediated protection of OPCs during glutamate challenge is attenuated by NBQX. Astrocytes were exposed to hyperoxia or normoxia (control) for 48 h before incubation with dye-labeled OPCs (red). Cocultures were challenged with 1 mm glutamate at the beginning of coculture or did not receive glutamate treatment. A–H, A2B5+ cells (green, A, C, E, G) were analyzed after 24 h and O4+ cells (green, B, D, F, H) after 48 h of coculture. A, B, A2B5+ and O4+ cells in control cocultures without glutamate challenge. C, D, Glutamate challenge does not significantly affect A2B5+ cells and O4+ cells in control cocultures. E, F, A significant decrease in the number of A2B5+ and O4+ cells was observed after glutamate treatment in cocultures with hyperoxia-treated astrocytes when compared to control cocultures after glutamate challenge. G, H, Addition of non-NMDA-receptor antagonist NBQX (100 μm) 1 h before glutamate challenge significantly attenuated the toxic effects on A2B5+/Dye+ (G) and (H) O4+/Dye+ cells. I, Values were normalized with DAPI and expressed as a percentage of control without glutamate challenge (Control − Glu) in each respective group (white bars, Dye+; gray bars, A2B5+Dye+; black bars, O4+Dye+). *p < 0.01; **p < 0.001 (unpaired student's t test vs Control − Glu); #p < 0.01, unpaired student's t test vs Hyperoxia + Glu); n = 4–5 for each group. Error bars indicate mean ± SD. Scale bar, 50 μm.
Our results obtained in astroglia–OPC coculture support the notion that hyperoxia diminishes extracellular glutamate uptake by a mechanism likely to involve reduced expression of GLAST, which in turn could inhibit the growth or survival of OPCs.
Discussion
In this study, we investigated cellular mechanisms underlying the delayed maturation of WM caused by hyperoxia exposure during a critical developmental time window. Our study is the first to (1) perform in vivo time course analysis of the oligodendroglial lineage after hyperoxia, (2) demonstrate recovery of WM oligodendroglial cells after hyperoxia exposure, (3) determine hyperoxia's effect on WM astrocyte phenotype and function, and (4) analyze the long-term consequences of neonatal hyperoxia. We demonstrate that hyperoxia exposure in newborn mice perturbs WM development by decreasing proliferation and increasing apoptosis in NG2+ oligodendroglia. Hyperoxia also alters astroglial GFAP and GLAST expression in vivo and decreases [3H]-d-aspartic acid uptake. Moreover, during recovery, animals exposed previously to hyperoxia exhibited compensatory (1) proliferation in WM NG2+ cells, (2) generation of new oligodendrocytes, and (3) MBP protein expression in the WM. However, despite the number of oligodendroglia, MBP levels, and [3H]-d-aspartic acid uptake returning to control levels in the WM by P15, impairment of WM integrity detected by DTI persisted into adulthood.
Animal models of premature brain injury have been shown to cause WM damage, hypomyelination, and altered WM integrity (Scafidi et al., 2009; Li et al., 2010). Several studies have also shown hyperoxia to cause injury to the developing WM in the rat (Felderhoff-Mueser et al., 2004; Gerstner et al., 2008). The loss of myelinating oligodendrocytes is thought to be a major contributing factor in chronic PWMI (Back et al., 2007; Khwaja and Volpe, 2008), and oligodendroglia exhibit a maturation-dependent vulnerability in vitro (Gerstner et al., 2006, 2008). We found significantly more NG2+O4−TUNEL+ cells after hyperoxia at P8, P10, and P12, without changes in the number of O4+NG2−TUNEL+ or NG2+O4+TUNEL+ cells. These data indicate that, in vivo, NG2+ progenitor cells, and not O4+ preoligodendrocytes, are highly susceptible to hyperoxia-induced apoptosis.
OPCs are the primary source of oligodendrocytes in the developing brain (Baumann and Pham-Dinh, 2001; Nishiyama et al., 2009) and in remyelinating WM lesions (Zhao et al., 2005; Franklin and Ffrench-Constant, 2008). The preterm infant is most vulnerable to PWMI when immature, premyelinating oligodendroglia, including OPCs, predominate in the developing WM (Back et al., 2001). Our observations in the proliferating NG2+ cell population are consistent with other studies that indicate that changes in oligodendroglia proliferation constitute mechanisms of injury (Robinson et al., 2005; Yan and Rivkees, 2006) and repair after such insults as ischemia or traumatic brain injury (Amat et al., 1998; Segovia et al., 2008) in the developing brain. Our finding that during recovery more NG2+ and NG2+BrdU+ cells were found only in the cingulum suggests that local NG2+ cell populations in the various WM regions respond differentially to injury, or that targeted recruitment of migratory OPCs into the cingulum occurs from germinal regions in close proximity, such as the subventricular zone.
The initial induction of cell death and reduced proliferation in the NG2+ cell population after hyperoxia is likely to be responsible for the decrease in WM CC1+ oligodendroglia observed at P8 and P10. This is because of fewer immature oligodendroglia being available to undergo maturation. The reduction in mature oligodendrocytes is, at least in part, responsible for the decrease in MBP expression observed at P8 and P10 in animals exposed to hyperoxia.
To evaluate the long-term impact of neonatal hyperoxia on WM integrity, we used DTI to determine FA in juvenile (P30) and adult mice (P60). Changes in FA have been associated with demyelination, axonal damage and behavioral abnormalities (Chahboune et al., 2009; Xie et al., 2010), and preterm infants exhibit a loss in WM volume and FA when compared to term infants (Dudin et al., 2007). Our DTI measurements in the CC demonstrate that neonatal hyperoxia during a critical developmental window produces a significant long-term decrease in WM diffusivity both at P30 and P60. These findings indicate that abnormalities in WM organization, including packing and alignment of WM fibers, can occur despite apparently normal numbers of CC1+ oligodendrocytes and levels of MBP, which are restored by P15. Further studies will determine the impact of these long-term WM deficiencies on functional properties of the underlying axons.
Astroglial reactivity plays a role in injury of the developing WM and was identified in ischemia (Biran et al., 2006), trauma (Rostworowski et al., 1997), and infection (Rousset et al., 2006). Although we observed no hyperoxia-induced changes in astrocyte number or cell death or proliferation in vivo, the astroglial response consisting of changes in GFAP and the glutamate transporter GLAST overlapped temporally with oligodendroglial changes and occurred in two phases: (1) an initial decrease in protein expression of GFAP and GLAST immediately after 48 h of hyperoxia at P8 and (2) increased levels of GFAP and restored GLAST expression after 4 d of recovery in room air at P12. The transporters GLAST and GLT-1 are coexpressed by both astrocytes and oligodendroglia in vivo and in vitro (Domercq et al., 2005; Regan et al., 2007), and GLT-1 has been shown recently to be important for glutamate homeostasis in cultured oligodendroglia (DeSilva et al., 2009). However, evidence suggests that only mature oligodendrocytes express functional glutamate uptake transporters in vivo (De Biase et al., 2010; Kukley et al., 2010), and as indicated by Table 2, the majority of the cells in the WM at P6 display an immature phenotype. Although we cannot rule out the contribution of oligodendroglial GLAST and GLT-1 in WM glutamate homeostasis, our data indicate that at P8 and P12, GLAST expression in the WM is localized predominantly in GFAP+GS+ astrocytes. Quantification also revealed a significant decrease in the number GFAP+GS+GLAST+ cells and GLAST protein levels in the WM after hyperoxia. No change in total GLT-1 expression or the number of GFAP+GS+GLT-1+ cells was found after hyperoxia.
[3H]-d-aspartic acid uptake assays conducted in WM tissue at P8 showed that hyperoxia caused a significant decrease in the rate of total and non-GLT-1-mediated sodium-dependent [3H]-d-aspartic acid uptake when compared with controls. Similar reductions in uptake were observed in control and hyperoxia groups with the application of the GLT-1 inhibitor DHK at P8 and P12 (Fig. 9), suggesting that hyperoxia did not alter levels of GLT-1 activity in the WM. Together, these observations support a non-GLT-1-mediated mechanism of injury.
Intriguingly, total and non-GLT-1-mediated [3H]-d-aspartic acid uptake was also significantly lower in the hyperoxia group at P12 and did not return to control levels until P15. This was despite a recovery in GLAST expression within the WM. Possible explanations for the decrease at P12 include posttranslational regulatory mechanisms such as (1) decreased cell surface expression of the transporter, (2) decreased Na,K-ATPase coupling (Rose et al., 2009), (3) altered interactions with the extracellular matrix (Ye and Sontheimer, 2002), and (4) oxidative stress-mediated modifications (Miralles et al., 2001). The cellular redox status can regulate glutamate uptake kinetics directly through changes in reactive cysteine residues in transporter structure, which allows maximal uptake activity only in the reduced state (Trotti et al., 1997).
Altered glutamate homeostasis has been implicated in many neurological disorders including epilepsy, Alzheimer's disease, and Parkinson's disease (Doble, 1999; Tilluex and Hermans, 2007; David et al., 2009). Glutamate has been shown to cause oligodendroglial damage (Domercq et al., 2005), changes in proliferation and apoptosis of NG2+ OPCs (Gallo et al., 1996; Yuan et al., 1998), axonal injury (Domercq et al., 2005), and disruption of axoglial junctions (Fu et al., 2009). In our glial coculture system, astrocytes exposed to hyperoxia were unable to protect A2B5+ and O4+ oligodendroglial cells against the toxicity of glutamate challenge. This vulnerability was eliminated in the presence of the non-NMDA antagonist NBQX. Therefore, it is possible that reduced glutamate uptake/clearance leads to higher amounts of glutamate in the extracellular space. This phenomenon may occur in isolation or in combination with enhanced cellular and/or axonal glutamate release throughout the WM. The resulting environment likely contributes to changes in the oligodendroglia population. Excitotoxicity has been associated with AMPA/kainite glutamate receptor activation in oligodendroglia (Alberdi et al., 2002; Sanchez-Gomez et al., 2003), and OPCs may exhibit enhanced vulnerability, because they express higher levels of AMPA/kainate glutamate receptors than pre-OLs or immature/mature oligodendrocytes (De Biase et al., 2010; Kukley et al., 2010).
In conclusion, our analysis shows that hyperoxia modulates glial interactions through a novel mechanism involving altered astrocyte GFAP and GLAST expression and decreased astrocyte-mediated glutamate uptake. These changes in astrocyte properties are associated with damage to the oligodendroglia lineage and delayed MBP expression. Despite apparent recovery in the glial population and in MBP levels, the disruption in oligodendroglia development and WM maturation during a critical period of vulnerability leads to long-term deficiencies in WM organization and integrity. A better understanding of the cellular and molecular events following hyperoxia will help define the role of glutamate in impaired WM development and PWMI. Future cell-based therapies aimed at improving WM integrity and function after hyperoxia may target these events to prevent damage and promote repair of the developing WM.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke (National Institutes of Health Grant R01 NS056427), the National Multiple Sclerosis Society (RG4019-A2 to V.G.; RG 3954 A1/2 to L.-J.C.), the Intellectual and Developmental Disabilities Research Center (Grant P30 HD40677), the Sanitaetsrat Dr. Emil Alexander Huebner and Gemahlin Foundation, and National Institutes of Health Grant P01 NS0626860 (V.G.; primary investigator, Flora Vaccarino, Yale University, New Haven, CT). J.R. was supported by Grant T32HD046388 (V.G.). We thank Dr. Ana Gadea for help with the coculture assays. We thank Dr. Michael Obladen, Dr. Karen Gale, and Dr. Christoph Buehrer for discussion and support. We thank Ying Cheng for breeding the CNP-EGFP mice.
References
- Alberdi E, Sanchez-Gomez MV, Marino A, Matute C. Ca2+ influx through AMPA or kainate receptors alone is sufficient to initiate excitotoxicity in cultured oligodendrocytes. Neurobiol Dis. 2002;9:234–243. doi: 10.1006/nbdi.2001.0457. [DOI] [PubMed] [Google Scholar]
- Amat JA, Farooq M, Ishiguro H, Norton WT. Cells of the oligodendrocyte lineage proliferate following cortical stab wounds: an in vitro analysis. Glia. 1998;22:64–71. doi: 10.1002/(sici)1098-1136(199801)22:1<64::aid-glia6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Swanson R. Astrocyte glutamate transport. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Arranz AM, Gottlieb M, Perez-Cerda F, Matute C. Increased expression of glutamate transporters in subcortical white matter after transient focal cerebral ischemia. Neurobiol Dis. 2010;37:156–165. doi: 10.1016/j.nbd.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Aylward GP. Cognitive and neurophysiological outcomes: More than just IQ scores. Ment Retard Dev Disabil Res Rev. 2002;8:234–240. doi: 10.1002/mrdd.10043. [DOI] [PubMed] [Google Scholar]
- Back SA. Perinatal white matter injury: the changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Retard Dev Disabil Res Rev. 2006;12:129–140. doi: 10.1002/mrdd.20107. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo N, Borenstein N, Levine J, Volpe JJ, Kinney H. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Riccle A, McClure MM. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke. 2007;38:724–730. doi: 10.1161/01.STR.0000254729.27386.05. [DOI] [PubMed] [Google Scholar]
- Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE. Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb use in the analysis of oligodendrocyte development. J Neurosci Res. 1989;24:548–557. doi: 10.1002/jnr.490240413. [DOI] [PubMed] [Google Scholar]
- Basser P, Pierpaoli C. Microstructural and physiological features of tissued elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;11:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Begni B, Brighina L, Fumagalli L, Andreoni S, Castelli E, Francesconi C, Del Bo R, Bresolin N, Ferrarese C. Altered glutamate uptake in peripheral tissues from Down syndrome patients. Neurosci Lett. 2003;343:73–76. doi: 10.1016/s0304-3940(03)00260-x. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE. Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol. 1999;9:293–298. doi: 10.1016/s0959-4388(99)80043-9. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bhutta AT, Cleves MA, Casey PH, Cradock MM, Anand KJ. Cognitive and behavioral outcomes of school-aged children who were born preterm. JAMA. 2002;288:728–737. doi: 10.1001/jama.288.6.728. [DOI] [PubMed] [Google Scholar]
- Biran V, Joly LM, Héron A, Vernet A, Véga C, Mariani J, Renolleau S, Charriaut-Marlangue C. Glial activation in white matter following ischemia in the neonatal P7 rat brain. Exp Neurol. 2006;199:103–112. doi: 10.1016/j.expneurol.2006.01.037. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;45:724–735. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2009;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo A, Sola AT, Baquero H, Neira F, Alvis R, Deulofeut R, Critz A. Pulse oxygen saturation levels and arterial oxygen tension values in newborns receiving oxygen therapy in the neonatal intensive care unit: is 85% to 93% an acceptable range? Pediatrics. 2008;121:882–889. doi: 10.1542/peds.2007-0117. [DOI] [PubMed] [Google Scholar]
- Cavaliere C, Cirillo G, Rosaria Bianco M, Rossi F, De Novellis V, Maione S, Papa M. Gliosis alters expression and uptake of spinal glial amino acid transporters in a mouse neuropathic pain model. Neuron Glia Biol. 2007;3:141–153. doi: 10.1017/S1740925X07000695. [DOI] [PubMed] [Google Scholar]
- Chahboune H, Ment LR, Stewart WB, Douglas L, Rothman DL, Vaccarino FM, Hyder F, Schwartz ML. Hypoxic injury during neonatal development in murine brain: correlation between in vivo DTI findings and behavioral assessment. Cereb Cortex. 2009;19:2891–2901. doi: 10.1093/cercor/bhp068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Hsu-Chou H, Lu JL, Chiang YC, Huang HM, Wang HL, Wu T, Liao JJ, Yeh TS. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology. 2005;49:703–714. doi: 10.1016/j.neuropharm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Cheong JL, Thompson DK, Wang HX, Hunt RW, Anderson PJ, Inder TE, Doyle LW. Abnormal white matter signal on MR imaging is related to abnormal tissue microstructure. Am J Neuroradiol. 2009;30:623–628. doi: 10.3174/ajnr.A1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MP, Lorenz JM, Jetton JR, Paneth N. Hypocapnia and other ventilation-related risk factors for cerebral palsy in low birth weight infants. Pediatr Res. 2001;50:712–719. doi: 10.1203/00006450-200112000-00014. [DOI] [PubMed] [Google Scholar]
- Counsell SJ, Allsop JM, Harrison MC, Larkman DJ, Kennea NL, Kapellou O. Diffusion-weighted imaging of the brain in preterm infants with focal and diffuse white matter abnormality. Pediatrics. 2003;112:1–7. doi: 10.1542/peds.112.1.1. [DOI] [PubMed] [Google Scholar]
- Curtis R, Cohen J, Fok-Seang J, Hanley MR, Gregson NA, Reynolds R, Wilkin GP. Development of macroglial cells in rat cerebellum. I. Use of antibodies to follow early in vivo development and migration of oligodendrocytes. J Neurocytol. 1988;17:43–54. doi: 10.1007/BF01735376. [DOI] [PubMed] [Google Scholar]
- Dallas M, Boycott HE, Atkinson L, Miller A, Boyle JP, Pearson HA, Peers C. Hypoxia suppresses glutamate transport in astrocytes. J Neurosci. 2007;27:3946–3955. doi: 10.1523/JNEUROSCI.5030-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio FE, Smith ME, Eng LF. Sequence of tissue responses in the early stages of experimental allergic encephalomyelitis (EAE): immunohistochemical, light microscopic, and ultrastructural observations in the spinal cord. Glia. 1990;3:229–240. doi: 10.1002/glia.440030402. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- David Y, Cacheaux LP, Ivens S, Lapilover E, Uwe H, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequences of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Wang H, Rosenberg PA, Volpe JJ, Jensen FE. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci U S A. 2004;18:7751–7756. doi: 10.1073/pnas.0307850101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Yue Q, Rosenberg PA, Volpe JJ, Jensen F. Oligodendrocyte excitotoxicity determined by local glutamate accumulation and mitochondrial function. J Neurochem. 2006;98:213–222. doi: 10.1111/j.1471-4159.2006.03861.x. [DOI] [PubMed] [Google Scholar]
- Deng W, Pleasure J, Pleasure D. Progress in periventricular leukomalacia. Arch Neurol. 2008;65:1291–1295. doi: 10.1001/archneur.65.10.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSilva TM, Kabakov AY, Goldhoff PE, Volpe JJ, Rosenberg PA. Regulation of glutamate transport in developing rat oligodendrocytes. J Neurosci. 2009;29:7898–7908. doi: 10.1523/JNEUROSCI.6129-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deulofeut R, Dudell G, Sola AT. Treatment-by-gender effect when aiming to avoid hyperoxia in preterm infants in the NICU. Acta Paediatr. 2007;96:990–994. doi: 10.1111/j.1651-2227.2007.00365.x. [DOI] [PubMed] [Google Scholar]
- Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- Domercq M, Etxebarria E, Perez-Samartin A, Matute C. Excitotoxic oligodendrocyte death and axonal damage induced by glutamate transporter inhibition. Glia. 2005;52:36–46. doi: 10.1002/glia.20221. [DOI] [PubMed] [Google Scholar]
- Dudin J, Lequin M, van Pul C, Buijs J, Conneman N, van Goudoever J, Govaert P. Fractional anisotropy in white matter tracts of very-low-birth-weight infants. Pediatr Radiol. 2007;37:1216–1223. doi: 10.1007/s00247-007-0626-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero CB, Mandell JW. A pharmacological activator of AMP-activated protein kinase (AMPK) induces astrocyte stellation. Brain Res. 2007;1168:1–10. doi: 10.1016/j.brainres.2007.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Sifringer M, Jarosz B, Korobowicz E, Mahler L, Piening T, Moysich A, Grune T, Thor F, Heumann R, Bührer C, Ikonomidou C. Oxygen causes cell death in the developing brain. Neurobiol Dis. 2004;17:273–282. doi: 10.1016/j.nbd.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Sifringer M, Polley O, Dzietko M, Leineweber B, Mahler L, Baier M, Bittigau P, Obladen M, Ikonomidou C, Buehrer C. Caspase-1–processed interleukins in hyperoxia-induced cell death in the developing brain. Ann Neurol. 2005;57:50–59. doi: 10.1002/ana.20322. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Fu Y, Sun W, Shi Y, Shi R, Cheng JX. Glutamate excitotoxicity inflicts paranodal myelin splitting and retraction. PLoS One. 2009;4:e6705. doi: 10.1371/journal.pone.0006705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukamachi S, Furuta A, Ikeda TI, Ikenove T, Kaneoka T, Rothstein JD, Iwaki T. Altered expression of glutamate transporter subtypes in rat model of neonatal cerebral hypoxia-ischemia. Dev Brain Res. 2001;132:131–139. doi: 10.1016/s0165-3806(01)00303-0. [DOI] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Armstrong RC. Developmental and growth factor induced regulation of nestin in oligodendrocyte lineage cells. J Neurosci. 1995;15:394–406. doi: 10.1523/JNEUROSCI.15-01-00394.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K1 channel block. J Neurosci. 1996;16:2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner B, Buhrer C, Rheinlander C, Polley O, Schuller A, Berns M, Obladen M, Felderhoff-Mueser U. Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia. J Neurosci Res. 2006;84:306–315. doi: 10.1002/jnr.20880. [DOI] [PubMed] [Google Scholar]
- Gerstner B, DeSilva TM, Genz K, Armstrong A, Brehmer F, Neve RL, Felderhoff-Mueser U, Volpe JJ, Rosenberg PA. Hyperoxia causes maturation-dependent cell death in the developing white matter. J Neurosci. 2008;28:1236–1245. doi: 10.1523/JNEUROSCI.3213-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Hoffmann JI. Basic science: the circulatory system. In: Rudolph CD, Rudolph AM, Hostetter MK, Lister G, Siegel NJ, editors. Rudolph's pediatrics. Ed 21. Mcgraw-Hill Professional: 2002. pp. 1745–1904. [Google Scholar]
- Horsfield MA, Jones DK. Applications of diffusion-weighted and diffusion tensor MRI to white matter diseases—a review. NMR Biomed. 2002;15:570–577. doi: 10.1002/nbm.787. [DOI] [PubMed] [Google Scholar]
- Hughes EG, Maguire JL, McMinn MT, Scholz RE, Sutherland ML. Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Brain Res Mol Brain Res. 2004;124:114–123. doi: 10.1016/j.molbrainres.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Huppi PS, Dubois J. Diffusion tensor imaging of brain development. Neonatal Med. 2006;11:489–497. doi: 10.1016/j.siny.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Inder T, Huppi P, Zientara G, Maier S, Jolesz F, di Salvo D, Robertson R, Barnes PD, Volpe JJ. Early detection of periventricular leukomalacia by diffusion weighted magnetic resonance imaging techniques. J Pediatr. 1999;134:631–634. doi: 10.1016/s0022-3476(99)70251-9. [DOI] [PubMed] [Google Scholar]
- Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008;93:153–161. doi: 10.1136/adc.2006.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Nishiyama A, Dietrich D. The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci. 2010;30:8320–8331. doi: 10.1523/JNEUROSCI.0854-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y, Shimamoto Y. Human tumor necrosis factor-a augments experimental allergic encephalomyelitis in rats. J Neuroimmunol. 1991;34:159–164. doi: 10.1016/0165-5728(91)90125-q. [DOI] [PubMed] [Google Scholar]
- Levi G, Gallo V, Ciotti MT. Bipotential precursors of putative fibrous astrocytes and oligodendrocytes in rat cerebellar cultures express distinct surface features and “neuron-like” GABA transport. Proc Natl Acad Sci U S A. 1986;83:1504–1508. doi: 10.1073/pnas.83.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JB, Kong J, Nadler M, Xu Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS) Glia. 1999;28:215–224. [PubMed] [Google Scholar]
- Li A, Ly S, Yu S, Zhang Y, Ma H, Zhao H, Piao H, Li S, Zhang N, Sun C. Simvastatin attenuates hypomyelination induced by hypoxia-ischemia in neonatal rats. 2010;32:945–952. doi: 10.1179/016164110X12670144737774. [DOI] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Liu X, Bolteus AJ, Balkin DM, Henschel O, Bordey A. GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia. 2006;54:394–410. doi: 10.1002/glia.20392. [DOI] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Ibarretxe G, Sanchez-Gomez MV. Excitotoxicity in glial cells. Eur J Pharmacology. 2002;447:239–246. doi: 10.1016/s0014-2999(02)01847-2. [DOI] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, Sánchez-Gómez MV, Pérez-Samartín A, Rodríguez-Antigüedad A, Pérez-Cerdá F. Exitotoxic damage to white matter. J Anat. 2007;210:693–702. doi: 10.1111/j.1469-7580.2007.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles VJ, Martınez-Lopez I, Zaragoza R, Borras E, Garcia C, Pallardo FV, Vina JR. Na+ dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) in primary astrocyte cultures: effect of oxidative stress. Brain Res. 2001;922:21–29. doi: 10.1016/s0006-8993(01)03124-9. [DOI] [PubMed] [Google Scholar]
- Miyake T, Kitamura T. Glutamine synthetase immunoreactivity in two types of mouse brain glial cells. Brain Res. 1992;586:53–60. doi: 10.1016/0006-8993(92)91370-t. [DOI] [PubMed] [Google Scholar]
- Montana V, Ni Y, Sunjara V, Hua X, Parpura V. Vesicular glutamate transporter-dependent glutamate release from astrocytes. J Neurosci. 2004;24:2633–2642. doi: 10.1523/JNEUROSCI.3770-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley M. Diffusion tensor imaging and aging—a review. NMR Biomed. 2002;15:553–560. doi: 10.1002/nbm.785. [DOI] [PubMed] [Google Scholar]
- Narumi S, Kimelberg HK, Bourke RS. Effects of norepinephrine on the morphology and some enzyme activities of primary monolayer cultures from rat brain. J Neurochem. 1978;31:1479–1490. doi: 10.1111/j.1471-4159.1978.tb06575.x. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 2001;33:72–86. [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajevic S, Pierpaoli C. Color schemes to represent the orientation of anisotropic tissues from diffusion tensor data: application to white matter fiber tract mapping in the human brain. Magn Reson Med. 1999;42:526–540. [PubMed] [Google Scholar]
- Pierson CR, Folkerth RD, Billards SS, Trachtenberg FL, Drinkwater ME, Volpe JJ. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007;114:619–631. doi: 10.1007/s00401-007-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S, Petelenz K, Li Q, Cohen ML, Dechant A, Tabrizi N, Bucek M, Lust D, Miller RH. Developmental changes induced by graded prenatal systemic hypoxic-ischemic insults in rats. Neurobiol Dis. 2005;18:568–581. doi: 10.1016/j.nbd.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR. Glutamate transporter coupling to Na,K-ATPase. J Neurosci. 2009;29:8143–8255. doi: 10.1523/JNEUROSCI.1081-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostworowski M, Balasingam V, Chabot S, Owens T, Yong VW. Astrogliosis in the neonatal and adult murine brain post-trauma: elevation of inflammatory cytokines and the lack of requirement for endogenous interferon-gamma. J Neurosci. 1997;17:3664–3674. doi: 10.1523/JNEUROSCI.17-10-03664.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, Saliba E. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. 2006;59:428–433. doi: 10.1203/01.pdr.0000199905.08848.55. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gomez MV, Alberdi E, Ibarretxe G, Torre I, Matute C. Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci. 2003;23:9519–9528. doi: 10.1523/JNEUROSCI.23-29-09519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi J, Fagel DM, Ment LR, Vaccarino FM. Modeling premature brain injury. Int J Dev Neurosci. 2009;27:863–871. doi: 10.1016/j.ijdevneu.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A, Waagepetersen HS. Role of astrocytes in glutamate homeostasis: implications for excitotoxicity. Neurotox Res. 2005;8:221–225. doi: 10.1007/BF03033975. [DOI] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS, Back SA. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:520–530. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro DL. Morphological and biochemical alterations in foetal rat brain cells cultured in the presence of monobutyryl cyclic AMP. Nature. 1973;241:203–204. doi: 10.1038/241203a0. [DOI] [PubMed] [Google Scholar]
- Silverstein FS, Buchanan K, Johnston MV. Perinatal hypoxia-ischemia disrupts striatal high-affinity [3H]glutamate uptake into synaptosomes. J Neurochem. 1986;47:1614–1619. doi: 10.1111/j.1471-4159.1986.tb00803.x. [DOI] [PubMed] [Google Scholar]
- Sullivan SM, Lee A, Bjorkman ST, Miller SM, Sullivann RK, Poronnik P, Colditz PB, Pow DV. Cytoskeletal anchoring of GLAST determines susceptibility to brain damage: an identified role for GFAP. J Biol Chem. 2007;282:29414–29423. doi: 10.1074/jbc.M704152200. [DOI] [PubMed] [Google Scholar]
- Taglialatela G, Perez-Polo JR, Rassin DK. Induction of apoptosis in the CNS during development by the combination of hyperoxia and inhibition of glutathione synthesis. Free Radic Biol Med. 1998;25:936–942. doi: 10.1016/s0891-5849(98)00131-2. [DOI] [PubMed] [Google Scholar]
- Tilluex S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85:2059–2070. doi: 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rizzini BL, Rossi D, Haugeto O, Racagni G, Danbolt NC, Volterra A. Neuronal and glial glutamate transporters posses an SH-based redox regulatory mechanism. Eur J Neurosci. 1997;9:1236–1243. doi: 10.1111/j.1460-9568.1997.tb01478.x. [DOI] [PubMed] [Google Scholar]
- Tzarouchi LC, Astrakas LG, Zikou A, Xydis V, Kosta P, Andronikou S, Argyropoulou MI. Periventricular leukomalacia in preterm children: assessment of grey and white matter and cerebrospinal fluid changes by MRI. Pediatr Radiol. 2009;39:1327–1332. doi: 10.1007/s00247-009-1389-0. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Hypoxic ischemic encephalopathy: periventricular leucomalacia. In: Volpe JJ, editor. Neurology of the newborn. Ed 5. Philadelphia: Elsevier; 2008. pp. 360–379. [Google Scholar]
- Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner T, Böntert M, Eyüpoglu I, Prass K, Prinz M, Klett FF, Heinze M, Bechmann I, Nitsch R, Kirchhoff F, Kettenmann H, Dirnagl U, Priller J. ) Bone marrow-derived cells expressing green fluorescent protein under the control of the glial fibrillary acidic protein promoter do not differentiate into astrocytes in vitro and in vivo. J Neurosci. 2003;23:5004–5011. doi: 10.1523/JNEUROSCI.23-12-05004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller ML, Stone IM, Goss A, Rau T, Rova C, Poulsen DJ. Selective overexpression of excitatory amino acid transporters 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience. 2008;155:1204–1211. doi: 10.1016/j.neuroscience.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wocadlo C, Rieger I. Motor impairment and low achievement in very preterm children at eight years of age. Early Hum Dev. 2008;84:769–776. doi: 10.1016/j.earlhumdev.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Xie M, Tobin JE, Budde MD, Chen CI, Trinkaus K, Cross AH, McDaniel DP, Song S-K, Armstrong RC. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol. 2010;69:704–716. doi: 10.1097/NEN.0b013e3181e3de90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Rivkees SA. Hypoglycemia influences oligodendrocyte development and myelin formation. Neuroreport. 2006;17:55–59. doi: 10.1097/01.wnr.0000192733.00535.b6. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Modulation of glial glutamate transport through cell interactions with the extracellular matrix. Int J Dev Neurosci. 2002;20:209–217. doi: 10.1016/s0736-5748(02)00048-5. [DOI] [PubMed] [Google Scholar]
- Yeh TH, Hwang HM, Chen JJ, Wu T, Li AH, Wang HL. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol Dis. 2005;18:473–483. doi: 10.1016/j.nbd.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Yuan X, Eisen AM, McBain CJ, Gallo V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development. 1998;125:2901–2914. doi: 10.1242/dev.125.15.2901. [DOI] [PubMed] [Google Scholar]
- Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res. 2002;70:529–545. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]
- Zhao C, Fancy SP, Magy L, Urwin JE, Franklin RJ. Stem cells, progenitors and myelin repair. J Anat. 2005;207:251–258. doi: 10.1111/j.1469-7580.2005.00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zugno AI, Oliveira DL, Scherer EB, Wajner M, Wofchuk S, Wyse AT. Guanidinoacetate inhibits glutamate uptake in rat striatum of rats at different ages. Neurochem Res. 2007;32:959–964. doi: 10.1007/s11064-006-9245-1. [DOI] [PubMed] [Google Scholar]